
Evaluation of the Potential of High-Performance Liquid Chromatography–Inductively Coupled Plasma–Mass Spectrometry for the Determination of Chemical Warfare Agents and Their Toxic Degradation Products


Abstract
:1. Introduction
2. HPLC-ICP-MS in the Determination of Organophosphorus Compounds
2.1. The Importance of Determining Organophosphorus Nerve Agents, Their Hydrolysis Products, and Their Metabolites
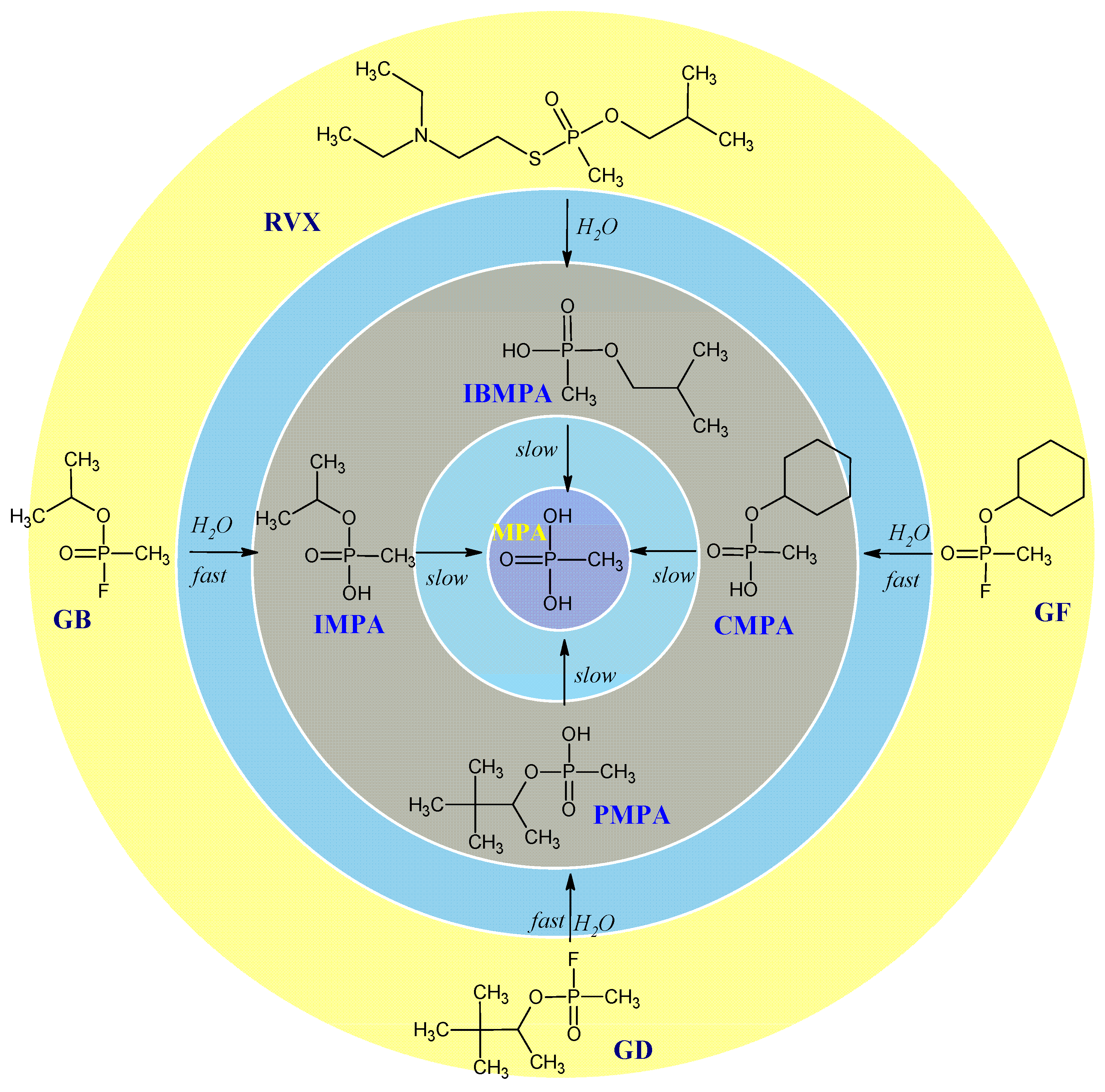
2.2. Hydrolysis of Organophosphorus Nerve Agents
2.3. The Use of the HPLC-ICP-MS Technique for the Determination of Organophosphorus Compounds
3. ICP-MS in the Determination of Organosulfur Compounds
3.1. Importance of Organosulfur Compound Determination
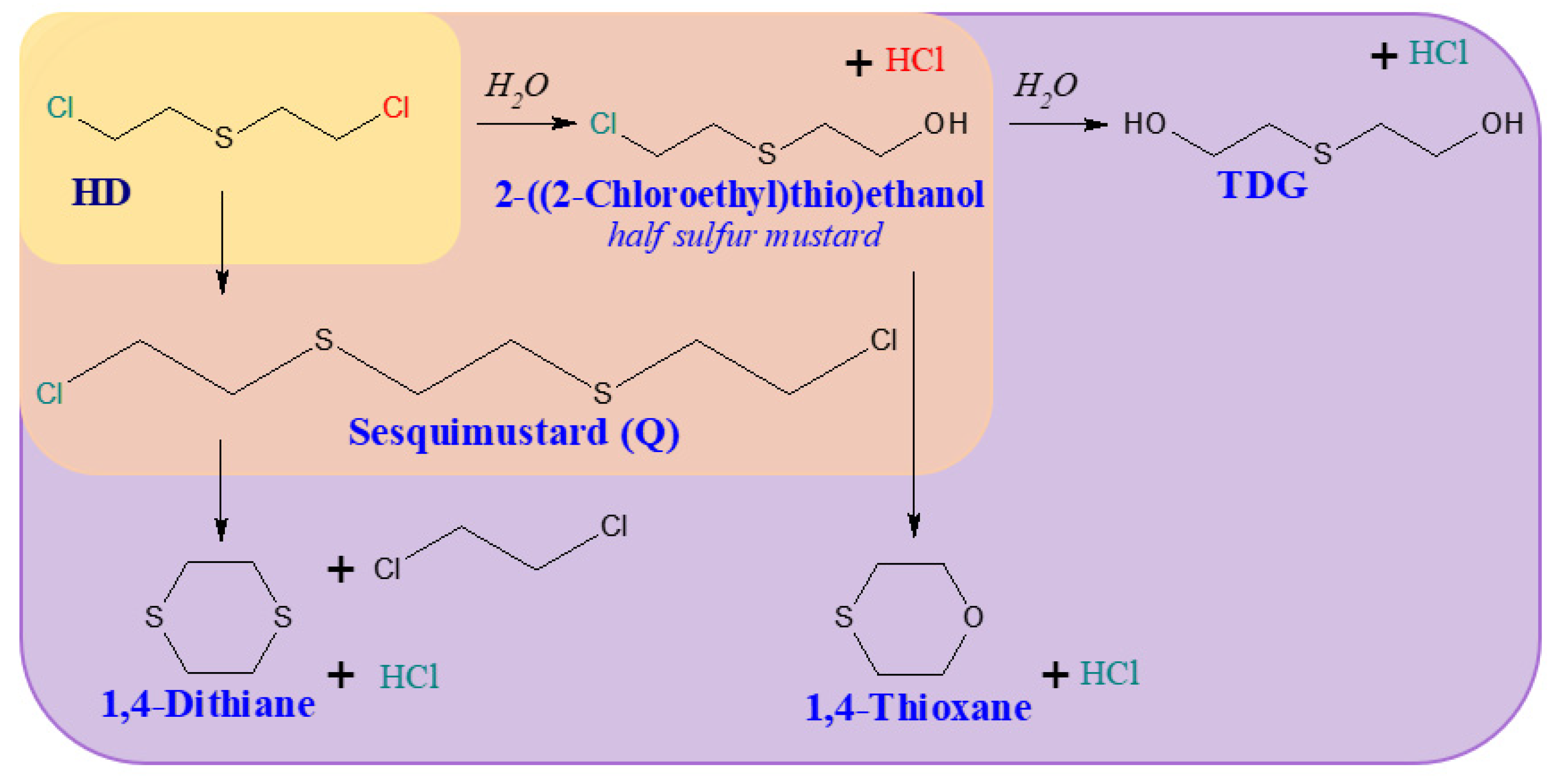
3.2. Degradation Reactions of Organosulfur Compounds
3.3. Use of the ICP-MS Technique to Determine Organosulfur Compounds
4. ICP-MS for the Determination of Organoarsenic Compounds
4.1. Significance of the Determination of Organoarsenic Compounds
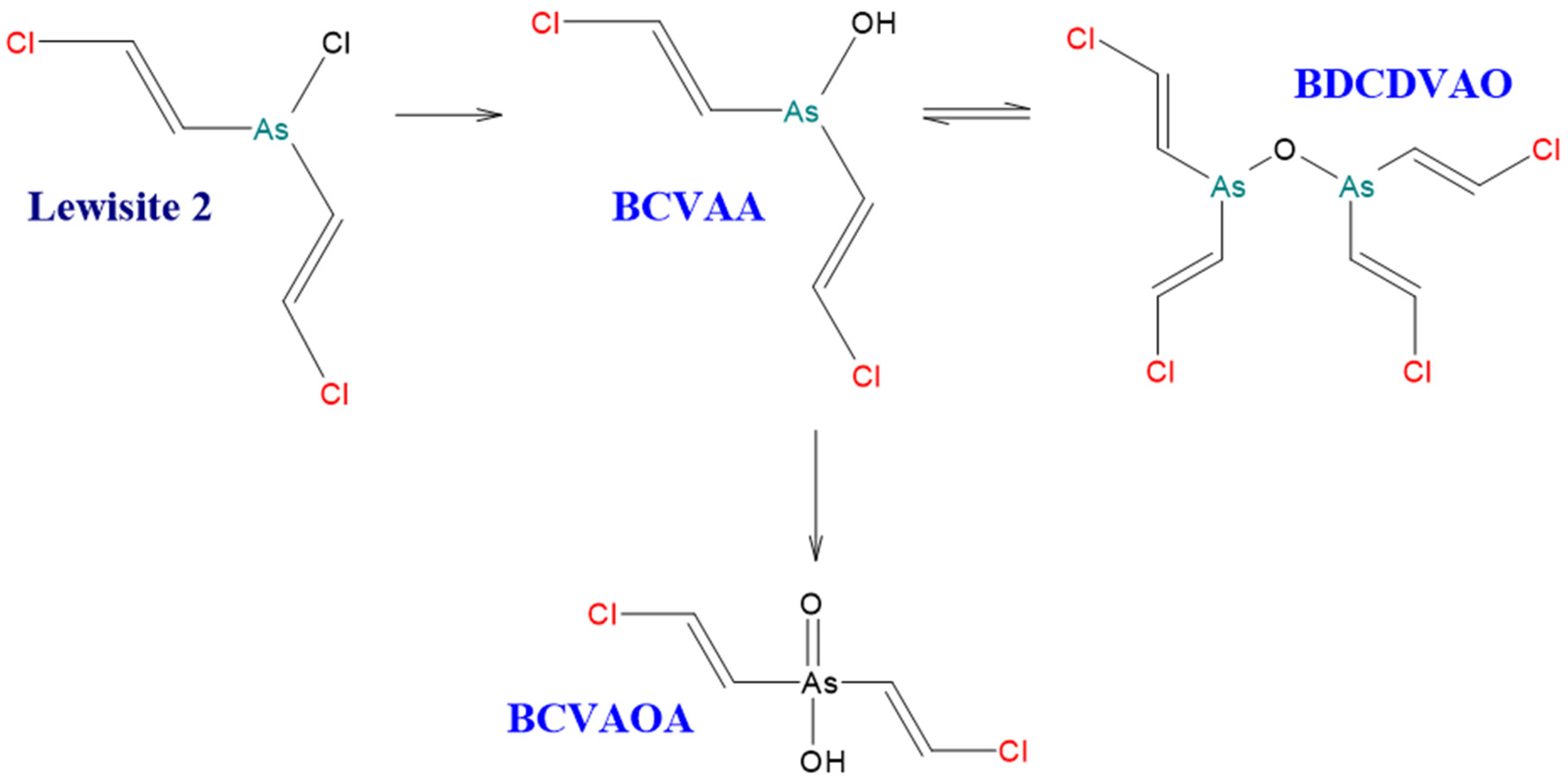
4.2. Degradation Reactions of Organoarsenic Compounds
4.3. The Use of the ICP-MS Technique for the Determination of Organoarsenic Compounds
5. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
Abbreviations and Acronyms
| Acronym and Abbreviation | Full Name | No CAS |
| AB | Arsenobetaine | 64436-13-1 |
| ACh | Acetylcholine | 51-84-3 |
| AChE | Acetylcholinesterase | 9000-81-1 |
| APCI | Atmospheric Pressure Chemical Ionization | - |
| BCVAA | Bis(2-chlorovinyl)arsinous acid | NF |
| BDPAO | Bis(diphenylarsine)oxide | 2215-16-9 |
| BHETBu | 1,4-bis(2-hydroxyethylthio)butane | 7425-93-6 |
| BHETE | 1,2-bis(2-hydroxyethylthio)ethane | 5244-34-8 |
| BHETM | Bis(2-hydroxyethylthio)methane | 44860-68-6 |
| BHETPr | 1,3-bis(2-hydroxyethylthio)propane | 16260-48-3 |
| CCT | Collision-reaction Cell Technology | - |
| CMPA | Cyclohexyl methylphosphonic acid | 1932-60-1 |
| CVAA | 2-chlorovinylarsenous acid | 85090-33-1 |
| CVAO | 2-chlorovinylarsine oxide | 3088-37-7 |
| CVAOA | 2-chlorovinylarsonic acid | 64038-44-4 |
| CWA | Chemical Warfare Agents | - |
| CWC | Chemical Weapons Convention | - |
| DEHP | Diethylhydrogen phosphate | 598-02-7 |
| DMA (III) | Dimethylarsinous acid | 55094-22-9 |
| DMAA | Dimethylarsinic acid | 75-60-5 |
| DMHP | Dimethylhydrogen phosphate | 813-78-5 |
| DMTA (III) | Dimethylthioarsinous acid | NF |
| DMTA (V) | Dimethyldithioarsinic acid (dimethylarsinodithioic acid) | NF |
| DPAA | Diphenylarsinic acid | 6217-24-9 |
| DPCA | Diphenylchloroarsine | 712-48-1 |
| DPCNA | Diphenylcyanoarsine | 23525-22-6 |
| DPMAO | Diphenylmethylarsine oxide | NF |
| DPTA | Diphenylthioarsinic acid | NF |
| DRC | Dynamic reaction cell | - |
| EDTA | Ethylenediaminetetraacetic acid | 60-00-4 |
| EHDAP | Ethyl hydrogen dimethylamidophosphate sodium salt | NF |
| EMPA | Ethyl methylphosphonic acid | 1832-53-7 |
| EMPTA | Ethyl methylphosphonothioic acid | 18005-40-8 |
| EPA | Ethylphosphonic acid | 6779-09-5 |
| EPCN | Ethylphosphorylcyanidate | NF |
| ESI | Electrospray ionization | - |
| GA | O-Ethyl N,N-dimethyl phosphoramidocyanidate (Tabun) | 77-81-6 |
| GB | Propan-2-yl methylphosphonofluoridate (Sarin) | 107-44-8 |
| GC | Gas Chromatography | - |
| GD | 3,3-Dimethylbutan-2-yl methylphosphonofluoridate (Soman) | 96-64-0 |
| GF | Cyclohexyl methylphosphonofluoridate (Cyclosarin) | 329-99-7 |
| GFAAS | Graphite Furnace Atomic Absorption Spectroscopy | - |
| HD | bis(2-chloroethyl)sulfide (Sulfur mustard) | 505-60-2 |
| HPLC | High-Performance Liquid Chromatography | - |
| IBHMP | Isobutyl hydrogen methylphosphonate | 1604-38-2 |
| IC50 | Median inhibitory concentration | - |
| ICP-MS | Inductively Coupled Plasma-Mass Spectrometry | - |
| IMPA | Isopropyl methylphosphonic acid | 1832-54-8 |
| IPHEP | Isopropyl hydrogen ethylphosphonate | 170135-50-9 |
| LC | Liquid Chromatography | - |
| LOD | Limit of detection | - |
| MMA (III) | Monomethylarsonous acid | 25400-23-1 |
| MMA (V) | Monomethylarsonic acid | 124-58-3 |
| MPA | Methylphosphonic acid | 993-13-5 |
| MS | Mass spectrometry | - |
| OES | Optical emission spectroscopy | - |
| OPNA | Organophosphorus Nerve Agents | - |
| OPNA DP | Organophosphorus Nerve Agents Degradation Products | - |
| ORS | Octopole Reaction System | - |
| PAA | Phenylarsonic acid | 98-05-5 |
| PAO | Phenylarsine oxide | 637-03-6 |
| PDCA | Phenyldichloroarsine | 696-28-6 |
| PDMAO | Phenyldimethylarsine oxide | NF |
| PMAA | Phenylmethylarsinic acid | NF |
| PMPA | Pinacolyl methylphosphonic acid | 616-52-4 |
| PPA | Propylphosphonic acid | 4672-38-2 |
| ppb | Parts per billion | - |
| ppm | Parts per million | - |
| ppt | Parts per trillion | - |
| PTE | Phosphotriesterase | - |
| QQQ | Triple quadrupole (mass spectrometer) | - |
| TBDMS | Tert-Butyldimethylsilyl ether | 124150-87-4 |
| TDG | Thiodiglycol | 111-48-8 |
| TMA | Tetramethylarsonium | NF |
| TMAO | Trimethylarsine oxide | 4964-14-1 |
| UV | Ultraviolet | - |
References
- Houk, R.S.; Fassel, V.A.; Flesch, G.D.; Svec, H.J.; Gray, A.L.; Taylor, C.E. Inductively Coupled Argon Plasma as an Ion Source for Mass Spectrometric Determination of Trace Elements. Anal. Chem. 1980, 52, 2283–2289. [Google Scholar] [CrossRef]
- Gray, A.L. Mass-spectrometric Analysis of Solutions Using an Atmospheric Pressure Ion Source. Analyst 1975, 100, 289–299. [Google Scholar] [CrossRef]
- Vanninen, P. (Ed.) Recommended Operating Procedures for Analysis in the Verification of Chemical Disarmament. In The Blue Book; University of Helsinki: Helsinki, Finland, 2017; ISBN 978-951-51-9958-4. [Google Scholar]
- Nawała, J.; Czupryński, K.; Popiel, S.; Dziedzic, D.; Bełdowski, J. Development of the HS-SPME-GC-MS/MS method for analysis of chemical warfare agent and their degradation products in environmental samples. Anal. Chim. Acta 2016, 933, 103–116. [Google Scholar] [CrossRef] [PubMed]
- Valdez, C.A.; Leif, R.N.; Hok, S.; Hart, B.R. Analysis of chemical warfare agents by gas chromatography-mass spectrometry: Methods for their direct detection and derivatization approaches for the analysis of their degradation products. Rev. Anal. Chem. 2018, 37, 20170007. [Google Scholar] [CrossRef]
- Wada, T.; Nagasawa, E.; Hanaoka, S. Simultaneous determination of degradation products related to chemical warfare agents by high-performance liquid chromatography/mass spectrometry. Appl. Organomet. Chem. 2006, 20, 573–579. [Google Scholar] [CrossRef]
- Muir, B.; Slater, B.J.; Cooper, D.B.; Timperley, C.M. Analysis of chemical warfare agents: I. Use of aliphatic thiols in the trace level determination of Lewisite compounds in complex matrices. J. Chromatogr. A 2004, 1028, 313–320. [Google Scholar] [CrossRef]
- Popiel, S.; Sankowska, M. Determination of chemical warfare agents and related compounds in environmental samples by solid-phase microextraction with gas chromatography. J. Chromatogr. A 2011, 1218, 8457–8479. [Google Scholar] [CrossRef]
- Terzic, O.; Swahn, I.; Cretu, G.; Palit, M.; Mallard, G. Gas chromatography-full scan mass spectrometry determination of traces of chemical warfare agents and their impurities in air samples by inlet based thermal desorption of sorbent tubes. J. Chromatogr. A 2012, 1225, 182–192. [Google Scholar] [CrossRef]
- Tørnes, J.A.; Opstad, A.M.; Johnsen, B.A. Determination of organoarsenic warfare agents in sediment samples from Skagerrak by gas chromatography-mass spectrometry. Sci. Total Environ. 2006, 356, 235–246. [Google Scholar] [CrossRef]
- Cheh, M.Y.; Chua, H.C.; Hopkins, F.B.; Riches, J.R.; Timperley, C.M.; Lee, H.S.N. Determination of lewisite constituents in aqueous samples using hollow-fibre liquid-phase microextraction followed by gas chromatography-mass spectrometry. Anal. Bioanal. Chem. 2014, 406, 5103–5110. [Google Scholar] [CrossRef]
- Capoun, T.; Krykorkova, J. Internal Standards for Quantitative Analysis of Chemical Warfare Agents by the GC/MS Method: Nerve Agents. J. Anal. Methods Chem. 2020, 2020, 8857210. [Google Scholar] [CrossRef] [PubMed]
- Wilbur, S. Applications of ICP-MS in Homeland Security. Agil. Technol. 2004, 36, 20–22. Available online: https://www.agilent.com/Library/applications/5989-0741EN.pdf (accessed on 20 April 2024).
- STANAG 4701(1); NATO Standard AEP-66, NATO Handbook for Sampling and Identification of Biological, Chemical and Radiological Agents (SIBCRA), Edition A Version 1. NATO Standardization Agency: Brussels, Belgium, 2015. Available online: http://nso.nato.int/nso/ (accessed on 12 July 2024).
- Blum, M.-M.; Mamidanna, R.V.S.M. Analytical chemistry and the Chemical Weapons Convention. Anal. Bioanal. Chem. 2014, 406, 5067–5069. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, S.; Gupta, R.D. Organophosphorus Nerve Agents: Types, Toxicity, and Treatments. J. Toxicol. 2020, 2020, 3007984. [Google Scholar] [CrossRef] [PubMed]
- DeLuca, M.A.; Chai, P.R.; Goralnick, E.; Erickson, T.B. Five Decades of Global Chemical Terror Attacks: Data Analysis to Inform Training and Preparedness. Disaster Med. Public Health Prep. 2021, 15, 750–761. [Google Scholar] [CrossRef]
- Chai, P.R.; Hayes, B.D.; Erickson, T.B.; Boyer, E.W. Novichok agents: A historical, current, and toxicological perspective. Toxicol. Commun. 2018, 2, 45–48. [Google Scholar] [CrossRef]
- Palkki, D.D.; Rubin, L. Saddam Hussein’s role in the gassing of Halabja. Nonprolif. Rev. 2021, 28, 115–129. [Google Scholar] [CrossRef]
- Hiltermann, J.R. A Poisonous Affair: America, Iraq, and the Gassing of Halabja; Cambridge University Press: Cambridge, UK, 2014; ISBN 9780521876865/0521876869. [Google Scholar]
- Sarin attacks in Japan: Acute and delayed health effects in survivors. In Handbook of Toxicology of Chemical Warfare Agents, 3rd ed.; Gupta, R.C. (Ed.) Academic Press: Cambridge, MA, USA; Elsevier: Amsterdam, The Netherlands, 2020; pp. 37–53. ISBN 9780128190906. [Google Scholar] [CrossRef]
- Okumura, T.; Nomura, T.; Suzuki, T.; Sugita, M.; Takeuchi, Y.; Naito, T.; Okumura, S.; Maekawa, H.; Takasu, N.; Miura, K.; et al. The Dark Morning: The Experiences and Lessons Learned from the Tokyo Subway Sarin Attack. In Chemical Warfare Agents; John Wiley & Sons: Hoboken, NJ, USA, 2007; pp. 277–285. ISBN 9780470013595. [Google Scholar] [CrossRef]
- OPCW. Note by the Technical Secretariat: Report of the OPCW Fact-Finding Mission in Syria Regarding Alleged Incidents in Ltamenah, the Syrian Arab Republic on 24 and 25 March 2018 (S/1636/2018). 2018. Available online: https://www.opcw.org/sites/default/files/documents/S_series/2018/en/s-1636-2018_e_.pdf (accessed on 6 June 2024).
- Tu, A.T. The use of VX as a terrorist agent: Action by Aum Shinrikyo of Japan and the death of Kim Jong-Nam in Malaysia: Four case studies. Glob. Secur. Health Sci. Policy 2020, 5, 48–56. [Google Scholar] [CrossRef]
- Nakagawa, T.; Tu, A.T. Murders with VX: Aum Shinrikyo in Japan and the assassination of Kim Jong-Nam in Malaysia. Forensic Toxicol. 2018, 36, 542–544. [Google Scholar] [CrossRef]
- Butler, D. Attacks in UK and Syria highlight growing need for chemical-forensics expertise. Nature 2018, 556, 285–286. [Google Scholar] [CrossRef]
- Kim, H.K. State Terrorism as a Mechanism for Acts of Violence against Individuals: Case Studies of Kim Jong-Nam, Skripal and Khashoggi Assassinations. J. East Asia Int. Law 2021, 14, 55–78. [Google Scholar] [CrossRef]
- Aroniadou-Anderjaska, V.; Apland, J.P.; Figueiredo, T.H.; De Araujo Furtado, M.; Braga, M.F. Acetylcholinesterase inhibitors (nerve agents) as weapons of mass destruction: History, mechanisms of action, and medical countermeasures. Neuropharmacology 2020, 181, 108298. [Google Scholar] [CrossRef] [PubMed]
- Black, R.M.; Muir, B. Derivatisation reactions in the chromatographic analysis of chemical warfare agents and their degradation products. J. Chromatogr. A 2003, 1000, 253–281. [Google Scholar] [CrossRef] [PubMed]
- Sulaiman, I.S.C.; Chieng, B.W.; Pojol, F.E.; Ong, K.K.; Rashid, J.I.A.; Yunus, W.M.Z.W.; Kasim, N.A.M.; Halim, N.A.; Noor, S.A.M.; Knight, V.F. A review on analysis methods for nerve agent hydrolysis products. Forensic Toxicol. 2020, 38, 297–313. [Google Scholar] [CrossRef]
- Richardson, D.D.; Caruso, J.A. Derivatization of organophosphorus nerve agent degradation products for gas chromatography with ICP-MS and TOF-MS detection. Anal. Bioanal. Chem. 2007, 388, 809–823. [Google Scholar] [CrossRef]
- Valdez, C.A.; Leif, R.N. Analysis of Organophosphorus-Based Nerve Agent Degradation Products by Gas Chromatography-Mass Spectrometry (GC-MS): Current Derivatization Reactions in the Analytical Chemist’s Toolbox. Molecules 2021, 26, 4631. [Google Scholar] [CrossRef]
- Richardson, D.D.; Sadi, B.B.M.; Caruso, J.A. Reversed phase ion-pairing HPLC-ICP-MS for analysis of organophosphorus chemical warfare agent degradation products. J. Anal. At. Spectrom. 2006, 21, 396–403. [Google Scholar] [CrossRef]
- Kubachka, K.M.; Richardson, D.D.; Heitkemper, D.T.; Caruso, J.A. Detection of chemical warfare agent degradation products in foods using liquid chromatography coupled to inductively coupled plasma mass spectrometry and electrospray ionization mass spectrometry. J. Chromatogr. A 2008, 1202, 124–131. [Google Scholar] [CrossRef]
- Zhang, Y.; Kubachka, K.M.; Caruso, J.A. Enhancing determination of organophosphate species in high inorganic phosphate matrices: Application to nerve agent degradation products. Anal. Methods 2010, 2, 1243. [Google Scholar] [CrossRef]
- Coleman, K. History of Chemical Warfare; Palgrave Macmillan: London, UK, 2005. [Google Scholar]
- Duchovic, R.J.; Vilensky, J.A. Mustard Gas: Its Pre-World War I History. J. Chem. Educ. 2007, 84, 944. [Google Scholar] [CrossRef]
- UN. Secretary-General, Report of the Mission Dispatched by the Secretary-General to Investigate Allegations of the Use of Chemical Weapons in the Conflict between the Islamic Republic of Iran and Iraq, S/20060, New York. 1988. Available online: https://digitallibrary.un.org/record/48547/files/S_20060-EN.pdf (accessed on 25 June 2024).
- Rabiee, M.H.; Ghanei, M.; Amini, H.; Akhlaghi, A. Mortality rate of people exposed to Mustard Gas during Iran-Iraq war in Sardasht, Iran: A 32 years retrospective cohort study. BMC Public Health 2022, 22, 1152. [Google Scholar] [CrossRef] [PubMed]
- Abolghasemi, H.; Radfar, M.H.; Rambod, M.; Salehi, P.; Ghofrani, H.; Soroush, M.R.; Falahaty, F.; Tavakolifar, Y.; Sadaghianifar, A.; Khademolhosseini, S.M.; et al. Childhood physical abnormalities following paternal exposure to sulfur mustard gas in Iran: A case-control study. Confl. Health 2010, 4, 13. [Google Scholar] [CrossRef] [PubMed]
- Knobloch, T.; Bełdowski, J.; Böttcher, C.; Söderström, M.; Rühl, N.P.; Sternheim, J. Baltic Marine Environment Protection Commission Chemical Munitions Dumped in the Baltic Sea Report of the ad hoc Expert Group to Update and Review the Existing Information on Dumped Chemical Munitions in the Baltic Sea (HELCOM MUNI), Balt. Sea Environ. Proc. Helsinki ISSN 0357-2994. 2013. Available online: https://helcom.fi/wp-content/uploads/2019/10/Chemical-Munitions-Dumped-in-the-Baltic-Sea-Report-of-the-ad-hoc-Expert-Group.pdf (accessed on 10 June 2024).
- Missiaen, T.; Söderström, M.; Popescu, I.; Vanninen, P. Evaluation of a chemical munition dumpsite in the Baltic Sea based on geophysical and chemical investigations. Sci. Total Environ. 2010, 408, 3536–3553. [Google Scholar] [CrossRef] [PubMed]
- Bełdowski, J.; Klusek, Z.; Szubska, M.; Turja, R.; Bulczak, A.I.; Rak, D.; Brenner, M.; Lang, T.; Kotwicki, L.; Grzelak, K.; et al. Chemical Munitions Search & Assessment-An evaluation of the dumped munitions problem in the Baltic Sea. Deep. Sea Res. Part II Top. Stud. Oceanogr. 2016, 128, 85–95. [Google Scholar] [CrossRef]
- Schlager, N.; Weisblatt, J.; Newton, D.E. Chemical Compounds, Thomson Gale. ISBN 1 4144 0467 0. 2006. Available online: https://rushim.ru/books/spravochniki/chemical-compounds.pdf (accessed on 10 June 2024).
- Hoenig, S.L. Compendium of Chemical Warfare Agents; Springer Science & Business Media, LLC.: Berlin/Heidelberg, Germany, 2007; ISBN 978-0-387-34626-7. [Google Scholar] [CrossRef]
- Røen, B.T.; Trace Determination of Sulphur Mustard and Related Compounds in Environmental Samples by Headspace-Trap GC-MS, Norwegian Defence Research Establishment (FFI), FFI-Rapport 2008/02247. ISBN 978-82-464-1469-0. 2008. Available online: https://ffi-publikasjoner.archive.knowledgearc.net/bitstream/handle/20.500.12242/2250/08-02247.pdf (accessed on 10 June 2024).
- Munro, N.B.; Talmage, S.S.; Griffin, G.D.; Waters, L.C.; Watson, A.P.; King, J.F.; Hauschild, V. The Sources, Fate, and Toxicity of Chemical Warfare Agent Degradation Products. Environ. Health Perspect. 1999, 107, 933–974. [Google Scholar] [CrossRef]
- Black, R.M.; Clarke, R.J.; Cooper, D.B.; Read, R.W.; Utley, D. Application of headspace analysis, solvent extraction, thermal desorption and gas chromatography-mass spectrometry to the analysis of chemical warfare samples containing sulphur mustard and related compounds. J. Chromatogr. A 1993, 637, 71–80. [Google Scholar] [CrossRef]
- Boyer, A.E.; Ash, D.; Barr, D.B.; Young, C.L.; Driskell, W.J.; Whitehead, R.D.; Ospina, M.; Preston, K.E.; Woolfitt, A.R.; Martinez, R.A.; et al. Quantitation of the sulfur mustard metabolites 1,1′-sulfonylbis[2-(methylthio)ethane] and thiodiglycol in urine using isotope-dilution gas chromatography-tandem mass spectrometry. J. Anal. Toxicol. 2004, 28, 327–332. [Google Scholar] [CrossRef]
- Riches, J.; Read, R.W.; Black, R.M. Analysis of the sulphur mustard metabolites thiodiglycol and thiodiglycol sulphoxide in urine using isotope-dilution gas chromatography-ion trap tandem mass spectrometry. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2007, 845, 114–120. [Google Scholar] [CrossRef]
- Popiel, S.; Nawała, J.; Dziedzic, D.; Söderström, M.; Vanninen, P. Determination of mustard gas hydrolysis products thiodiglycol and thiodiglycol sulfoxide by gas chromatography-tandem mass spectrometry after trifluoroacetylation. Anal. Chem. 2014, 86, 5865–5872. [Google Scholar] [CrossRef]
- Kroening, K.K.; Richardson, D.D.; Afton, S.; Caruso, J.A. Screening hydrolysis products of sulfur mustard agents by high-performance liquid chromatography with inductively coupled plasma mass spectrometry detection. Anal. Bioanal. Chem. 2009, 393, 1949–1956. [Google Scholar] [CrossRef]
- Garnaga, G.; Wyse, E.; Azemard, S.; Stankevičius, A.; de Mora, S. Arsenic in sediments from the southeastern Baltic Sea. Environ. Pollut. 2006, 144, 855–861. [Google Scholar] [CrossRef] [PubMed]
- Knobloch, T.; Bełdowski, J.; Böttcher, C.; Söderström, M.; Rühl, N.-P.; Sternheim, J. Chemical Munitions Dumped in the Baltic Sea. Report of the ad hoc Expert Group to Update and Review the Existing Information on Dumped Chemical Munitions in the Baltic Sea (HELCOM MUNI) HELCOM. Available online: https://helcom.fi/wp-content/uploads/2019/08/Dumped-chemical-munitions-in-the-Baltic-Sea.pdf (accessed on 20 August 2024).
- Bausinger, T.; Preuß, J. Environmental remnants of the first World War: Soil contamination of a burning ground for arsenical ammunition. Bull. Environ. Contam. Toxicol. 2005, 74, 1045–1052. [Google Scholar] [CrossRef] [PubMed]
- Hempel, M.; Daus, B.; Vogt, C.; Weiss, H. Natural attenuation potential of phenylarsenicals in anoxic groundwaters. Environ. Sci. Technol. 2009, 43, 6989–6995. [Google Scholar] [CrossRef]
- Ishizaki, M.; Yanaoka, T.; Nakamura, M.; Hakuta, T.; Ueno, S.; Komuro, M.; Shibata, M.; Kitamura, T.; Honda, A.; Doy, M.; et al. Detection of Bis(diphenyl wrsine)oxide, Diphenylarsinic Acid and Phenylarsonic Acid, Compounds Probably Derived from Chemical Warfare Agents, in Drinking Well Water. J. Health Sci. 2005, 52, 130–137. [Google Scholar] [CrossRef]
- Kinoshita, K.; Shida, Y.; Sakuma, C.; Ishizaki, M.; Kiso, K.; Shikino, O.; Ito, H.; Morita, M.; Ochi, T.; Kaise, T. Determination of diphenylarsinic acid and phenylarsonic acid, the degradation products of organoarsenic chemical warfare agents, in well water by HPLC-ICP-MS. Appl. Organomet. Chem. 2005, 19, 287–293. [Google Scholar] [CrossRef]
- Nawala, J.; Gordon, D.; Dziedzic, D.; Rodziewicz, P.; Popiel, S. Why does Clark I remain in the marine environment for a long time? Sci. Total Environ. 2021, 774, 145675. [Google Scholar] [CrossRef]
- Brzeziński, T.; Czub, M.; Nawała, J.; Gordon, D.; Dziedzic, D.; Dawidziuk, B.; Popiel, S.; Maszczyk, P. The effects of chemical warfare agent Clark I on the life histories and stable isotopes composition of Daphnia magna. Environ. Pollut. 2020, 266, 115142. [Google Scholar] [CrossRef]
- Jackson, B.P.; Liba, A.; Nelson, J. Advantages of reaction cell ICP-MS on doubly charged interferences for arsenic and selenium analysis in foods. J. Anal. At. Spectrom. 2015, 30, 1179–1183. [Google Scholar] [CrossRef]
- Yost, R.A. The triple quadrupole: Innovation, serendipity and persistence. J. Mass. Spectrom. Adv. Clin. Lab. 2022, 24, 90–99. [Google Scholar] [CrossRef]
- Kinoshita, K.; Shikino, O.; Seto, Y.; Kaise, T. Determination of degradation compounds derived from Lewisite by high performance liquid chromatography/inductively coupled plasma-mass spectrometry. Appl. Organomet. Chem. 2006, 20, 591–596. [Google Scholar] [CrossRef]
- Kinoshita, K.; Noguchi, A.; Ishii, K.; Tamaoka, A.; Ochi, T.; Kaise, T. Urine analysis of patients exposed to phenylarsenic compounds via accidental pollution. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2008, 867, 179–188. [Google Scholar] [CrossRef]
- Daus, B.; Mattusch, J.; Wennrich, R.; Weiss, H. Analytical investigations of phenyl arsenicals in groundwater. Talanta 2008, 75, 376–379. [Google Scholar] [CrossRef] [PubMed]
- Stetson, S.J.; Erickson, M.L.; Brenner, J.; Berquist, E.C.; Kanagy, C.; Whitcomb, S.; Lawrence, C. Stability of inorganic and methylated arsenic species in laboratory standards, surface water and groundwater under three different preservation regimes. J. Appl. Geochem. 2021, 125, 13. [Google Scholar] [CrossRef]
- Hisatomi, S.; Guan, L.; Nakajima, M.; Fujii, K.; Nonaka, M.; Harada, N. Formation of diphenylthioarsinic acid from diphenylarsinic acid under anaerobic sulfate-reducing soil conditions. J. Hazard. Mater. 2013, 262, 25–30. [Google Scholar] [CrossRef]
- Suzuki, K.T.; Mandal, B.K.; Katagiri, A.; Sakuma, Y.; Kawakami, A.; Ogra, Y.; Yamaguchi, K.; Sei, Y.; Yamanaka, K.; Anzai, K.; et al. Dimethylthioarsenicals as arsenic metabolites and their chemical preparations. Chem. Res. Toxicol. 2004, 17, 914–921. [Google Scholar] [CrossRef]
- Baygildiev, T.M.; Rodin, I.A.; Stavrianidi, A.N.; Braun, A.V.; Akhmerova, D.I.; Shpigun, O.A.; Rybalchenko, I.V. Time-efficient LC/MS/MS determination of low concentrations of methylphosphonic acid. Inorg. Mater. 2017, 53, 1382–1385. [Google Scholar] [CrossRef]
- Lee, J.Y.; Lee, Y.H. Rapid screening and determination of nerve agent metabolites in human urine by LC-MS/MS. J. Anal. Chem. 2014, 69, 909–916. [Google Scholar] [CrossRef]
- Baygildiev, T.; Vokuev, M.; Braun, A.; Rybalchenko, I.; Rodin, I. Monitoring of hydrolysis products of mustard gas, some sesqui- and oxy-mustards and other chemical warfare agents in a plant material by HPLC-MS/MS. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2021, 1162, 122452. [Google Scholar] [CrossRef]
- OPCW. Chemical Weapon Convention on the Prohibition of the Development, Production, Stockpiling and Use of Chemical Weapons and on their Destruction. In The Legal Texts, 2nd ed.; Tabassi, L.W., Ed.; TMC Asser Press: Hague, The Netherlands, 2009. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Analyte | LOD for HPLC-ICP-MS [pg/mL] | LOD for GC-ICP-MS [pg/mL] |
|---|---|---|
| EMPA | 263 | 34.2 |
| IMPA | 183 | 20.9 |
| MPA | 139 | 49.6 |
| No. | Type of Column | Mobile Phase | Flow [mL/min] | Vinjection [μL] | Analytes | LOD [ng/mL] | Separation Time [min] |
|---|---|---|---|---|---|---|---|
| 1. | Dionex IonPac AS7 4.1 mm × 250 mm, guard column | 0.4 mM acetic acid/sodium acetate, 5 mM HNO3 (pH∼2.4) in DDW | 1.0 | 25 | EMPA | d.n.a. | 5 |
| IMPA | d.n.a. | ||||||
| MPA | d.n.a. | ||||||
| 2. | Hamilton PRP-X100 4.6 mm × 100 mm 5 μm | A = 0.5% H2CO3 (pH∼2.3), 5% MeOH in DDW B = 0.3 M NH4HCO3 (pH∼2.3), 22% MeOH in DDW | 1.0 | 100 | EDHAP | 21.7 | 25 |
| MPA | 18.3 | ||||||
| EPA | 19.9 | ||||||
| DMHP | 10.0 | ||||||
| PPA | 22.9 | ||||||
| EMPA | 20.4 | ||||||
| IMPA | 19.5 | ||||||
| DEHP | 26.6 | ||||||
| IPHEP | 61.5 | ||||||
| IBHMP | 81.1 | ||||||
| 3. | Hamilton PRP-X100 2.1 mm× 150 mm 5 μm | A = 10 mM (NH4)2CO3 (pH∼8.5) in DDW B = 50 mM (NH4)2CO3 (pH∼8.5) in DDW | 0.5 | 100 | EDHAP | 88.1 | 30 |
| MPA | 7.4 | ||||||
| EPA | 7.4 | ||||||
| DMHP | 8.9 | ||||||
| PPA | 14.5 | ||||||
| EMPA | 11.2 | ||||||
| IMPA | 28.6 |
| STRUCTURE | 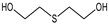 | 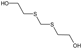 | 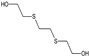 | 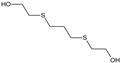 | 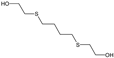 |
|---|---|---|---|---|---|
| ACRONYM | TDG | BHETM | BHETE | BHETPr | BHETBu |
| COMPOUND NAME | Thiodiglycol | Bis(2-hydroxyethylthio)methane | 1,2-bis(2-hydroxyethylthio)ethane | 1,3-bis(2-hydroxyethylthio)propane | 1,4-bis(2-hydroxyethylthio)butane |
| LOD [NG/ML] STUDY | 4.60 | 35.50 | 79.30 | 98.50 | 73.20 |
| LOQ [NG/ML] RIVER WATER SAMPLES | 750 | 690 | 1000 | 1160 | 3300 |
| Publication [64] | Publication [63] | ||||
|---|---|---|---|---|---|
| Mechanism | Hydrophobicity | Dipole–Dipole Interaction | Separation of Functional Groups | ||
| Column | C4 | CN | NH2 | C8 | |
| Organic solvents | H2O (HNO3)/C2H5OH/ACN = 80:15:5, | citrate buffer/CH3OH/ACN = 70:20:10, | phosphate buffer/ACN = 50:50 | 0.1% HCOOH–CH3CN = 80:20 | |
| pH | 1.5 | 5.5 | 2.5 | 2.0 | |
| Tcolumn [°C] | 40 | 40 | 40 | 40 | |
| Flow [mL/min] | 0.3 | 0.3 | 0.1 | 0.2 | |
| Vinjection [μL] | 20 | 10 | no satisfactory separation; the NH2 column was not used for further research | 20 | |
| LOD [ng/mL As] | PAA | 0.25 | 1.0 | 0.3 | |
| PMAA | 0.25 | 0.5 | - | ||
| DPAA | 0.5 | - | 0.5 | ||
| PDMAO | 0.1 | 0.5 | - | ||
| DMPAO | 0.3 | 1.0 | - | ||
| No. | Analyte | LOD [ppb = ng/mL] | |||
|---|---|---|---|---|---|
| HPLC-MS/MS | Ref. | HPLC-ICP-MS | Ref. | ||
| 1. | MPA | 10 | [69] | 0.14 (a) | [33] |
| 2. | EMPA | 5 * 1 ** | [70] | 0.03 (a) | [33] |
| 3. | IMPA | 5 * 1 ** | [70] | 0.02 (a) | [33] |
| 4. | TDG | 40 | [71] | 4.6 | [52] |
| 5. | CVAOA | 500 | [6] | 0.1 | [63] |
| 6. | PAA | 50 | [6] | 0.25 | [64] |
| 7. | PMAA | 1 | [6] | 0.25 | [64] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kuligowska, M.; Neffe, S. Evaluation of the Potential of High-Performance Liquid Chromatography–Inductively Coupled Plasma–Mass Spectrometry for the Determination of Chemical Warfare Agents and Their Toxic Degradation Products. Molecules 2024, 29, 5031. https://doi.org/10.3390/molecules29215031
Kuligowska M, Neffe S. Evaluation of the Potential of High-Performance Liquid Chromatography–Inductively Coupled Plasma–Mass Spectrometry for the Determination of Chemical Warfare Agents and Their Toxic Degradation Products. Molecules. 2024; 29(21):5031. https://doi.org/10.3390/molecules29215031
Chicago/Turabian StyleKuligowska, Monika, and Slawomir Neffe. 2024. "Evaluation of the Potential of High-Performance Liquid Chromatography–Inductively Coupled Plasma–Mass Spectrometry for the Determination of Chemical Warfare Agents and Their Toxic Degradation Products" Molecules 29, no. 21: 5031. https://doi.org/10.3390/molecules29215031
APA StyleKuligowska, M., & Neffe, S. (2024). Evaluation of the Potential of High-Performance Liquid Chromatography–Inductively Coupled Plasma–Mass Spectrometry for the Determination of Chemical Warfare Agents and Their Toxic Degradation Products. Molecules, 29(21), 5031. https://doi.org/10.3390/molecules29215031