
Inhibition of VHL by VH298 Accelerates Pexophagy by Activation of HIF-1α in HeLa Cells
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
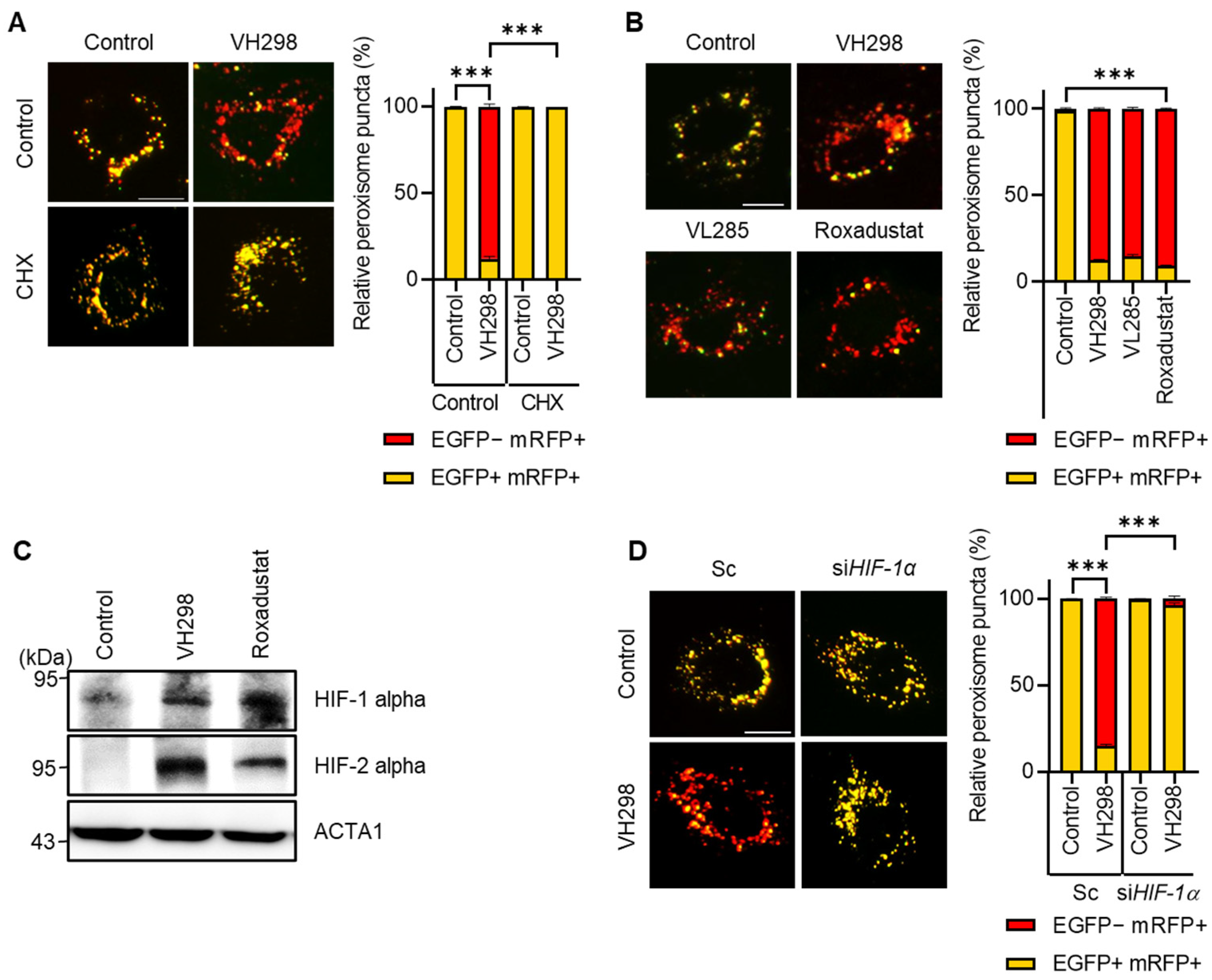
2. Results
2.1. Inhibition of VHL Promotes Pexophagy
2.2. Depletion of ATG5 or NBR1 Impedes VH298-Induced Peroxisomal Autophagy in HeLa Cells
2.3. VH298 Promotes Pexophagy by Enhancing the Transcriptional Activation of HIF-1α in RPE Cells
3. Discussion
4. Materials and Methods
4.1. Reagents, siRNAs and Plasmids
4.2. Cell Culture and Establishment of Stable Cell Lines
4.3. Confocal Microscopy
4.4. Determination of Pexophagic Cells
4.5. Quantification of ABCD3-UB Colocalization
4.6. Western Blotting
4.7. Immunoprecipitation
4.8. Quantification and Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Jo, D.S.; Park, N.Y.; Cho, D.H. Peroxisome quality control and dysregulated lipid metabolism in neurodegenerative diseases. Exp. Mol. Med. 2020, 52, 1486–1495. [Google Scholar] [CrossRef]
- Titorenko, V.I.; Rachubinski, R.A. The life cycle of the peroxisome. Nat. Rev. Mol. Cell Biol. 2001, 2, 357–368. [Google Scholar] [CrossRef] [PubMed]
- Fransen, M.; Nordgren, M.; Wang, B.; Apanasets, O. Role of peroxisomes in ROS/RNS-metabolism: Implications for human disease. Biochim. Biophys. Acta 2012, 1822, 1363–1373. [Google Scholar] [CrossRef] [PubMed]
- Uzor, N.E.; McCullough, L.D.; Tsvetkov, A.S. Peroxisomal Dysfunction in Neurological Diseases and Brain Aging. Front. Cell Neurosci. 2020, 14, 44. [Google Scholar] [CrossRef] [PubMed]
- Delille, H.K.; Bonekamp, N.A.; Schrader, M. Peroxisomes and disease—An overview. Int. J. Biomed. Sci. 2006, 2, 308–314. [Google Scholar] [PubMed]
- Kim, J.; Bai, H. Peroxisomal Stress Response and Inter-Organelle Communication in Cellular Homeostasis and Aging. Antioxidants 2022, 11, 192. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.J.; Aitchison, J.D. Peroxisomes take shape. Nat. Rev. Mol. Cell Biol. 2013, 14, 803–817. [Google Scholar] [CrossRef]
- Weller, S.; Gould, S.J.; Valle, D. Peroxisome biogenesis disorders. Annu. Rev. Genom. Hum. Genet. 2003, 4, 165–211. [Google Scholar] [CrossRef]
- Braverman, N.E.; Raymond, G.V.; Rizzo, W.B.; Moser, A.B.; Wilkinson, M.E.; Stone, E.M.; Steinberg, S.J.; Wangler, M.F.; Rush, E.T.; Hacia, J.G.; et al. Peroxisome biogenesis disorders in the Zellweger spectrum: An overview of current diagnosis, clinical manifestations, and treatment guidelines. Mol. Genet. Metab. 2016, 117, 313–321. [Google Scholar] [CrossRef]
- Braverman, N.; Dodt, G.; Gould, S.J.; Valle, D. Disorders of peroxisome biogenesis. Hum. Mol. Genet. 1995, 4, 1791–1798. [Google Scholar] [CrossRef]
- Di Cara, F.; Andreoletti, P.; Trompier, D.; Vejux, A.; Bulow, M.H.; Sellin, J.; Lizard, G.; Cherkaoui-Malki, M.; Savary, S. Peroxisomes in Immune Response and Inflammation. Int. J. Mol. Sci. 2019, 20, 3877. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J.; Abdel-Aziz, A.K.; Abdelfatah, S.; Abdellatif, M.; Abdoli, A.; Abel, S.; Abeliovich, H.; Abildgaard, M.H.; Abudu, Y.P.; Acevedo-Arozena, A.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (4th edition)(1). Autophagy 2021, 17, 1–382. [Google Scholar] [CrossRef]
- Parzych, K.R.; Klionsky, D.J. An overview of autophagy: Morphology, mechanism, and regulation. Antioxid. Redox Signal 2014, 20, 460–473. [Google Scholar] [CrossRef] [PubMed]
- Vargas, J.N.S.; Hamasaki, M.; Kawabata, T.; Youle, R.J.; Yoshimori, T. The mechanisms and roles of selective autophagy in mammals. Nat. Rev. Mol. Cell Biol. 2023, 24, 167–185. [Google Scholar] [CrossRef]
- Yoshii, S.R.; Mizushima, N. Monitoring and Measuring Autophagy. Int. J. Mol. Sci. 2017, 18, 1865. [Google Scholar] [CrossRef]
- Sargent, G.; van Zutphen, T.; Shatseva, T.; Zhang, L.; Di Giovanni, V.; Bandsma, R.; Kim, P.K. PEX2 is the E3 ubiquitin ligase required for pexophagy during starvation. J. Cell Biol. 2016, 214, 677–690. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.H.; Chang, Y.J.; Lin, S.; Yang, W.Y. Hsc70/Stub1 promotes the removal of individual oxidatively stressed peroxisomes. Nat. Commun. 2020, 11, 5267. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Chen, X.; Liu, Q.; Zhong, G.; Zhuang, M. Ubiquitin ligase MARCH5 localizes to peroxisomes to regulate pexophagy. J. Cell Biol. 2022, 221, 3156. [Google Scholar] [CrossRef]
- Walter, K.M.; Schonenberger, M.J.; Trotzmuller, M.; Horn, M.; Elsasser, H.P.; Moser, A.B.; Lucas, M.S.; Schwarz, T.; Gerber, P.A.; Faust, P.L.; et al. Hif-2alpha promotes degradation of mammalian peroxisomes by selective autophagy. Cell Metab. 2014, 20, 882–897. [Google Scholar] [CrossRef]
- Law, K.B.; Bronte-Tinkew, D.; Di Pietro, E.; Snowden, A.; Jones, R.O.; Moser, A.; Brumell, J.H.; Braverman, N.; Kim, P.K. The peroxisomal AAA ATPase complex prevents pexophagy and development of peroxisome biogenesis disorders. Autophagy 2017, 13, 868–884. [Google Scholar] [CrossRef]
- Zhang, J.; Tripathi, D.N.; Jing, J.; Alexander, A.; Kim, J.; Powell, R.T.; Dere, R.; Tait-Mulder, J.; Lee, J.H.; Paull, T.T.; et al. ATM functions at the peroxisome to induce pexophagy in response to ROS. Nat. Cell Biol. 2015, 17, 1259–1269. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.H.; Jo, D.S.; Park, N.Y.; Bae, J.E.; Kim, J.B.; Lee, H.J.; Kim, S.H.; Kim, S.H.; Lee, S.; Son, M.; et al. Inhibition of BRD4 Promotes Pexophagy by Increasing ROS and ATM Activation. Cells 2022, 11, 2839. [Google Scholar] [CrossRef]
- Riccio, V.; Demers, N.; Hua, R.; Vissa, M.; Cheng, D.T.; Strilchuk, A.W.; Wang, Y.; McQuibban, G.A.; Kim, P.K. Deubiquitinating enzyme USP30 maintains basal peroxisome abundance by regulating pexophagy. J. Cell Biol. 2019, 218, 798–807. [Google Scholar] [CrossRef] [PubMed]
- Deosaran, E.; Larsen, K.B.; Hua, R.; Sargent, G.; Wang, Y.; Kim, S.; Lamark, T.; Jauregui, M.; Law, K.; Lippincott-Schwartz, J.; et al. NBR1 acts as an autophagy receptor for peroxisomes. J. Cell Sci. 2013, 126, 939–952. [Google Scholar] [CrossRef]
- Germain, K.; Kim, P.K. Pexophagy: A Model for Selective Autophagy. Int. J. Mol. Sci. 2020, 21, 578. [Google Scholar] [CrossRef]
- Shaid, S.; Brandts, C.H.; Serve, H.; Dikic, I. Ubiquitination and selective autophagy. Cell Death Differ. 2013, 20, 21–30. [Google Scholar] [CrossRef]
- Gatica, D.; Lahiri, V.; Klionsky, D.J. Cargo recognition and degradation by selective autophagy. Nat. Cell Biol. 2018, 20, 233–242. [Google Scholar] [CrossRef] [PubMed]
- Jin, A.; Lee, J.N.; Kim, M.S.; Kwak, S.; Kim, S.J.; Song, K.; Choe, S.K.; Park, R. 2,2’-dipyridyl induces pexophagy. Biochem. Biophys. Res. Commun. 2016, 469, 941–947. [Google Scholar] [CrossRef]
- Hirota, Y.; Yamashita, S.; Kurihara, Y.; Jin, X.; Aihara, M.; Saigusa, T.; Kang, D.; Kanki, T. Mitophagy is primarily due to alternative autophagy and requires the MAPK1 and MAPK14 signaling pathways. Autophagy 2015, 11, 332–343. [Google Scholar] [CrossRef]
- Su, K.; Li, Z.; Yu, Y.; Zhang, X. The prolyl hydroxylase inhibitor roxadustat: Paradigm in drug discovery and prospects for clinical application beyond anemia. Drug Discov. Today 2020, 25, 1262–1269. [Google Scholar] [CrossRef]
- Chen, W.; Hill, H.; Christie, A.; Kim, M.S.; Holloman, E.; Pavia-Jimenez, A.; Homayoun, F.; Ma, Y.; Patel, N.; Yell, P.; et al. Targeting renal cell carcinoma with a HIF-2 antagonist. Nature 2016, 539, 112–117. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.; Du, X.; Rizzi, J.P.; Liberzon, E.; Chakraborty, A.A.; Gao, W.; Carvo, I.; Signoretti, S.; Bruick, R.K.; Josey, J.A.; et al. On-target efficacy of a HIF-2alpha antagonist in preclinical kidney cancer models. Nature 2016, 539, 107–111. [Google Scholar] [CrossRef] [PubMed]
- Dengler, V.L.; Galbraith, M.; Espinosa, J.M. Transcriptional regulation by hypoxia inducible factors. Crit. Rev. Biochem. Mol. Biol. 2014, 49, 1–15. [Google Scholar] [CrossRef]
- Semenza, G.L. Hypoxia-inducible factors in physiology and medicine. Cell 2012, 148, 399–408. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; Yu, H.; Yan, N.; Lai, K.; Xiang, M. Hypoxia-Inducible Factor-1alpha Target Genes Contribute to Retinal Neuroprotection. Front. Cell Neurosci. 2017, 11, 20. [Google Scholar] [CrossRef] [PubMed]
- Mu, Y.; Maharjan, Y.; Dutta, R.K.; Kim, H.; Wei, X.; Kim, J.H.; Kim, D.; Park, C.; Park, R. Dimethyloxaloylglycine induces pexophagy in a HIF-2alpha dependent manner involving autophagy receptor p62. Biochem. Biophys. Res. Commun. 2020, 525, 46–52. [Google Scholar] [CrossRef]
- Hoppe, G.; Yoon, S.; Gopalan, B.; Savage, A.R.; Brown, R.; Case, K.; Vasanji, A.; Chan, E.R.; Silver, R.B.; Sears, J.E. Comparative systems pharmacology of HIF stabilization in the prevention of retinopathy of prematurity. Proc. Natl. Acad. Sci. USA 2016, 113, E2516–E2525. [Google Scholar] [CrossRef]
- Yu, J.; Wang, S.; Shi, W.; Zhou, W.; Niu, Y.; Huang, S.; Zhang, Y.; Zhang, A.; Jia, Z. Roxadustat prevents Ang II hypertension by targeting angiotensin receptors and eNOS. JCI Insight 2021, 6, e133690. [Google Scholar] [CrossRef]
- Bruick, R.K. Expression of the gene encoding the proapoptotic Nip3 protein is induced by hypoxia. Proc. Natl. Acad. Sci. USA 2000, 97, 9082–9087. [Google Scholar] [CrossRef]
- Esteban-Martinez, L.; Boya, P. BNIP3L/NIX-dependent mitophagy regulates cell differentiation via metabolic reprogramming. Autophagy 2018, 14, 915–917. [Google Scholar] [CrossRef]
- Guo, Y. Role of HIF-1a in regulating autophagic cell survival during cerebral ischemia reperfusion in rats. Oncotarget 2017, 8, 98482–98494. [Google Scholar] [CrossRef]
- Wang, K.; Chen, Y.S.; Chien, H.W.; Chiou, H.L.; Yang, S.F.; Hsieh, Y.H. Melatonin inhibits NaIO(3)-induced ARPE-19 cell apoptosis via suppression of HIF-1alpha/BNIP3-LC3B/mitophagy signaling. Cell Biosci. 2022, 12, 133. [Google Scholar] [CrossRef] [PubMed]
- Wilhelm, L.P.; Zapata-Munoz, J.; Villarejo-Zori, B.; Pellegrin, S.; Freire, C.M.; Toye, A.M.; Boya, P.; Ganley, I.G. BNIP3L/NIX regulates both mitophagy and pexophagy. EMBO J. 2022, 41, e111115. [Google Scholar] [CrossRef]
- Wang, C.; Youle, R. Mitochondria and peroxisomes are NIXed for clearance. EMBO J. 2022, 41, e112918. [Google Scholar] [CrossRef] [PubMed]
- Chandel, N.S.; Tuveson, D.A. The promise and perils of antioxidants for cancer patients. N. Engl. J. Med. 2014, 371, 177–178. [Google Scholar] [CrossRef]
- Daskalaki, I.; Gkikas, I.; Tavernarakis, N. Hypoxia and Selective Autophagy in Cancer Development and Therapy. Front. Cell Dev. Biol. 2018, 6, 104. [Google Scholar] [CrossRef]
- Cho, D.H.; Kim, Y.S.; Jo, D.S.; Choe, S.K.; Jo, E.K. Pexophagy: Molecular Mechanisms and Implications for Health and Diseases. Mol. Cells 2018, 41, 55–64. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, Y.H.; Park, N.Y.; Jo, D.S.; Bae, J.-E.; Kim, J.B.; Park, K.; Jeong, K.; Kim, P.; Yeom, E.; Cho, D.-H. Inhibition of VHL by VH298 Accelerates Pexophagy by Activation of HIF-1α in HeLa Cells. Molecules 2024, 29, 482. https://doi.org/10.3390/molecules29020482
Kim YH, Park NY, Jo DS, Bae J-E, Kim JB, Park K, Jeong K, Kim P, Yeom E, Cho D-H. Inhibition of VHL by VH298 Accelerates Pexophagy by Activation of HIF-1α in HeLa Cells. Molecules. 2024; 29(2):482. https://doi.org/10.3390/molecules29020482
Chicago/Turabian StyleKim, Yong Hwan, Na Yeon Park, Doo Sin Jo, Ji-Eun Bae, Joon Bum Kim, Kyuhee Park, Kwiwan Jeong, Pansoo Kim, Eunbyul Yeom, and Dong-Hyung Cho. 2024. "Inhibition of VHL by VH298 Accelerates Pexophagy by Activation of HIF-1α in HeLa Cells" Molecules 29, no. 2: 482. https://doi.org/10.3390/molecules29020482