

A Simple Analysis of the Second (Extra) Disulfide Bridge of VHHs

 , , , and
, , , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
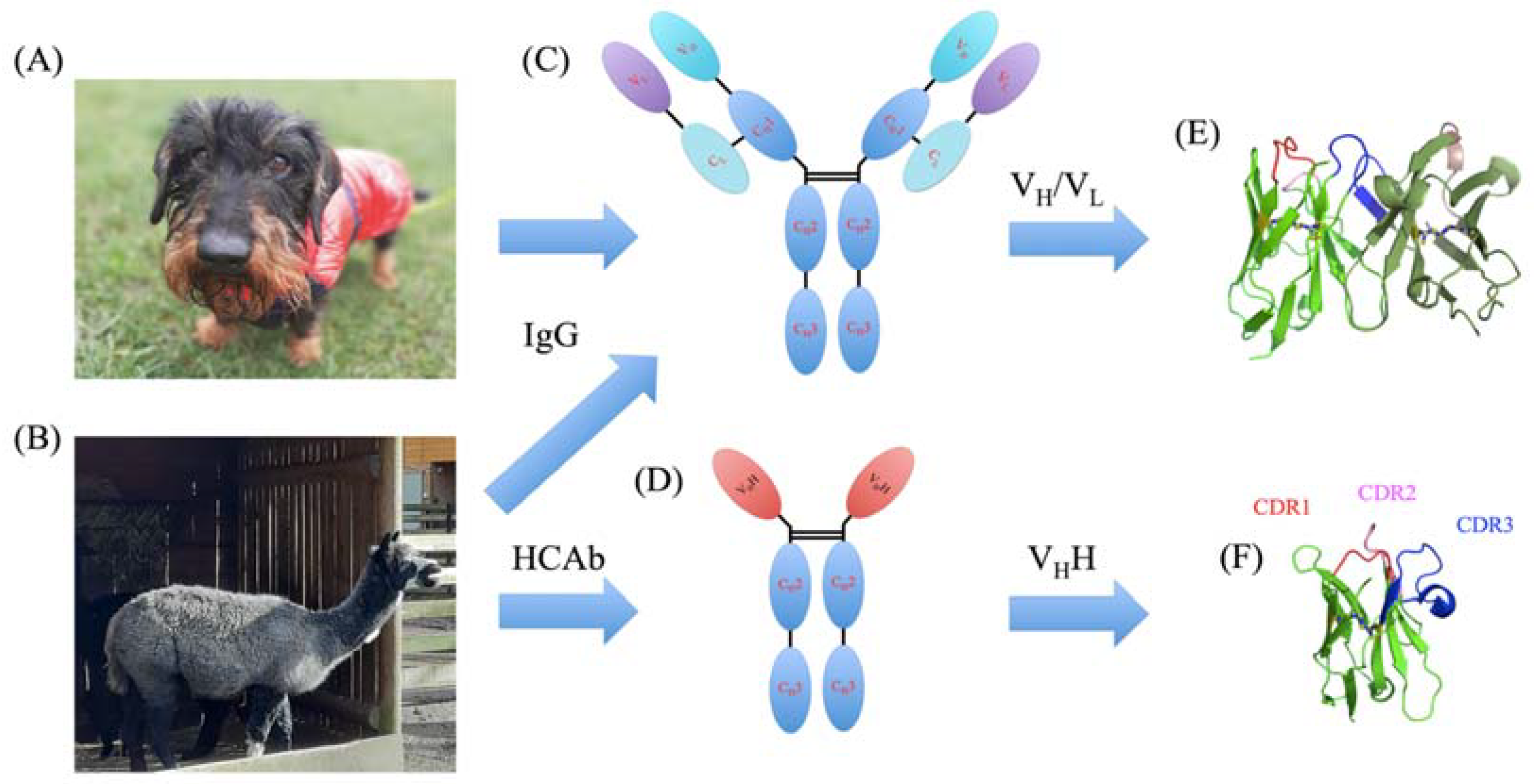
1. Introduction
2. Results
2.1. Systems
2.2. Global Analyses
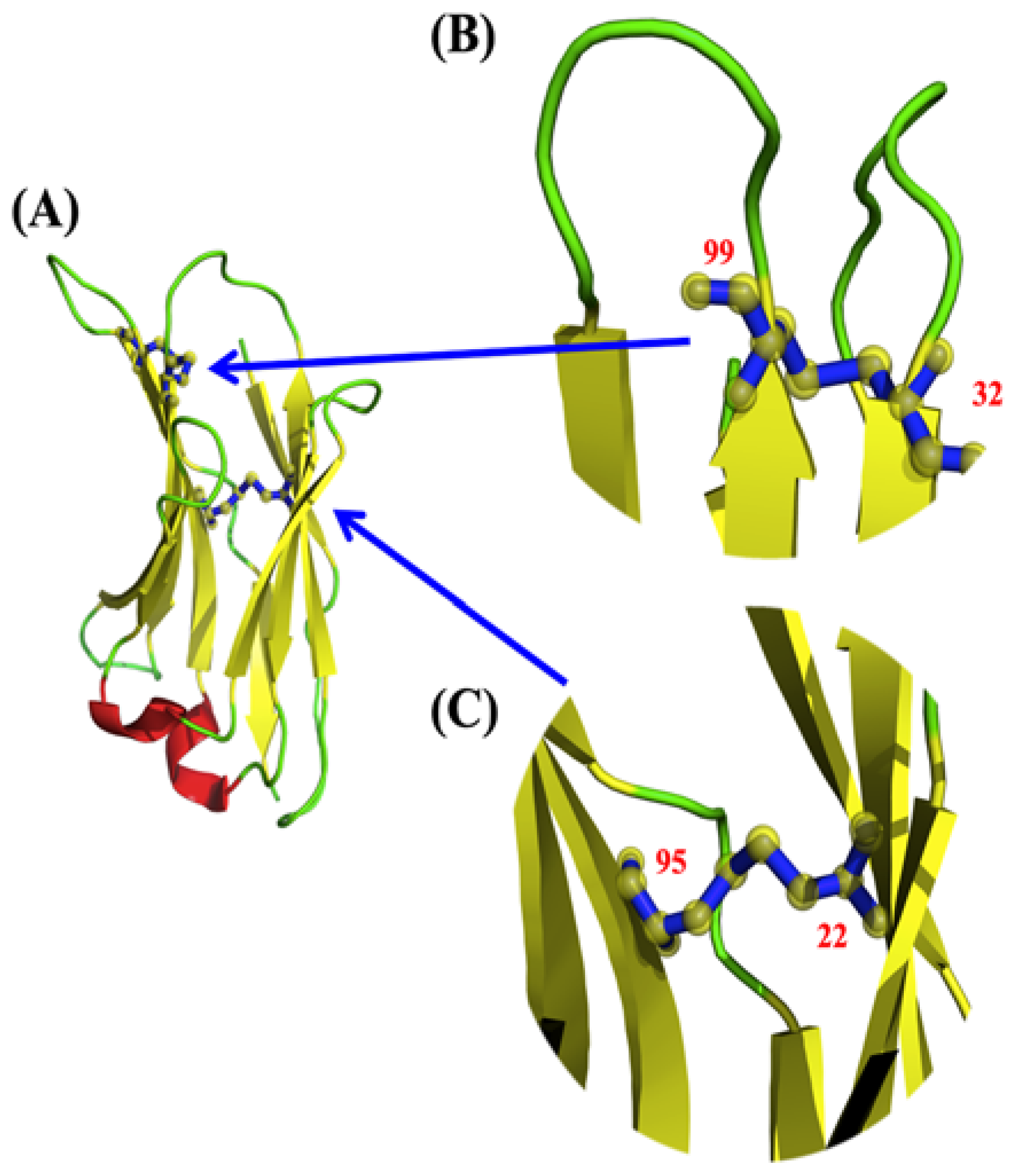
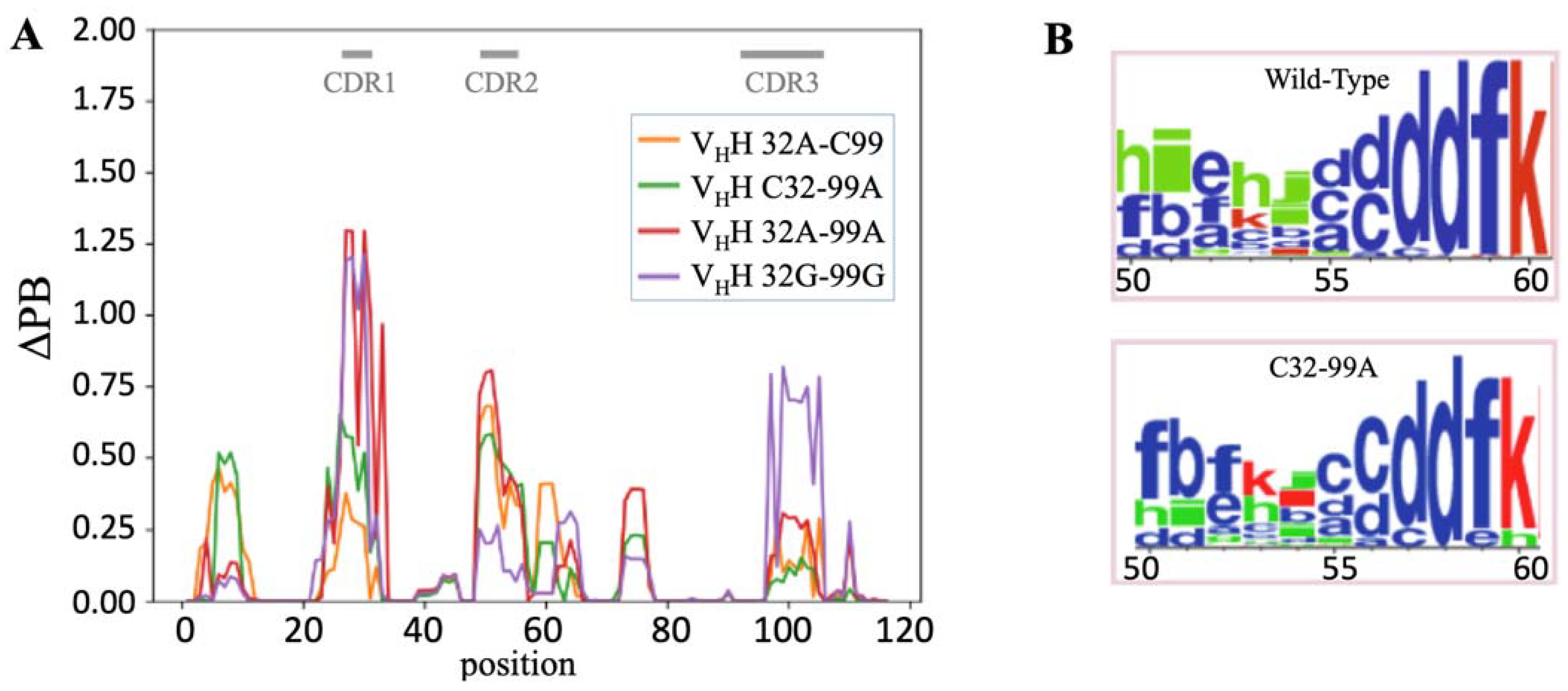
2.3. Local Analyses
2.4. Comparison of Local Protein Conformations
3. Discussion
4. Materials and Methods
4.1. VHH Structure
4.2. Molecular Dynamics
4.3. MDs Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hamers-Casterman, C.; Atarhouch, T.; Muyldermans, S.; Robinson, G.; Hamers, C.; Songa, E.B.; Bendahman, N.; Hamers, R. Naturally occurring antibodies devoid of light chains. Nature 1993, 363, 446–448. [Google Scholar] [CrossRef] [PubMed]
- Roovers, R.C.; van Dongen, G.A.; van Bergen en Henegouwen, P.M. Nanobodies in therapeutic applications. Curr. Opin. Mol. Ther. 2007, 9, 327–335. [Google Scholar] [PubMed]
- Siontorou, C.G. Nanobodies as novel agents for disease diagnosis and therapy. Int. J. Nanomed. 2013, 8, 4215–4227. [Google Scholar] [CrossRef]
- Wernery, U. Camelid immunoglobulins and their importance for the new-born—A review. J. Vet. Med. Ser. B Infect. Dis. Vet. Public Health 2001, 48, 561–568. [Google Scholar]
- Janssens, R.; Dekker, S.; Hendriks, R.W.; Panayotou, G.; van Remoortere, A.; San, J.K.; Grosveld, F.; Drabek, D. Generation of heavy-chain-only antibodies in mice. Proc. Natl. Acad. Sci. USA 2006, 103, 15130–15135. [Google Scholar] [CrossRef]
- De Genst, E.; Saerens, D.; Muyldermans, S.; Conrath, K. Antibody repertoire development in camelids. Dev. Comp. Immunol. 2006, 30, 187–198. [Google Scholar] [CrossRef]
- Omidfar, K.; Rasaee, M.J.; Kashanian, S.; Paknejad, M.; Bathaie, Z. Studies of thermostability in camelus bactrianus (bactrian camel) single-domain antibody specific for the mutant epidermal-growth-factor receptor expressed by pichia. Biotechnol. Appl. Biochem. 2007, 46, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Jin, B.K.; Odongo, S.; Radwanska, M.; Magez, S. Nanobodies®: A review of diagnostic and therapeutic applications. Int. J. Mol. Sci. 2023, 24, 5994. [Google Scholar] [CrossRef]
- Tanaka, T.; Lobato, M.N.; Rabbitts, T.H. Single domain intracellular antibodies: A minimal fragment for direct in vivo selection of antigen-specific intrabodies. J. Mol. Biol. 2003, 331, 1109–1120. [Google Scholar] [CrossRef]
- Delano, W.L. The Pymol Molecular Graphics System on World Wide Web. 2013. Available online: http://www.pymol.org (accessed on 11 February 2022).
- The Pymol Molecular Graphics System, version 1.7.2.2; Schrödinger, LLC: New York, NY, USA, 2015.
- Pymol, version 2.4.0; Schrödinger, LLC: New York, NY, USA, 2020.
- Mitchell, L.S.; Colwell, L.J. Comparative analysis of nanobody sequence and structure data. Proteins 2018, 86, 697–706. [Google Scholar] [CrossRef]
- Guilbaud, A.; Pecorari, F. Construction of synthetic VHH libraries in ribosome display format. Methods Mol. Biol. 2023, 2681, 19–31. [Google Scholar] [PubMed]
- De Greve, H. Production of designer VHH-based antibodies in plants. Methods Mol. Biol. 2022, 2446, 205–230. [Google Scholar] [PubMed]
- Asada, T.; Takagi, D.; Nakai, M.; Abe, S.; Yuasa, K. Secretory production of a camelid single-domain antibody (VHH, nanobody) by the Serratia marcescens lip system in Escherichia coli. Biochem. Biophys. Res. Commun. 2021, 549, 105–112. [Google Scholar] [CrossRef]
- Jia, Q.; Ren, H.; Zhang, S.; Yang, H.; Gao, S.; Fan, R. Preparation and application of Clostridium perfringens alpha toxin nanobodies. Vet. Sci. 2024, 11, 381. [Google Scholar] [CrossRef]
- Saerens, D.; Pellis, M.; Loris, R.; Pardon, E.; Dumoulin, M.; Matagne, A.; Wyns, L.; Muyldermans, S.; Conrath, K. Identification of a universal VHH framework to graft non-canonical antigen-binding loops of camel single-domain antibodies. J. Mol. Biol. 2005, 352, 597–607. [Google Scholar] [CrossRef]
- Wang, M.; Ying, T.; Wu, Y. Single-domain antibodies as therapeutics for solid tumor treatment. Acta Pharm. Sin. B 2024, 14, 2854–2868. [Google Scholar] [CrossRef]
- Kinoshita, S.; Nakakido, M.; Mori, C.; Kuroda, D.; Caaveiro, J.M.M.; Tsumoto, K. Molecular basis for thermal stability and affinity in a VHH: Contribution of the framework region and its influence in the conformation of the CDR3. Protein Sci. Publ. Protein Soc. 2022, 31, e4450. [Google Scholar] [CrossRef] [PubMed]
- Scully, M.; Cataland, S.R.; Peyvandi, F.; Coppo, P.; Knöbl, P.; Kremer Hovinga, J.A.; Metjian, A.; de la Rubia, J.; Pavenski, K.; Callewaert, F.; et al. Caplacizumab treatment for acquired thrombotic thrombocytopenic purpura. N. Engl. J. Med. 2019, 380, 335–346. [Google Scholar] [CrossRef]
- Völker, L.A.; Kaufeld, J.; Balduin, G.; Merkel, L.; Kühne, L.; Eichenauer, D.A.; Osterholt, T.; Hägele, H.; Kann, M.; Grundmann, F.; et al. Impact of first-line use of caplacizumab on treatment outcomes in immune thrombotic thrombocytopenic purpura. J. Thromb. Haemost. 2023, 21, 559–572. [Google Scholar] [CrossRef]
- Scully, M.; de la Rubia, J.; Pavenski, K.; Metjian, A.; Knöbl, P.; Peyvandi, F.; Cataland, S.; Coppo, P.; Kremer Hovinga, J.A.; Minkue Mi Edou, J.; et al. Long-term follow-up of patients treated with caplacizumab and safety and efficacy of repeat caplacizumab use: Post-hercules study. J. Thromb. Haemost. 2022, 20, 2810–2822. [Google Scholar] [CrossRef]
- Keam, S.J. Ozoralizumab: First approval. Drugs 2023, 83, 87–92. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, Y.; Miyazaki, Y.; Kawanishi, M.; Yamasaki, H.; Takeuchi, T. Long-term safety and efficacy of anti-TNF multivalent VHH antibodies ozoralizumab in patients with rheumatoid arthritis. RMD Open 2024, 10, e004480. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.H.; Wang, B.Y.; Chen, L.J.; Fu, W.J.; Xu, J.; Liu, J.; Jin, S.W.; Chen, Y.X.; Cao, X.M.; Yang, Y.; et al. Four-year follow-up of LCAR-B38M in relapsed or refractory multiple myeloma: A phase 1, single-arm, open-label, multicenter study in china (LEGEND-2). J. Hematol. Oncol. 2022, 15, 86. [Google Scholar] [CrossRef]
- Mullard, A. FDA approves second BCMA-targeted CAR-T cell therapy. Nat. Rev. Drug Discov. 2022, 21, 249. [Google Scholar] [CrossRef] [PubMed]
- Milstein, C. The disulphide bridges of immunoglobulin kappa-chains. Biochem. J. 1966, 101, 338–351. [Google Scholar] [CrossRef]
- Pinck, J.R.; Milstein, C. Disulphide bridges of a human immunoglobulin g protein. Nature 1967, 216, 941–942. [Google Scholar] [CrossRef] [PubMed]
- Edelman, G.M.; Cunningham, B.A.; Gall, W.E.; Gottlieb, P.D.; Rutishauser, U.; Waxdal, M.J. The covalent structure of an entire gammag immunoglobulin molecule. Proc. Natl. Acad. Sci. USA 1969, 63, 78–85. [Google Scholar] [CrossRef]
- Amzel, L.M.; Poljak, R.J. Three-dimensional structure of immunoglobulins. Annu. Rev. Biochem. 1979, 48, 961–997. [Google Scholar] [CrossRef]
- Proba, K.; Honegger, A.; Plückthun, A. A natural antibody missing a cysteine in VH: Consequences for thermodynamic stability and folding. J. Mol. Biol. 1997, 265, 161–172. [Google Scholar] [CrossRef]
- Lefranc, M.P.; Pommié, C.; Kaas, Q.; Duprat, E.; Bosc, N.; Guiraudou, D.; Jean, C.; Ruiz, M.; Da Piédade, I.; Rouard, M.; et al. IMGT unique numbering for immunoglobulin and T cell receptor constant domains and Ig superfamily C-like domains. Dev. Comp. Immunol. 2005, 29, 185–203. [Google Scholar] [CrossRef]
- Liu, H.; May, K. Disulfide bond structures of IgG molecules: Structural variations, chemical modifications and possible impacts to stability and biological function. mAbs 2012, 4, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Conrath, K.; Vincke, C.; Stijlemans, B.; Schymkowitz, J.; Decanniere, K.; Wyns, L.; Muyldermans, S.; Loris, R. Antigen binding and solubility effects upon the veneering of a camel VHH in framework-2 to mimic a VH. J. Mol. Biol. 2005, 350, 112–125. [Google Scholar] [CrossRef]
- Nguyen, V.K.; Hamers, R.; Wyns, L.; Muyldermans, S. Camel heavy-chain antibodies: Diverse germline VHH and specific mechanisms enlarge the antigen-binding repertoire. EMBO J. 2000, 19, 921–930. [Google Scholar] [CrossRef]
- Conrath, K.E.; Wernery, U.; Muyldermans, S.; Nguyen, V.K. Emergence and evolution of functional heavy-chain antibodies in camelidae. Dev. Comp. Immunol. 2003, 27, 87–103. [Google Scholar] [CrossRef] [PubMed]
- Eskier, N.E.; Eskier, D.; Firuzan, E.; Uzunlar, S.K. Physicochemical differences between camelid single-domain antibodies and mammalian antibodies. Turk. J. Biol. 2023, 47, 423–436. [Google Scholar] [CrossRef]
- Liu, Y.; Yi, L.; Li, Y.; Wang, Z.; Jirimutu. Characterization of heavy-chain antibody gene repertoires in bactrian camels. J. Genet. Genom. 2023, 50, 38–45. [Google Scholar] [CrossRef]
- Xu, M.; Zhao, Z.; Deng, P.; Sun, M.; Chiu, C.K.C.; Wu, Y.; Wang, H.; Bi, Y. Functional divergence in the affinity and stability of non-canonical cysteines and non-canonical disulfide bonds: Insights from a VHH and VNAR study. Int. J. Mol. Sci. 2024, 25, 9801. [Google Scholar] [CrossRef] [PubMed]
- Harmsen, M.M.; Ruuls, R.C.; Nijman, I.J.; Niewold, T.A.; Frenken, L.G.; de Geus, B. Llama heavy-chain V regions consist of at least four distinct subfamilies revealing novel sequence features. Mol. Immunol. 2000, 37, 579–590. [Google Scholar] [CrossRef]
- Li, X.; Duan, X.; Yang, K.; Zhang, W.; Zhang, C.; Fu, L.; Ren, Z.; Wang, C.; Wu, J.; Lu, R.; et al. Comparative analysis of immune repertoires between bactrian camel’s conventional and heavy-chain antibodies. PLoS ONE 2016, 11, e0161801. [Google Scholar] [CrossRef]
- Melarkode Vattekatte, A.; Shinada, N.K.; Narwani, T.J.; Noel, F.; Bertrand, O.; Meyniel, J.P.; Malpertuy, A.; Gelly, J.C.; Cadet, F.; de Brevern, A.G. Discrete analysis of camelid variable domains: Sequences, structures, and in-silico structure prediction. PeerJ 2020, 8, e8408. [Google Scholar] [CrossRef]
- Mendoza, M.N.; Jian, M.; King, M.T.; Brooks, C.L. Role of a noncanonical disulfide bond in the stability, affinity, and flexibility of a VHH specific for the Listeria virulence factor InlB. Protein Sci. 2020, 29, 1004–1017. [Google Scholar] [CrossRef]
- Liu, H.; Schittny, V.; Nash, M.A. Removal of a conserved disulfide bond does not compromise mechanical stability of a VHH antibody complex. Nano Lett. 2019, 19, 5524–5529. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.; Ó’Fágáin, C.; O’Kennedy, R. Antibody stability: A key to performance—Analysis, influences and improvement. Biochimie 2020, 177, 213–225. [Google Scholar] [CrossRef]
- Thornton, J.M. Disulphide bridges in globular proteins. J. Mol. Biol. 1981, 151, 261–287. [Google Scholar] [CrossRef]
- Thangudu, R.R.; Manoharan, M.; Srinivasan, N.; Cadet, F.; Sowdhamini, R.; Offmann, B. Analysis on conservation of disulphide bonds and their structural features in homologous protein domain families. BMC Struct. Biol. 2008, 8, 55. [Google Scholar] [CrossRef] [PubMed]
- Govaert, J.; Pellis, M.; Deschacht, N.; Vincke, C.; Conrath, K.; Muyldermans, S.; Saerens, D. Dual beneficial effect of interloop disulfide bond for single domain antibody fragments. J. Biol. Chem. 2012, 287, 1970–1979. [Google Scholar] [CrossRef]
- de Brevern, A.G.; Etchebest, C.; Hazout, S. Bayesian probabilistic approach for predicting backbone structures in terms of protein blocks. Proteins 2000, 41, 271–287. [Google Scholar] [CrossRef] [PubMed]
- Barnoud, J.; Santuz, H.; Craveur, P.; Joseph, A.P.; Jallu, V.; de Brevern, A.G.; Poulain, P. Pbxplore: A tool to analyze local protein structure and deformability with protein blocks. PeerJ 2017, 5, e4013. [Google Scholar] [CrossRef]
- Jallu, V.; Poulain, P.; Fuchs, P.F.; Kaplan, C.; de Brevern, A.G. Modeling and molecular dynamics of HPA-1a and -1b polymorphisms: Effects on the structure of the β3 subunit of the αiibβ3 integrin. PLoS ONE 2012, 7, e47304. [Google Scholar] [CrossRef]
- Craveur, P.; Joseph, A.P.; Esque, J.; Narwani, T.J.; Noël, F.; Shinada, N.; Goguet, M.; Leonard, S.; Poulain, P.; Bertrand, O.; et al. Protein flexibility in the light of structural alphabets. Front. Mol. Biosci. 2015, 2, 20. [Google Scholar] [CrossRef]
- Melarkode Vattekatte, A.; Narwani, T.J.; Floch, A.; Maljković, M.; Bisoo, S.; Shinada, N.K.; Kranjc, A.; Gelly, J.C.; Srinivasan, N.; Mitić, N.; et al. A structural entropy index to analyse local conformations in intrinsically disordered proteins. J. Struct. Biol. 2020, 210, 107464. [Google Scholar]
- Venkatachalam, C.M. Stereochemical criteria for polypeptides and proteins. V. Conformation of a system of three linked peptide units. Biopolymers 1968, 6, 1425–1436. [Google Scholar] [CrossRef]
- Venkatachalam, C.M.; Ramachandran, G.N. Conformation of polypeptide chains. Annu. Rev. Biochem. 1969, 38, 45–82. [Google Scholar] [CrossRef]
- Vattekatte, A.M.; Diharce, J.; Rebehmed, J.; Cadet, F.; Gardebien, F.; Etchebest, C.; de Brevern, A.G. General trends of the Camelidae antibody VHHs domain dynamics. Int. J. Mol. Sci. 2023, 24, 4511. [Google Scholar] [CrossRef]
- Goguet, M.; Narwani, T.J.; Petermann, R.; Jallu, V.; de Brevern, A.G. In silico analysis of glanzmann variants of Calf-1 domain of αIIbβ3 integrin revealed dynamic allosteric effect. Sci. Rep. 2017, 7, 8001. [Google Scholar] [CrossRef]
- Crooks, G.E.; Hon, G.; Chandonia, J.M.; Brenner, S.E. Weblogo: A sequence logo generator. Genome Res. 2004, 14, 1188–1190. [Google Scholar] [CrossRef]
- Martins, C.; Diharce, J.; Nadaradjane, A.A.; de Brevern, A.G. Evaluation of the potential impact of in silico humanization on VHH dynamics. Int. J. Mol. Sci. 2023, 24, 14586. [Google Scholar] [CrossRef] [PubMed]
- Vishwakarma, P.; Vattekatte, A.M.; Shinada, N.; Diharce, J.; Martins, C.; Cadet, F.; Gardebien, F.; Etchebest, C.; Nadaradjane, A.A.; de Brevern, A.G. VHH structural modelling approaches: A critical review. Int. J. Mol. Sci. 2022, 23, 3721. [Google Scholar] [CrossRef] [PubMed]
- Melarkode Vattekatte, A.; Cadet, F.; Gelly, J.C.; de Brevern, A.G. Insights into comparative modeling of VHH domains. Int. J. Mol. Sci. 2021, 22, 9771. [Google Scholar] [CrossRef]
- Nadaradjane, A.A.; Diharce, J.; Rebehmed, J.; Cadet, F.; Gardebien, F.; Gelly, J.-C.; Etchebest, C.; de Brevern, A.G. Quality assessment of VHH models. JBSD 2023, 41, 13287–133301. [Google Scholar] [CrossRef] [PubMed]
- Verkhivker, G. Allosteric determinants of the SARS-CoV-2 spike protein binding with nanobodies: Examining mechanisms of mutational escape and sensitivity of the omicron variant. Int. J. Mol. Sci. 2022, 23, 2172. [Google Scholar] [CrossRef] [PubMed]
- Verkhivker, G. Conformational flexibility and local frustration in the functional states of the SARS-CoV-2 spike B.1.1.7 and B.1.351 variants: Mutation-induced allosteric modulation mechanism of functional dynamics and protein stability. Int. J. Mol. Sci. 2022, 23, 1646. [Google Scholar] [CrossRef] [PubMed]
- Omotuyi, O.; Olubiyi, O.; Nash, O.; Afolabi, E.; Oyinloye, B.; Fatumo, S.; Femi-Oyewo, M.; Bogoro, S. SARS-CoV-2 omicron spike glycoprotein receptor binding domain exhibits super-binder ability with ACE2 but not convalescent monoclonal antibody. Comput. Biol. Med. 2022, 142, 105226. [Google Scholar] [CrossRef]
- Fernández-Quintero, M.L.; DeRose, E.F.; Gabel, S.A.; Mueller, G.A.; Liedl, K.R. Nanobody paratope ensembles in solution characterized by md simulations and nmr. Int. J. Mol. Sci. 2022, 23, 5419. [Google Scholar] [CrossRef]
- Verkhivker, G.M.; Agajanian, S.; Oztas, D.Y.; Gupta, G. Atomistic simulations and in silico mutational profiling of protein stability and binding in the SARS-CoV-2 spike protein complexes with nanobodies: Molecular determinants of mutational escape mechanisms. ACS Omega 2021, 6, 26354–26371. [Google Scholar] [CrossRef]
- Soler, M.A.; Medagli, B.; Wang, J.; Oloketuyi, S.; Bajc, G.; Huang, H.; Fortuna, S.; de Marco, A. Effect of humanizing mutations on the stability of the llama single-domain variable region. Biomolecules 2021, 11, 163. [Google Scholar] [CrossRef]
- Ikeuchi, E.; Kuroda, D.; Nakakido, M.; Murakami, A.; Tsumoto, K. Delicate balance among thermal stability, binding affinity, and conformational space explored by single-domain VHH antibodies. Sci. Rep. 2021, 11, 20624. [Google Scholar] [CrossRef]
- Higashida, R.; Matsunaga, Y. Enhanced conformational sampling of nanobody CDR H3 loop by generalized replica-exchange with solute tempering. Life 2021, 11, 1428. [Google Scholar] [CrossRef] [PubMed]
- Bekker, G.J.; Ma, B.; Kamiya, N. Thermal stability of single-domain antibodies estimated by molecular dynamics simulations. Protein Sci. 2019, 28, 429–438. [Google Scholar] [CrossRef]
- Su, Z.Y.; Wang, Y.T. A molecular dynamics simulation of the human lysozyme—Camelid VHH HL6 antibody system. Int. J. Mol. Sci. 2009, 10, 1719–1727. [Google Scholar] [CrossRef]
- Hasannejad-Asl, B.; Hashemzadeh, H.; Pooresmaeil, F.; Dabiri, M.; Pooresmaeil, M.-R.; Ahmadvand, D.; Hosseini, A. Molecular dynamics simulation of the brain-isolated single-domain antibody/nanobody from camels through in vivo phage display screening. Front. Mol. Biosci. 2024, 11, 1414119. [Google Scholar] [CrossRef]
- Gómez-Mulas, A.; Salido, E.; Pey, A.L. Thermodynamic versus kinetic basis for the high conformational stability of nanobodies for therapeutic applications. bioRxiv 2024. [Google Scholar] [CrossRef]
- Kunz, P. Assessing the aggregation propensity of single-domain antibodies upon heat-denaturation employing the ΔTm shift. Methods Mol. Biol. 2022, 2446, 233–244. [Google Scholar] [PubMed]
- Dumoulin, M.; Conrath, K.; Van Meirhaeghe, A.; Meersman, F.; Heremans, K.; Frenken, L.G.; Muyldermans, S.; Wyns, L.; Matagne, A. Single-domain antibody fragments with high conformational stability. Protein Sci. 2002, 11, 500–515. [Google Scholar] [CrossRef]
- Pérez, J.M.; Renisio, J.G.; Prompers, J.J.; van Platerink, C.J.; Cambillau, C.; Darbon, H.; Frenken, L.G. Thermal unfolding of a llama antibody fragment: A two-state reversible process. Biochemistry 2001, 40, 74–83. [Google Scholar] [CrossRef] [PubMed]
- Kunz, P.; Flock, T.; Soler, N.; Zaiss, M.; Vincke, C.; Sterckx, Y.; Kastelic, D.; Muyldermans, S.; Hoheisel, J.D. Exploiting sequence and stability information for directing nanobody stability engineering. Biochim. Biophys. Acta Gen. Subj. 2017, 1861, 2196–2205. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The protein data bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef]
- Williams, C.J.; Headd, J.J.; Moriarty, N.W.; Prisant, M.G.; Videau, L.L.; Deis, L.N.; Verma, V.; Keedy, D.A.; Hintze, B.J.; Chen, V.B.; et al. Molprobity: More and better reference data for improved all-atom structure validation. Protein Sci. 2018, 27, 293–315. [Google Scholar] [CrossRef]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J. Gromacs: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef]
- Huang, J.; MacKerell, A.D., Jr. CHARMM36 all-atom additive protein force field: Validation based on comparison to NMR data. J. Comput. Chem. 2013, 34, 2135–2145. [Google Scholar] [CrossRef]
- Narwani, T.J.; Craveur, P.; Shinada, N.K.; Floch, A.; Santuz, H.; Vattekatte, A.M.; Srinivasan, N.; Rebehmed, J.; Gelly, J.C.; Etchebest, C.; et al. Discrete analyses of protein dynamics. J. Biomol. Struct. Dyn. 2020, 38, 2988–3002. [Google Scholar] [CrossRef] [PubMed]
- Kabsch, W.; Sander, C. Dictionary of protein secondary structure: Pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 1983, 22, 2577–2637. [Google Scholar] [CrossRef] [PubMed]
- Touw, W.G.; Baakman, C.; Black, J.; te Beek, T.A.; Krieger, E.; Joosten, R.P.; Vriend, G. A series of pdb-related databanks for everyday needs. Nucleic Acids Res. 2015, 43, D364–D368. [Google Scholar] [CrossRef]
- de Brevern, A.G. Analysis of protein disorder predictions in the light of a protein structural alphabet. Biomolecules 2020, 10, 1080. [Google Scholar] [CrossRef]
- Offmann, B.; Tyagi, M.; de Brevern, A.G. Local protein structures. Curr. Bioinform. 2007, 3, 165–202. [Google Scholar] [CrossRef]
- Joseph, A.P.; Agarwal, G.; Mahajan, S.; Gelly, J.C.; Swapna, L.S.; Offmann, B.; Cadet, F.; Bornot, A.; Tyagi, M.; Valadié, H. A short survey on protein blocks. Biophys. Rev. 2010, 2, 137–145. [Google Scholar] [CrossRef]
- Joseph, A.P.; Srinivasan, N.; de Brevern, A.G. Improvement of protein structure comparison using a structural alphabet. Biochimie 2011, 93, 1434–1445. [Google Scholar] [CrossRef]
- Léonard, S.; Joseph, A.P.; Srinivasan, N.; Gelly, J.C.; de Brevern, A.G. Mulpba: An efficient multiple protein structure alignment method based on a structural alphabet. J. Biomol. Struct. Dyn. 2014, 32, 661–668. [Google Scholar] [CrossRef] [PubMed]
- Radjasandirane, R.; de Brevern, A.G. Structural and dynamic differences between calreticulin mutants associated with essential thrombocythemia. Biomolecules 2023, 13, 509. [Google Scholar] [CrossRef]
- Vander Meersche, Y.; Cretin, G.; Gheeraert, A.; Gelly, J.C.; Galochkina, T. ATLAS: Protein flexibility description from atomistic molecular dynamics simulations. Nucleic Acids Res. 2024, 52, D384–D392. [Google Scholar] [CrossRef]
- Ladislav, M.; Cerny, J.; Krusek, J.; Horak, M.; Balik, A.; Vyklicky, L. The LILI motif of M3-S2 linkers is a component of the NMDA receptor channel gate. Front. Mol. Neurosci. 2018, 11, 113. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martins, C.; Gardebien, F.; Nadaradjane, A.A.; Diharce, J.; de Brevern, A.G. A Simple Analysis of the Second (Extra) Disulfide Bridge of VHHs. Molecules 2024, 29, 4863. https://doi.org/10.3390/molecules29204863
Martins C, Gardebien F, Nadaradjane AA, Diharce J, de Brevern AG. A Simple Analysis of the Second (Extra) Disulfide Bridge of VHHs. Molecules. 2024; 29(20):4863. https://doi.org/10.3390/molecules29204863
Chicago/Turabian StyleMartins, Carla, Fabrice Gardebien, Aravindan Arun Nadaradjane, Julien Diharce, and Alexandre G. de Brevern. 2024. "A Simple Analysis of the Second (Extra) Disulfide Bridge of VHHs" Molecules 29, no. 20: 4863. https://doi.org/10.3390/molecules29204863
APA StyleMartins, C., Gardebien, F., Nadaradjane, A. A., Diharce, J., & de Brevern, A. G. (2024). A Simple Analysis of the Second (Extra) Disulfide Bridge of VHHs. Molecules, 29(20), 4863. https://doi.org/10.3390/molecules29204863