
Visible-Light-Promoted Tandem Skeletal Rearrangement/Dearomatization of Heteroaryl Enallenes

 , and
, and
Abstract
1. Introduction
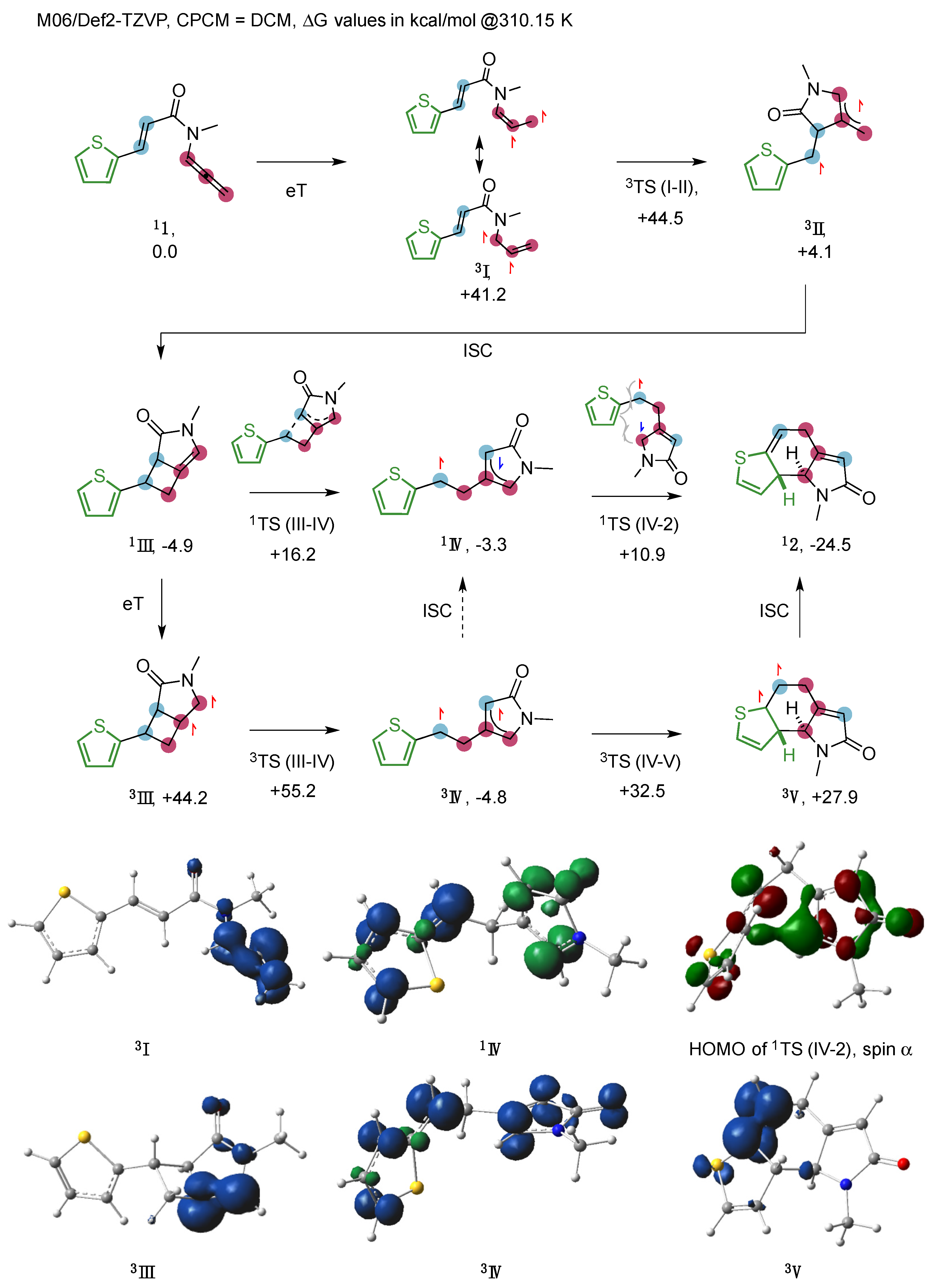
2. Results
3. Discussion
4. Materials and Methods
4.1. General Procedure for the Alkyne Isomerization Leading to Allenes 1 (GP-1)
4.1.1. (E)-N-Benzyl-N-(propa-1,2-dien-1-yl)-3-(thiophen-2-yl)acrylamide
4.1.2. (E)-N-(2-Bromobenzyl)-N-(propa-1,2-dien-1-yl)-3-(thiophen-2-yl)acrylamide
4.1.3. (E)-N-(Benzo[d][1,3]dioxol-5-ylmethyl)-N-(propa-1,2-dien-1-yl)-3-(thiophen-2-yl)acrylamide
4.1.4. (E)-N-Methyl-N-(propa-1,2-dien-1-yl)-3-(thiophen-2-yl)acrylamide
4.1.5. (E)-N-Benzyl-3-(furan-2-ySl)-N-(propa-1,2-dien-1-yl)acrylamide
4.1.6. (E)-N-Benzyl-3-(furan-2-yl)-N-(propa-1,2-dien-1-yl)acrylamide-2-d
4.1.7. (E)-N-(4-Fluorobenzyl)-3-(furan-2-yl)-N-(propa-1,2-dien-1-yl)acrylamide
4.1.8. (E)-3-(Furan-2-yl)-N-(3-methoxybenzyl)-N-(propa-1,2-dien-1-yl)acrylamide
4.1.9. (E)-N-Cyclopropyl-3-(furan-2-yl)-N-(propa-1,2-dien-1-yl)acrylamide
4.1.10. (E)-3-(Benzo[b]thiophen-2-yl)-N-benzyl-N-(propa-1,2-dien-1-yl)acrylamide
4.1.11. (E)-3-(Benzo[b]thiophen-2-yl)-N-(3-methylbenzyl)-N-(propa-1,2-dien-1-yl)acrylamide
4.1.12. (E)-3-(Benzofuran-2-yl)-N-benzyl-N-(propa-1,2-dien-1-yl)acrylamide
4.1.13. (E)-N-Benzyl-3-(3-methylthiophen-2-yl)-N-(propa-1,2-dien-1-yl)acrylamide
4.1.14. (E)-N-Benzyl-N-(propa-1,2-dien-1-yl)-3-(thiophen-3-yl)acrylamide
4.1.15. (E)-N-Benzyl-3-(1-benzyl-1H-pyrrol-2-yl)-N-(propa-1,2-dien-1-yl)acrylamide
4.1.16. (E)-N-Benzyl-3-(1-methyl-1H-indol-2-yl)-N-(propa-1,2-dien-1-yl)acrylamide
4.2. General Procedure for the Photocatalytic Reaction Leading to Products 2 (GP-2)
4.2.1. 1-Benzyl-1,4,8a,8b-tetrahydro-2H-thieno [2,3-g]indol-2-one
4.2.2. 1-(2-Bromobenzyl)-1,4,8a,8b-tetrahydro-2H-thieno [2,3-g]indol-2-one
4.2.3. 1-(Benzo[d][1,3]dioxol-5-ylmethyl)-1,4,8a,8b-tetrahydro-2H-thieno [2,3-g]indol-2-one
4.2.4. 1-Methyl-1,4,8a,8b-tetrahydro-2H-thieno [2,3-g]indol-2-one
4.2.5. 1-Benzyl-1,4,8a,8b-tetrahydro-2H-furo [2,3-g]indol-2-one
4.2.6. 1-Benzyl-1,4,8a,8b-tetrahydro-2H-furo [2,3-g]indol-2-one-3-d
4.2.7. 1-(4-Fluorobenzyl)-8a,8b-dihydro-1H-furo [2,3-g]indol-2(4H)-one
4.2.8. 1-(3-Methoxybenzyl)-8a,8b-dihydro-1H-furo [2,3-g]indol-2(4H)-one
4.2.9. 1-Cyclopropyl-8a,8b-dihydro-1H-furo [2,3-g]indol-2(4H)-one
4.2.10. 1-Benzyl-10b,10c-dihydro-1H-benzo [4,5]thieno [2,3-g]indol-2(4H)-one
4.2.11. 1-(3-Methylbenzyl)-1,4,10b,10c-tetrahydro-2H-benzo [4,5]thieno [2,3-g]indol-2-one
4.2.12. 1-Benzyl-10b,10c-dihydro-1H-benzofuro [2,3-g]indol-2(4H)-one
4.2.13. 1-Benzyl-8a-methyl-8a,8b-dihydro-1H-thieno [2,3-g]indol-2(4H)-one
4.2.14. 1,6-Dibenzyl-4,5,6,8-tetrahydropyrrolo [3,4-f]indol-7(1H)-one
4.2.15. 2-Benzyl-5-methyl-1,2,5,10-tetrahydropyrrolo [3,4-b]carbazol-3(4H)-one
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lovering, F.; Bikker, J.; Humblet, C. Escape from Flatland: Increasing Saturation as an Approach to Improving Clinical Success. J. Med. Chem. 2009, 52, 6752–6756. [Google Scholar] [CrossRef]
- Wei, W.; Cherukupalli, S.; Jing, L.; Liu, X.; Zhan, P. Fsp3: A new parameter for drug-likeness. Drug Discov. Today 2020, 25, 1839–1845. [Google Scholar] [CrossRef]
- Huffman, B.J.; Chen, S.; Schwarz, L.J.; Plata, E.R.; Chin, E.N.; Lairson, L.L.; Houk, K.N.; Shenvi, R.A. Electronic complementarity permits hindered butenolide heterodimerization and discovery of novel cGAS/STING pathway antagonists. Nat. Chem. 2020, 12, 310–317. [Google Scholar] [CrossRef]
- Chen, Q.-C.; Kress, S.; Molinelli, R.; Wuttig, A. Interfacial tuning of electrocatalytic Ag surfaces for fragment-based electrophile coupling. Nat. Catal. 2024. [Google Scholar] [CrossRef]
- Remy, R.; Bochet, C.G. Arene–Alkene Cycloaddition. Chem. Rev. 2016, 116, 9816–9849. [Google Scholar] [CrossRef] [PubMed]
- Wertjes, W.C.; Southgate, E.H.; Sarlah, D. Recent advances in chemical dearomatization of nonactivated arenes. Chem. Soc. Rev. 2018, 47, 7996–8017. [Google Scholar] [CrossRef]
- Zhu, M.; Zhang, X.; Zheng, C.; You, S.-L. Energy-Transfer-Enabled Dearomative Cycloaddition Reactions of Indoles/Pyrroles via Excited-State Aromatics. Acc. Chem. Res. 2022, 55, 2510–2525. [Google Scholar] [CrossRef] [PubMed]
- Bariwal, J.; Voskressensky, L.G.; Van der Eycken, E.V. Recent Advances in Spirocyclization of Indole Derivatives. Chem. Soc. Rev. 2018, 47, 3831–3848. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.-Z.; Feng, Z.; Zhang, X.; You, S.-L. Visible-Light Induced Dearomatization Reactions. Chem. Soc. Rev. 2022, 51, 2145–2170. [Google Scholar] [CrossRef] [PubMed]
- Palai, A.; Rai, P.; Maji, B. Rejuvenation of dearomative cycloaddition reactions via visible light energy transfer catalysis. Chem. Sci. 2023, 14, 12004–12025. [Google Scholar] [CrossRef] [PubMed]
- Rai, P.; Maji, K.; Jana, S.K.; Maji, B. Intermolecular Dearomative [4+2] Cycloaddition of Naphthalenes via Visible-Light Energy-Transfer-Catalysis. Chem. Sci. 2022, 13, 12503–12510. [Google Scholar] [CrossRef]
- Wang, W.; Cai, Y.; Guo, R.; Brown, M.K. Synthesis of Complex Bicyclic Scaffolds by Intermolecular Photosensitized Dearomative Cycloadditions of Activated Alkenes and Naphthalenes. Chem. Sci. 2022, 13, 13582–13587. [Google Scholar] [CrossRef]
- Ma, J.; Chen, S.; Bellotti, P.; Guo, R.; Schäfer, F.; Heusler, A.; Zhang, X.; Daniliuc, C.; Brown, M.K.; Houk, K.N.; et al. Photochemical Intermolecular Dearomative Cycloaddition of Bicyclic Azaarenes with Alkenes. Science 2021, 371, 1338–1345. [Google Scholar] [CrossRef] [PubMed]
- Southgate, E.H.; Pospech, J.; Fu, J.; Holycross, D.R.; Sarlah, D. Dearomative dihydroxylation with arenophiles. Nat. Chem. 2016, 8, 922–928. [Google Scholar] [CrossRef]
- Chiminelli, M.; Serafino, A.; Ruggeri, D.; Marchiò, L.; Bigi, F.; Maggi, R.; Malacria, M.; Maestri, G. Visible-Light Promoted Intramolecular para-Cycloaddition on Simple Aromatics. Angew. Chem. Int. Ed. 2023, 62, e202216817. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Strieth-Kalthoff, F.; Dalton, T.; Freitag, M.; Schwarz, L.J.; Bergander, K.; Daniliuc, C.; Glorius, F. Direct Dearomatization of Pyridines via an Energy-Transfer-Catalyzed Intramolecular [4+2] Cycloaddition. Chem 2019, 5, 2854–2864. [Google Scholar] [CrossRef]
- Wang, M.; Lu, P. Catalytic approaches to assemble cyclobutane motifs in natural product synthesis. Org. Chem. Front. 2018, 5, 254–259. [Google Scholar] [CrossRef]
- Sarkar, D.; Bera, N.; Ghosh, S. [2+2] Photochemical Cycloaddition in Organic Synthesis. Eur. J. Org. Chem. 2020, 2020, 1310–1326. [Google Scholar] [CrossRef]
- Alcaide, B.; Almendros, P.; Aragoncillo, C. Exploiting [2+2] cycloaddition chemistry: Achievements with allenes. Chem. Soc. Rev. 2010, 39, 783–816. [Google Scholar] [CrossRef]
- Serafino, A.; Balestri, D.; Marchiò, L.; Malacria, M.; Derat, E.; Maestri, G. Orthogonal Syntheses of 3.2.0 Bicycles from Enallenes Promoted by Visible Light. Org. Lett. 2020, 22, 6354–6359. [Google Scholar] [CrossRef]
- Jovanovic, M.; Jovanovic, P.; Tasic, G.; Simic, M.; Maslak, V.; Rakic, S.; Rodic, M.; Vlahovic, F.; Petkovic, M.; Savic, V. Regio- and stereoselective, intramolecular [2+2] cycloaddition of allenes, promoted by visible light photocatalysis. Adv. Synth. Catal. 2023, 365, 2516–2523. [Google Scholar] [CrossRef]
- Ha, S.; Lee, Y.; Kwak, Y.; Mishra, A.; Yu, E.; Ryou, B.; Park, C.-M. Alkyne–Alkene [2+2] cycloaddition based on visible light photocatalysis. Nat. Commun. 2020, 11, 2509. [Google Scholar] [CrossRef]
- Serafino, A.; Chiminelli, M.; Balestri, D.; Marchiò, L.; Bigi, F.; Maggi, R.; Malacria, M.; Maestri, G. Dimerizing Cascades of Enallenamides Reveal the Visible-Light-Promoted Activation of Cumulated C–C Double Bonds. Chem. Sci. 2022, 13, 2632–2639. [Google Scholar] [CrossRef]
- Lanzi, M.; Santacroce, V.; Balestri, D.; Marchiò, L.; Bigi, F.; Maggi, R.; Malacria, M.; Maestri, G. Visible-Light-Promoted Polycyclizations of Dienynes. Angew. Chem. Int. Ed. 2019, 58, 6703–6707. [Google Scholar] [CrossRef]
- Raviola, C.; Carrera, C.; Serra, M.; Palmieri, A.; Lupidi, G.; Maestri, G.; Protti, S. Visible-Light-DrivenCompetitiveStereo-andRegioisomerizationof (E)-β-Nitroenones. ChemPhotoChem 2021, 5, 871–875. [Google Scholar] [CrossRef]
- Chiminelli, M.; Scarica, G.; Balestri, D.; Marchiò, L.; Della Ca’, N.; Maestri, G. The visible-light-promoted intermolecular para-cycloadditions of allenamides on naphthalene. Tetrahedron Chem 2023, 8, 100053. [Google Scholar] [CrossRef]
- Strieth-Kalthoff, F.; Glorius, F. Triplet Energy Transfer Photocatalysis: Unlocking the Next Level. Chem 2020, 6, 1888–1903. [Google Scholar] [CrossRef]
- Harper, K.C.; Moschetta, E.G.; Bordawekar, S.V.; Wittenberger, S.J. A Laser Driven Flow Chemistry Platform for Scaling Photochemical Reactions with Visible Light. ACS Cent. Sci. 2019, 5, 109–115. [Google Scholar] [CrossRef] [PubMed]
- Kocsis, L.S.; Kagalwala, H.N.; Mutto, S.; Godugu, B.; Bernhard, S.; Tantillo, D.J.; Brummond, K.M. Mechanistic Insight into the Dehydro-Diels-Alder Reaction of Styrene-Ynes. J. Org. Chem. 2015, 80, 11686–11698. [Google Scholar] [CrossRef] [PubMed]
- Vessally, E. Aromatic stability energy studies on five-membered heterocyclic C4H4M (M = O, S, Se, Te, NH, PH, AsH and SbH): DFT calculations. J. Struct. Chem. 2008, 49, 979–985. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. The M06 Suite of Density Functionals for Main Group Thermochemistry, Thermochemical Kinetics, Noncovalent Interactions, Excited States, and Transition Elements: Two New Functionals and Systematic Testing of Four M06-Class Functionals and 12 Other Functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced Basis Sets of Split Valence, Triple Zeta Valence and Quadruple Zeta Valence Quality for H to Rn: Design and Assessment of Accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef] [PubMed]
- Barone, V.; Cossi, M. Quantum Calculation of Molecular Energies and Energy Gradients in Solution by a Conductor Solvent Model. J. Phys. Chem. A 1998, 102, 1995–2001. [Google Scholar] [CrossRef]
- Großkopf, J.; Kratz, T.; Rigotti, T.; Bach, T. Enantioselective Photochemical Reactions Enabled by Triplet Energy Transfer. Chem. Rev. 2022, 122, 1626–1653. [Google Scholar] [CrossRef]
- Neveselý, T.; Wienhold, M.; Molloy, J.J.; Gilmour, R. Advances in the E → Z Isomerization of Alkenes Using Small Molecule Photocatalysts. Chem. Rev. 2022, 122, 2650–2694. [Google Scholar] [CrossRef] [PubMed]
- Strieth-Kalthoff, F.; James, M.J.; Teders, M.; Pitzer, L.; Glorius, F. Energy Transfer Catalysis Mediated by Visible Light: Principles, Applications, Directions. Chem. Soc. Rev. 2018, 47, 7190–7202. [Google Scholar] [CrossRef]
- Silvi, M.; Arceo, E.; Jurberg, I.D.; Cassani, C.; Melchiorre, P. Enantioselective organocatalytic alkylation of aldehydes and enals driven by the direct photoexcitation of enamines. J. Am. Chem. Soc. 2015, 137, 6120–6123. [Google Scholar] [CrossRef]
- Macklin, T.K.; Micalizio, G.C. Convergent and stereospecific synthesis of complex skipped polyenes and polyunsaturated fatty acids. Nat. Chem. 2010, 2, 638–643. [Google Scholar] [CrossRef]
- Kim, J.H.; Ruffoni, A.; Al-Faiyz, Y.S.S.; Sheikh, N.S.; Leonori, D. Divergent Strain-Release Amino-Functionalization of [1.1.1]Propellane with Electrophilic Nitrogen-Radicals. Angew. Chem. Int. Ed. 2020, 59, 8225–8231. [Google Scholar] [CrossRef]
- Guo, R.; Chang, Y.-C.; Herter, L.; Salome, C.; Braley, S.E.; Fessard, T.C.; Brown, M.K. Strain-Release [2π + 2σ] Cycloadditions for the Synthesis of Bicyclo[2.1.1]hexanes Initiated by Energy Transfer. J. Am. Chem. Soc. 2022, 144, 7988–7994. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. (Eds.) Gaussian 16, Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | ||
|---|---|---|
| Entry a | Deviation from Optimal | Yield of 2a b |
| 1 | -- | 66 |
| 2 | 0.2 M on 1a | 39 |
| 3 | at 25 °C | 60 |
| 4 | DCE, at 25 °C | 35 |
| 5 | THF, at 25 °C | 24 |
| 6 | toluene, at 25 °C | 26 |
| 7 | DMF, at 25 °C | 37 |
| 8 | Ru(bpy)3Cl2 as PC, DMF, at 25 °C | 24 |
| 9 | Ir(ppy)3 as PC, DMF, at 25 °C | 32 |
| 10 c | 10 mol% TXT as PC, DMF, at 25 °C | 14 |
| 11 | w/o light or PC | -- |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chiminelli, M.; Scarica, G.; Serafino, A.; Marchiò, L.; Viscardi, R.; Maestri, G. Visible-Light-Promoted Tandem Skeletal Rearrangement/Dearomatization of Heteroaryl Enallenes. Molecules 2024, 29, 595. https://doi.org/10.3390/molecules29030595
Chiminelli M, Scarica G, Serafino A, Marchiò L, Viscardi R, Maestri G. Visible-Light-Promoted Tandem Skeletal Rearrangement/Dearomatization of Heteroaryl Enallenes. Molecules. 2024; 29(3):595. https://doi.org/10.3390/molecules29030595
Chicago/Turabian StyleChiminelli, Maurizio, Gabriele Scarica, Andrea Serafino, Luciano Marchiò, Rosanna Viscardi, and Giovanni Maestri. 2024. "Visible-Light-Promoted Tandem Skeletal Rearrangement/Dearomatization of Heteroaryl Enallenes" Molecules 29, no. 3: 595. https://doi.org/10.3390/molecules29030595
APA StyleChiminelli, M., Scarica, G., Serafino, A., Marchiò, L., Viscardi, R., & Maestri, G. (2024). Visible-Light-Promoted Tandem Skeletal Rearrangement/Dearomatization of Heteroaryl Enallenes. Molecules, 29(3), 595. https://doi.org/10.3390/molecules29030595