Computer-Aided Strategy on 5-(Substituted benzylidene) Thiazolidine-2,4-Diones to Develop New and Potent PTP1B Inhibitors: QSAR Modeling, Molecular Docking, Molecular Dynamics, PASS Predictions, and DFT Investigations

 , , and
, , and
Abstract
1. Introduction
2. Results and Discussions
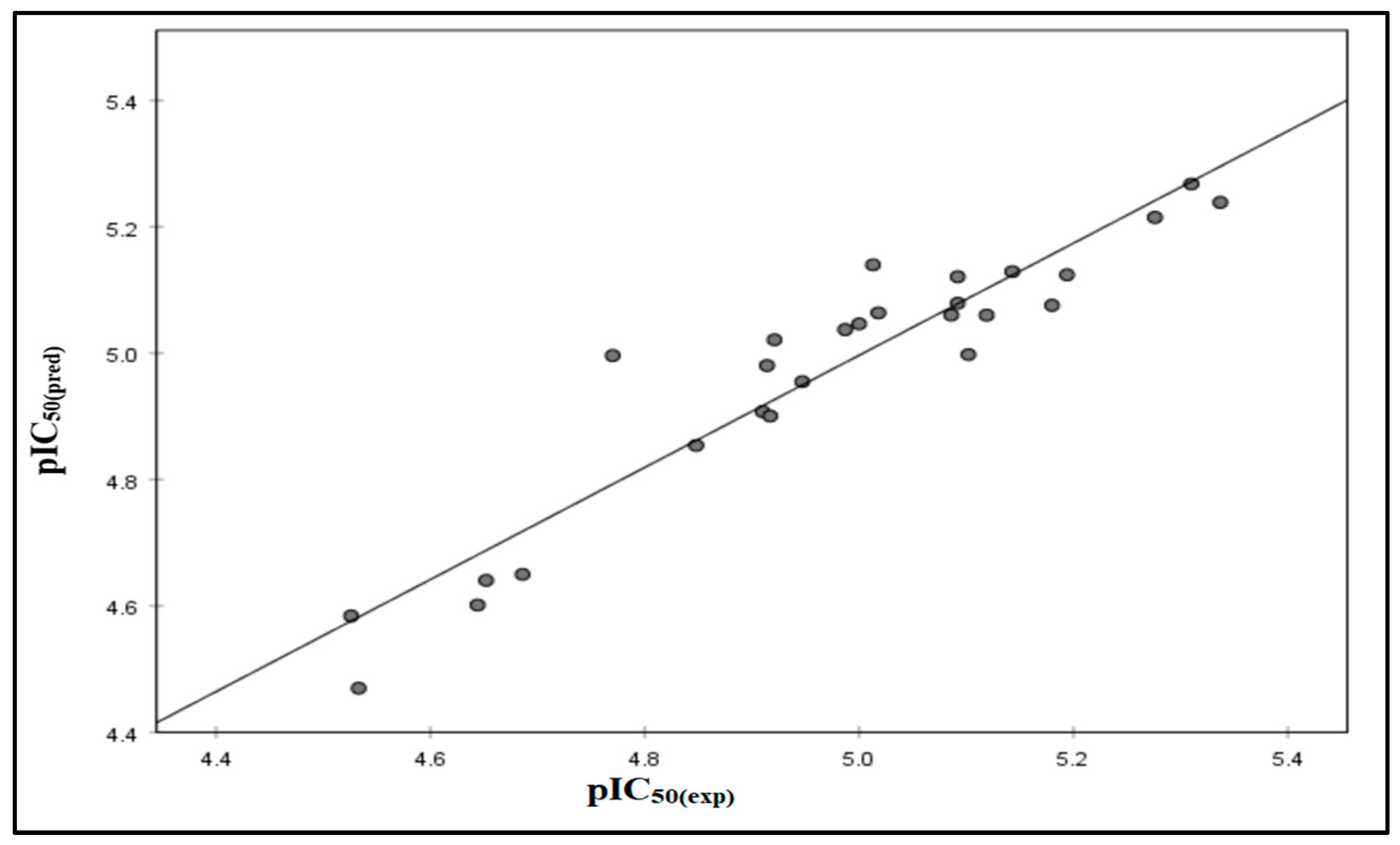
2.1. QSAR Results
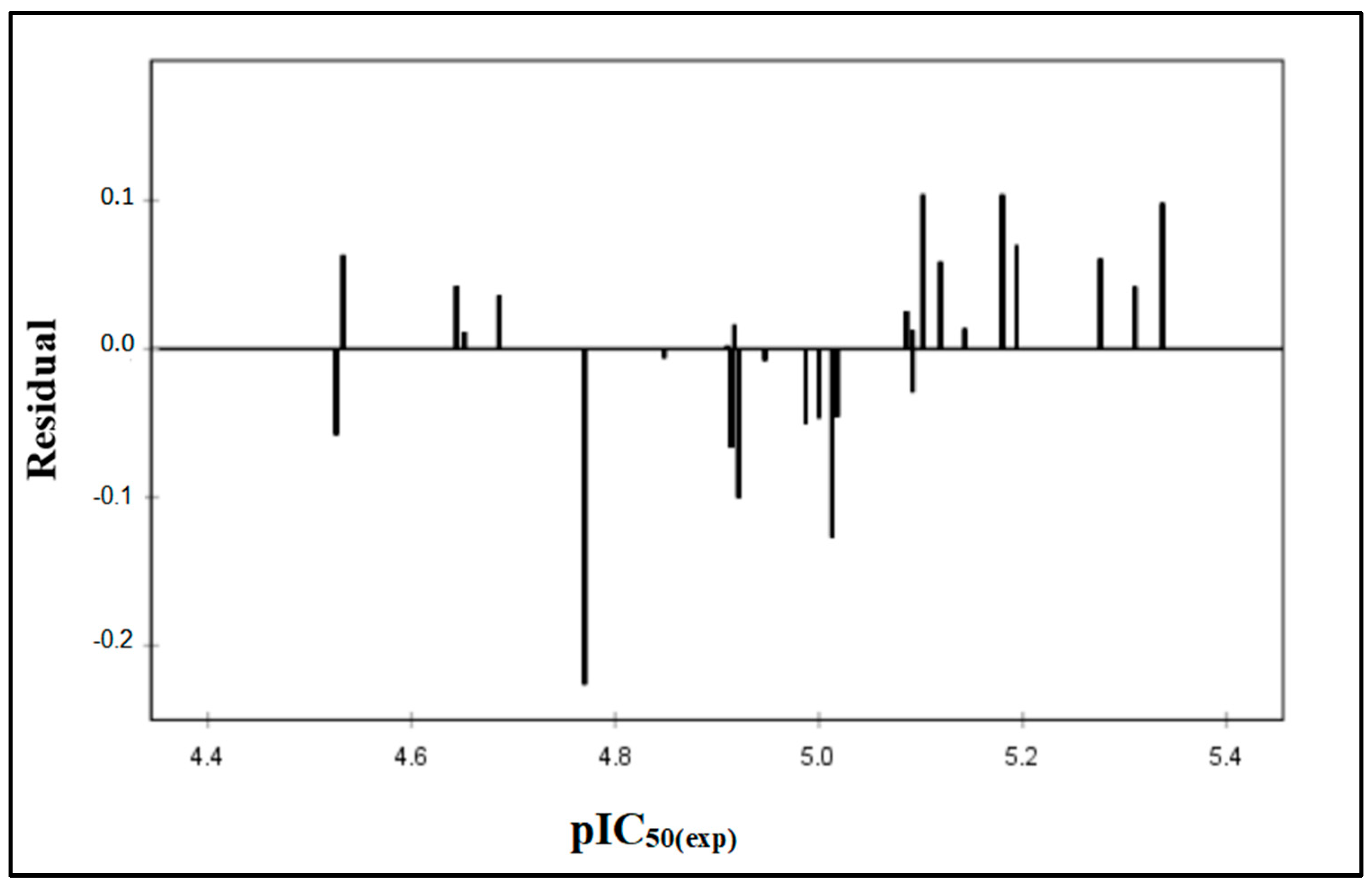
2.2. QSAR Validation
2.3. Applicability Domain
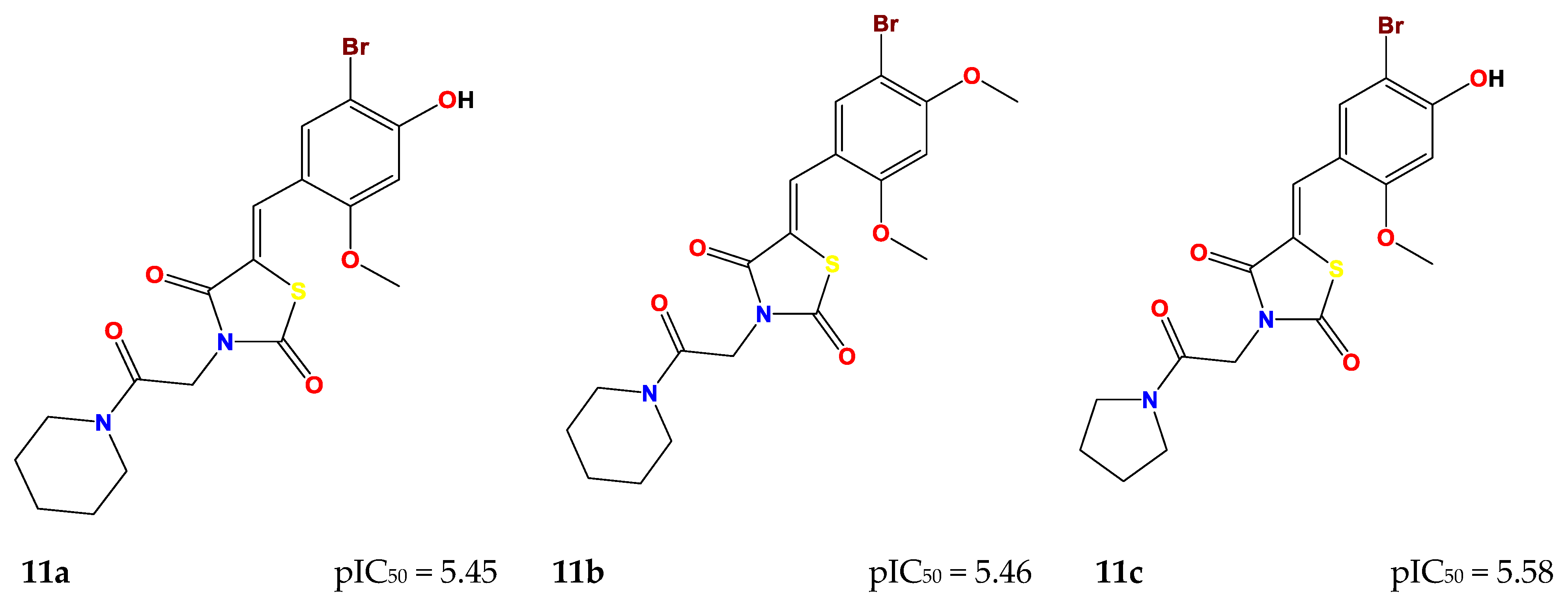
2.4. Design of New Compounds
2.5. Molecular Docking
2.5.1. Docking Validation Protocol
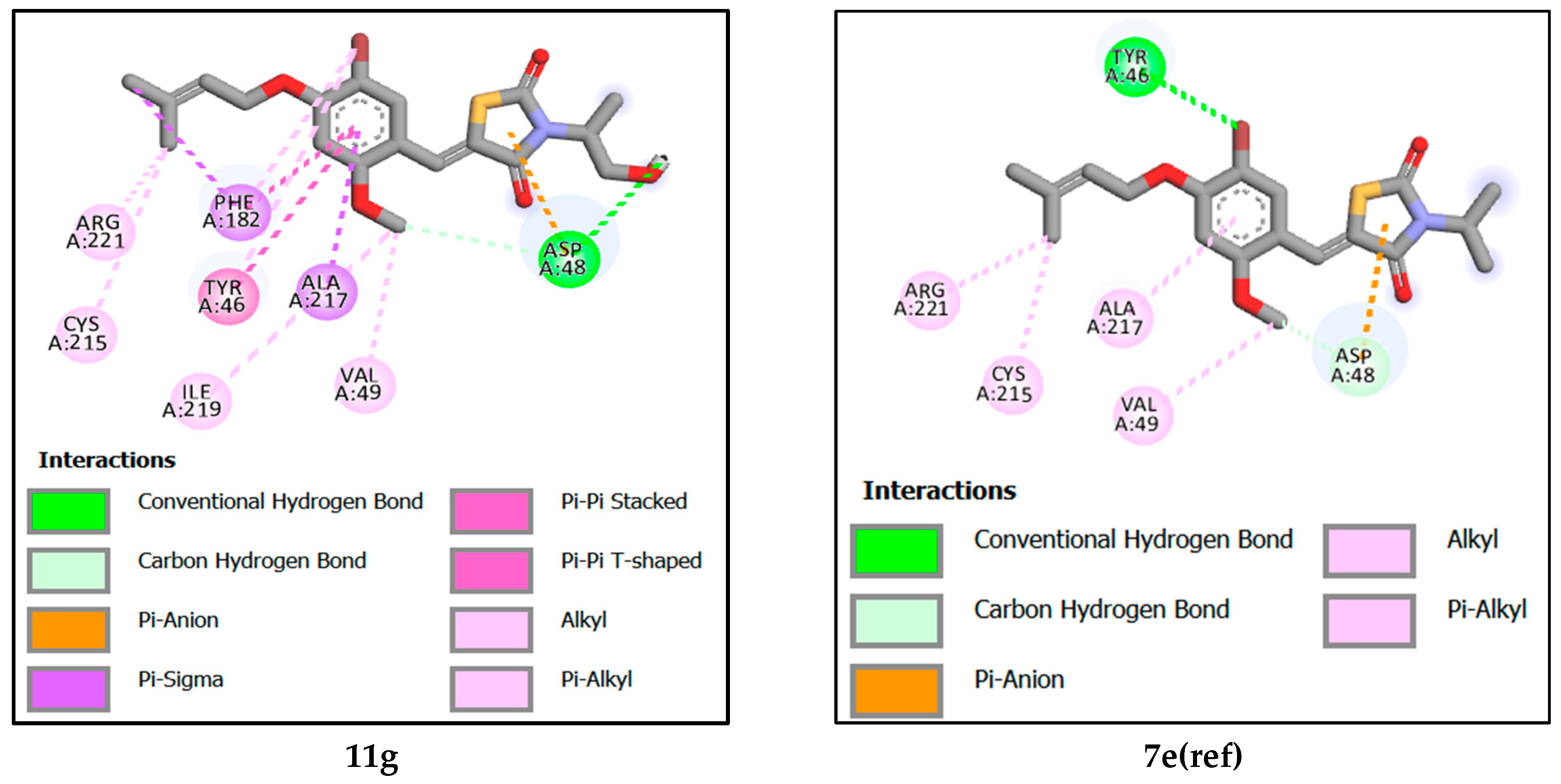
2.5.2. Analysis of Interactions between Newly Designed Ligands and Protein Active Site
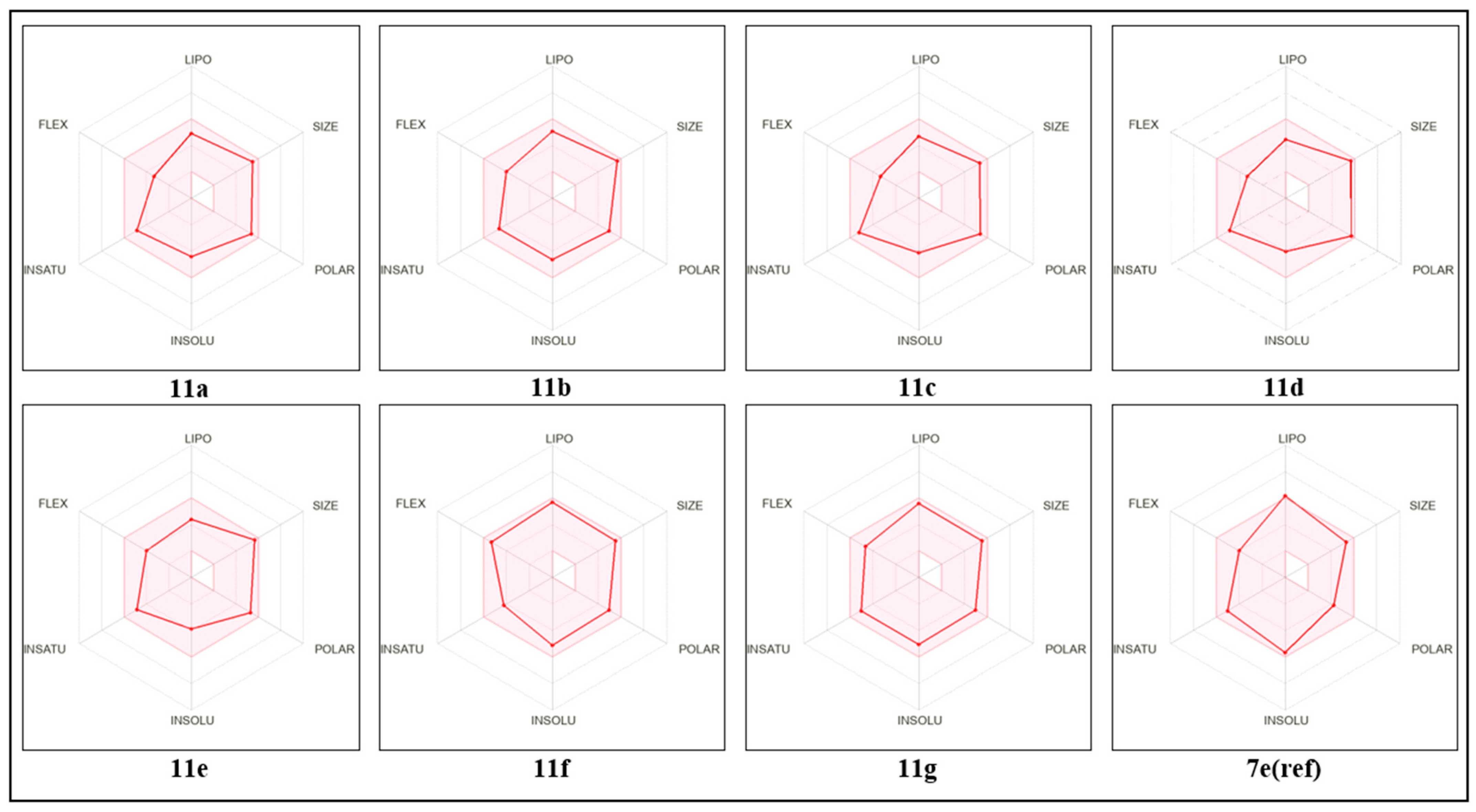
2.6. ADME-Tox Prediction and Bioavailability
2.7. Biological Activities Using PASS
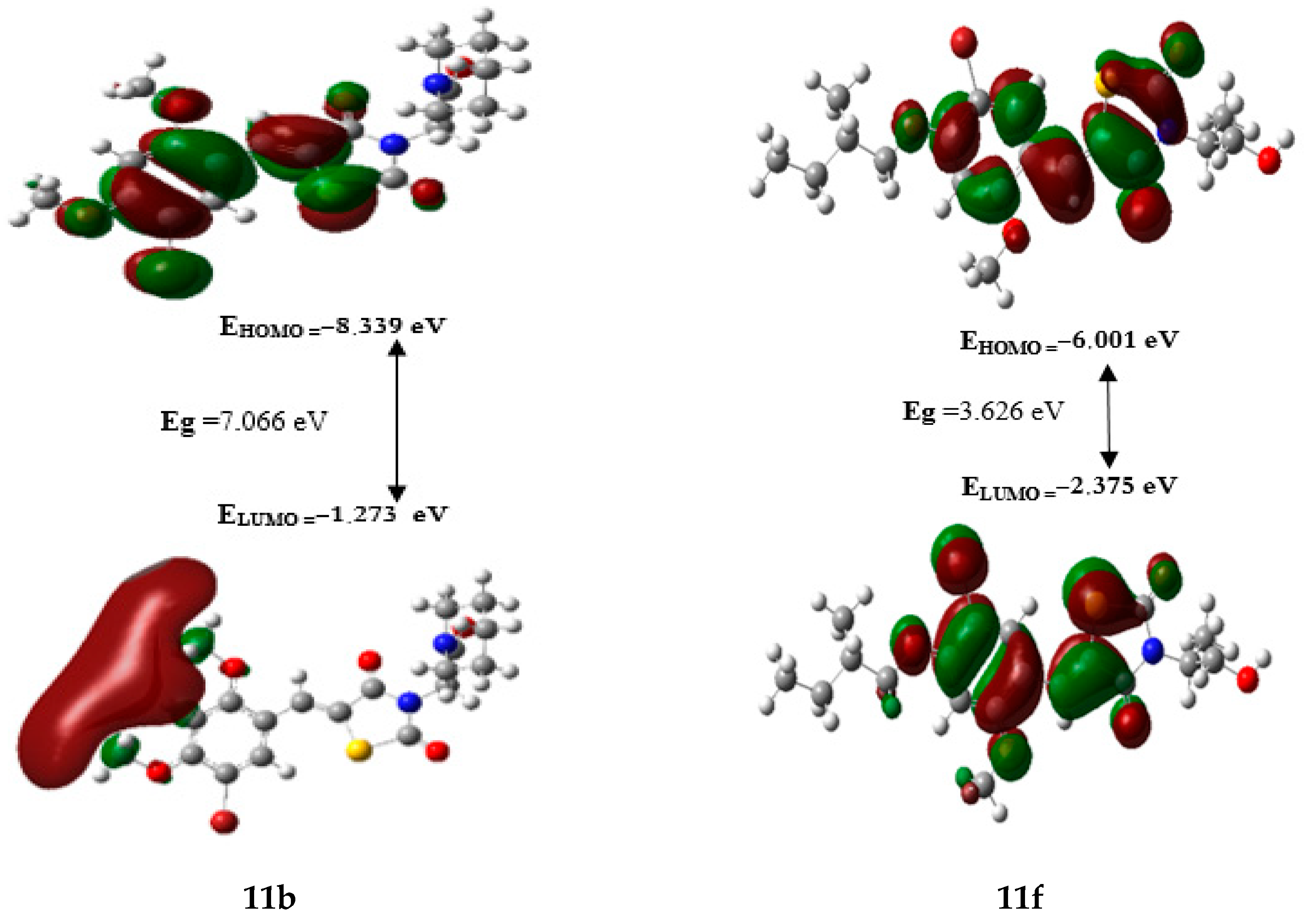
2.8. Frontier Orbital Energies and Global Reactivity Parameters Using DFT
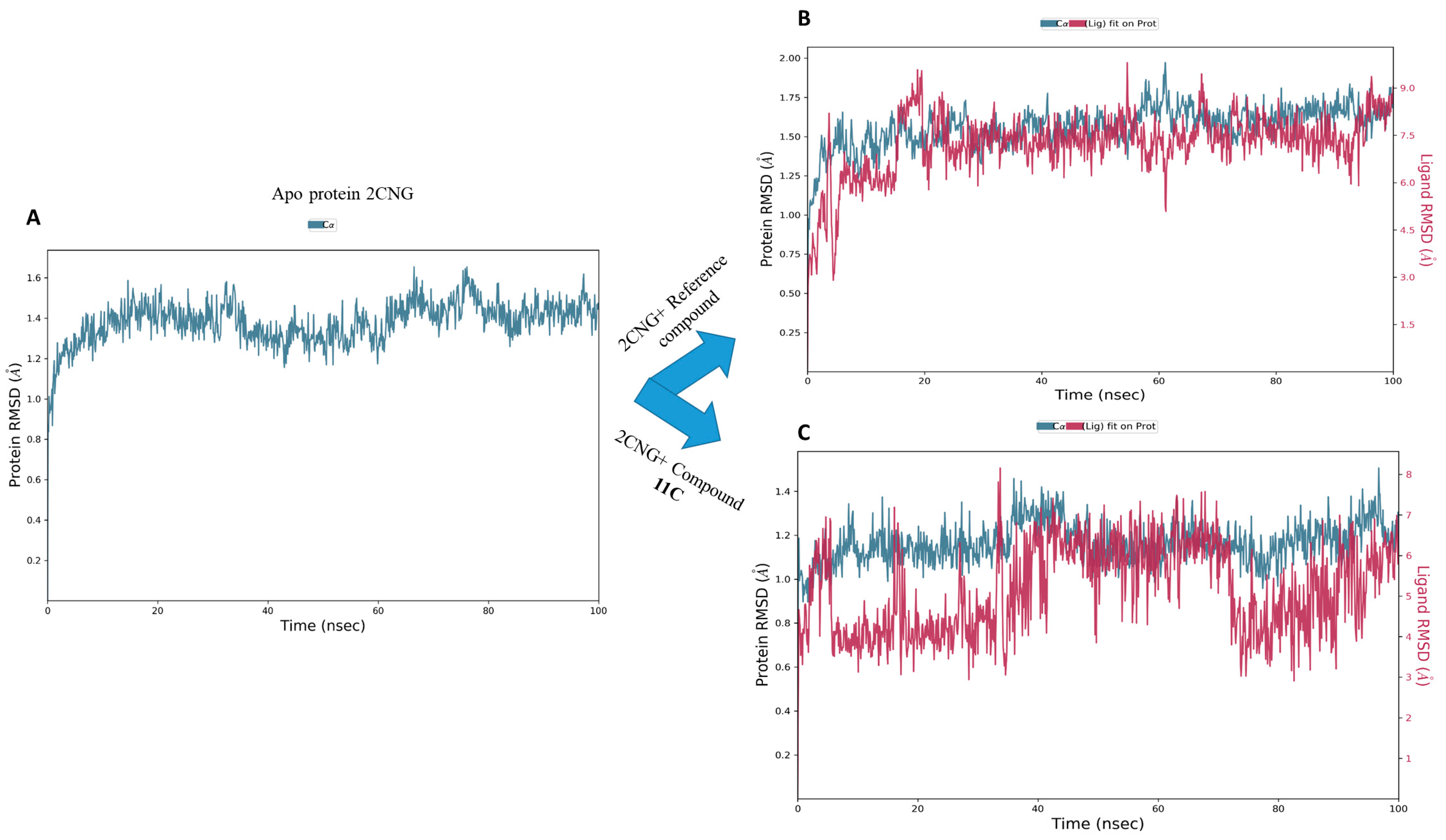
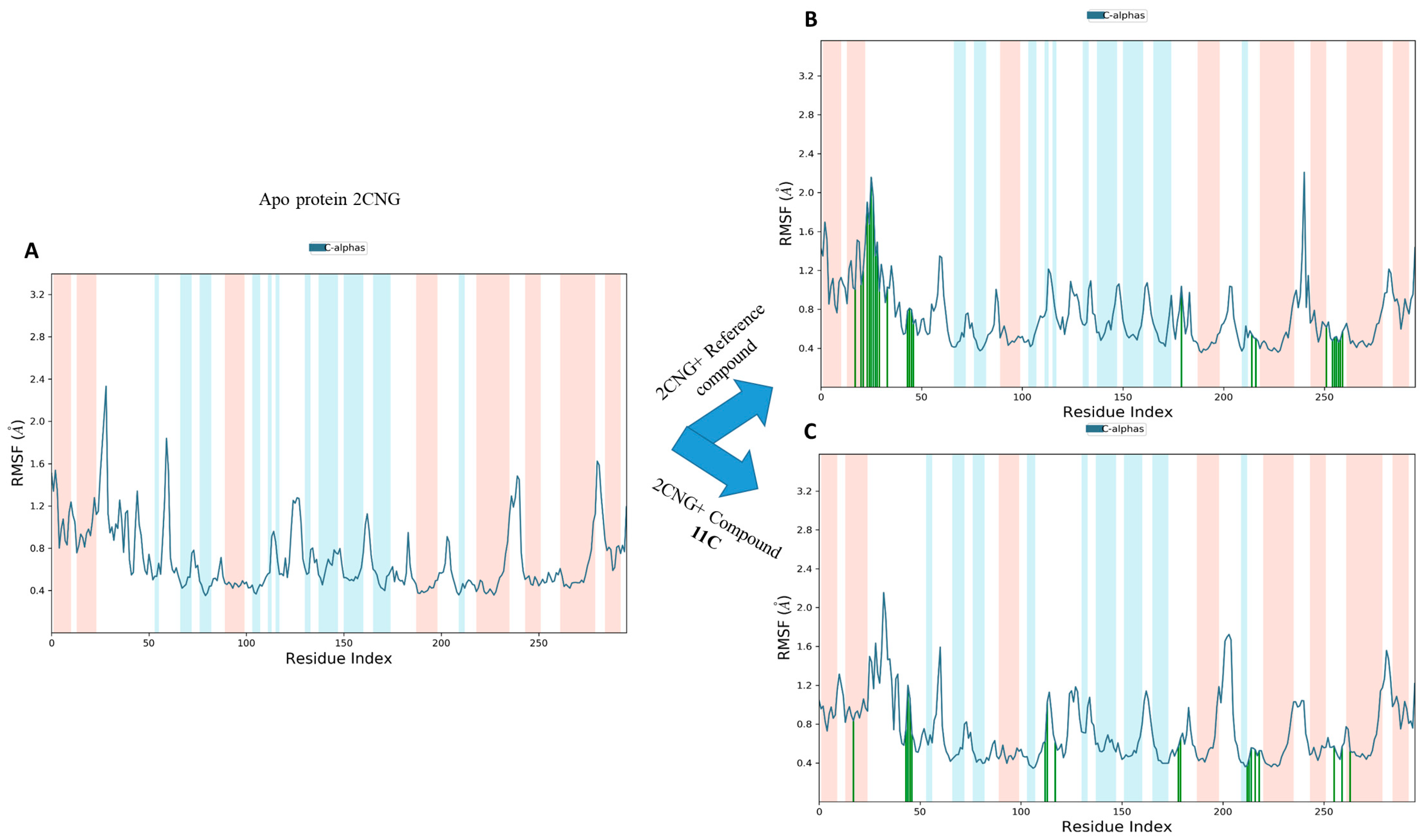
2.9. Molecular Dynamics Results
2.9.1. Root Mean Square Deviation Analysis (RMSD)
2.9.2. Root Mean Square Fluctuation (RMSF) Analysis
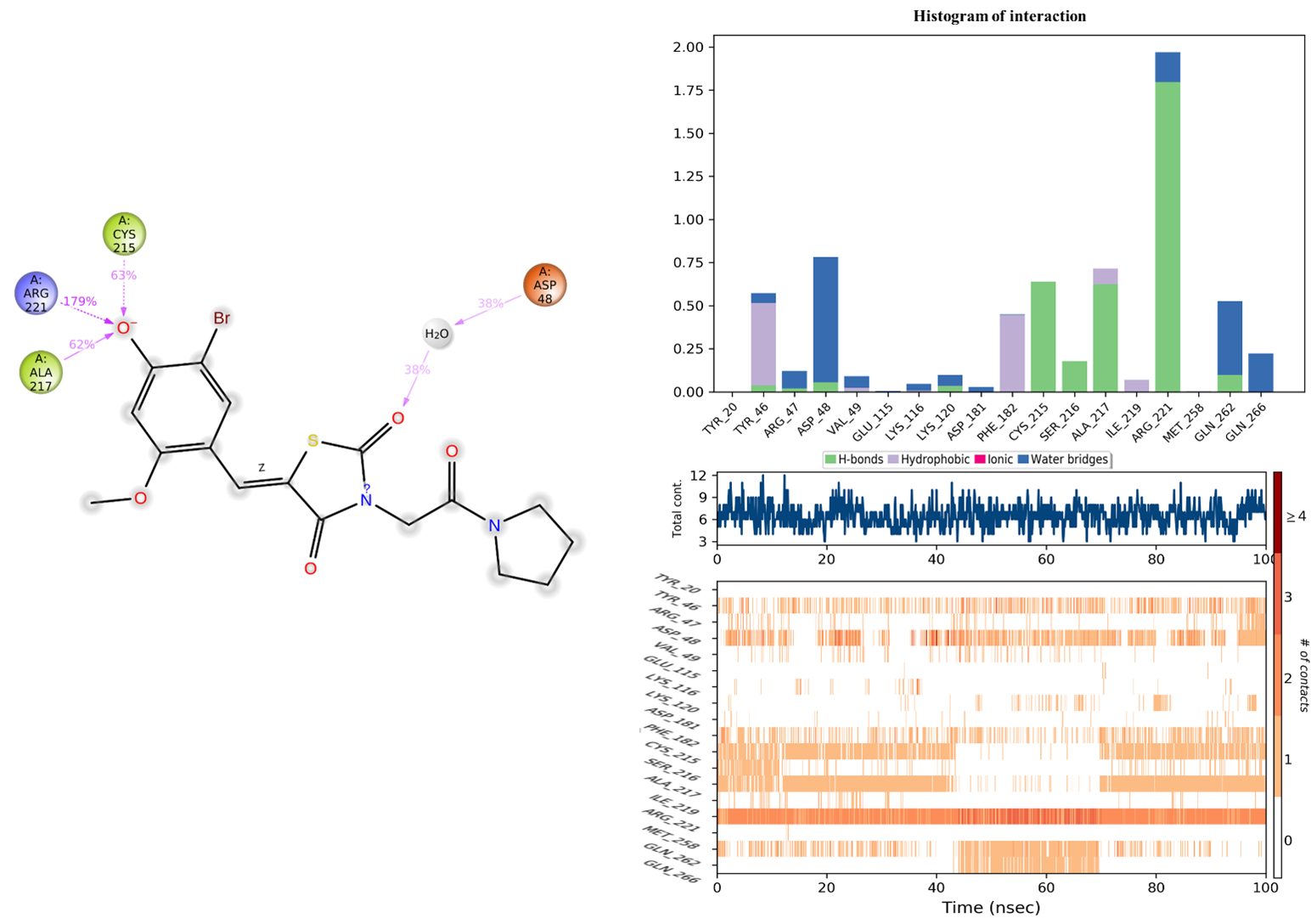
2.9.3. Protein–Ligand Interaction Analysis
3. Materials
3.1. QSAR Analysis
3.2. Computation of Molecular Descriptors
3.3. Y-Randomization
3.4. Applicability Domain
3.5. Molecular Docking
3.6. Molecular Dynamics Simulation
3.7. In Silico and ADME and Drug-Likeness Prediction
3.8. PASS Prediction
3.9. Assessing Chemical Reactivity through DFT Calculations
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Eleftheriou, P.; Geronikaki, A.; Petrou, A. PTP1b inhibition, a promising approach for the treatment of diabetes type II. Curr. Top. Med. Chem. 2019, 19, 246–263. [Google Scholar] [CrossRef] [PubMed]
- Mikłosz, A.; Chabowski, A. Adipose-derived mesenchymal stem cells therapy as a new treatment option for diabetes mellitus. J. Clin. Endocrinol. Metab. 2023, 108, 1889–1897. [Google Scholar] [CrossRef] [PubMed]
- Santwana, P.; Nayak, A.K.; Behera, A. Type II diabetes mellitus: A review on recent drug based therapeutics. Biomed. Pharmacother. 2020, 131, 110708. [Google Scholar] [CrossRef]
- Rocha, S.; Lucas, M.; Silva, V.L.; Gomes, P.M.; Silva, A.M.; Araújo, A.N.; Aniceto, N.; Guedes, R.C.; Corvo, M.L.; Fernandes, E.; et al. Pyrazoles as novel protein tyrosine phosphatase 1B (PTP1B) inhibitors: An in vitro and in silico study. Int. J. Biol. Macromol. 2021, 181, 1171–1182. [Google Scholar] [CrossRef] [PubMed]
- Kahn, S.E. The importance of β-cell failure in the development and progression of type 2 diabetes. J. Clin. Endocrinol. Metab. 2001, 86, 4047–4058. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Liu, Z.; Lee, W.; Kim, S.N.; Yoon, G.; Cheon, S.H. Design, synthesis and docking study of 5-(substituted benzylidene) thiazolidine-2, 4-dione derivatives as inhibitors of protein tyrosine phosphatase 1B. Bioorganic Med. Chem. Lett. 2014, 24, 3337–3340. [Google Scholar] [CrossRef] [PubMed]
- Mokdad, A.H.; Ford, E.S.; Bowman, B.A.; Dietz, W.H.; Vinicor, F.; Bales, V.S.; Marks, J.S. Prevalence of obesity, diabetes, and obesity-related health risk factors, 2001. JAMA 2003, 289, 76–79. [Google Scholar] [CrossRef] [PubMed]
- Johnson, T.O.; Ermolieff, J.; Jirousek, M.R. Protein tyrosine phosphatase 1B inhibitors for diabetes. Nat. Rev. Drug Discov. 2002, 1, 696–709. [Google Scholar] [CrossRef]
- Tsamos, G.; Vasdeki, D.; Koufakis, T.; Michou, V.; Makedou, K.; Tzimagiorgis, G. Therapeutic Potentials of Reducing Liver Fat in Non-Alcoholic Fatty Liver Disease: Close Association with Type 2 Diabetes. Metabolites 2023, 13, 517. [Google Scholar] [CrossRef]
- Tamrakar, A.K.; Maurya, C.K.; Rai, A.K. PTP1B inhibitors for type 2 diabetes treatment: A patent review (2011–2014). Expert Opin. Ther. Pat. 2014, 24, 1101–1115. [Google Scholar] [CrossRef]
- Egawa, K.; Maegawa, H.; Shimizu, S.; Morino, K.; Nishio, Y.; Bryer-Ash, M.; Cheung, A.T.; Kolls, J.K.; Kikkawa, R.; Kashiwagi, A. Protein-tyrosine phosphatase-1B negatively regulates insulin signaling in l6 myocytes and Fao hepatoma cells. J. Biol. Chem. 2001, 276, 10207–10211. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Zhang, Z.Y. PTP1B as a drug target: Recent developments in PTP1B inhibitor discovery. Drug Discov. Today 2007, 12, 373–381. [Google Scholar] [CrossRef] [PubMed]
- Vilar, S.; Cozza, G.; Moro, S. Medicinal chemistry and the molecular operating environment (MOE): Application of QSAR and molecular docking to drug discovery. Curr. Top. Med. Chem. 2008, 8, 1555–1572. [Google Scholar] [CrossRef] [PubMed]
- Ghaleb, A.; Aouidate, A.; Ayouchia, H.B.E.; Aarjane, M.; Anane, H.; Stiriba, S.E. In silico molecular investigations of pyridine N-Oxide compounds as potential inhibitors of SARS-CoV-2: 3D QSAR, molecular docking modeling, and ADMET screening. J. Biomol. Struct. Dyn. 2022, 40, 143–153. [Google Scholar] [CrossRef] [PubMed]
- Devore, J.L.; Peck, R. Introductory Statistics, 2nd ed.; West Pub.: St. Paul, MN, USA, 1994. [Google Scholar]
- Sobhi, W.; Attoui, A.; Lemaoui, T.; Erto, A.; Benguerba, Y. In silico drug discovery of Acetylcholinesterase and Butyrylcholinesterase enzymes inhibitors based on Quantitative Structure-Activity Relationship (QSAR) and drug-likeness evaluation. J. Mol. Struct. 2021, 1229, 129845. [Google Scholar] [CrossRef]
- Belaidi, S.; Kerassa, A.; Lanez, T.; Cinar, M. Structure–Activity Relationships and Quantitative Structure—Activity Relationships Modeling of Some 3-(aryl)-N-(aryl)-1, 2, 4-Oxadiazol-5-Amine Derivatives as Anti-Proliferative Agents. J. Comput. Theor. Nanosci. 2015, 12, 2127–2133. [Google Scholar] [CrossRef]
- Puratchikody, A.; Doble, M. Antinociceptive and antiinflammatory activities and QSAR studies on 2-substituted-4, 5-diphenyl-1H-imidazoles. Bioorganic Med. Chem. 2007, 15, 1083–1090. [Google Scholar] [CrossRef] [PubMed]
- Purkayastha, S.K.; Jha, T.; Pal, D.K.; De, A.U. Possible antineoplastic agents: Part XIII. Synthesis, biological evaluation and QSAR studies of some 1-(substituted benzenesulphonyl)-5-oxopyrrolidine-2-carboxylic acid derivatives. Anti-Cancer Drug Des. 1993, 8, 95–100. [Google Scholar]
- Srivastava, A.K.; Shukla, N. QSAR based modeling on a series of lactam fused chroman derivatives as selective 5-HT transporters. J. Saudi Chem. Soc. 2012, 16, 405–412. [Google Scholar] [CrossRef]
- Zitouni, K.; Belaidi, S.; Kerassa, A. Conformational analysis and qsar modeling of 14-membered macrolide analogues against mycobacterium tuberculosis. J. Fundam. Appl. Sci. 2020, 12, 1035–1066. [Google Scholar] [CrossRef]
- Jalali-Heravi, M.; Kyani, A. Use of computer-assisted methods for the modeling of the retention time of a variety of volatile organic compounds: A PCA-MLR-ANN approach. J. Chem. Inf. Comput. Sci. 2004, 44, 1328–1335. [Google Scholar] [CrossRef] [PubMed]
- Sangande, F.; Julianti, E.; Tjahjono, D.H. Ligand-based pharmacophore modeling, molecular docking, and molecular dynamic studies of dual tyrosine kinase inhibitor of EGFR and VEGFR2. Int. J. Mol. Sci. 2020, 21, 7779. [Google Scholar] [CrossRef] [PubMed]
- Abchir, O.; Daoui, O.; Nour, H.; Yamari, I.; Elkhattabi, S.; Errougui, A.; Chtita, S. Cannabis constituents as potential candidates against diabetes mellitus disease using moleculardocking, dynamics simulations and ADMET investigations. Sci. Afr. 2023, 21, e01745. [Google Scholar] [CrossRef]
- Westermaier, Y.; Barril, X.; Scapozza, L. Virtual screening: An in silico tool for interlacing the chemical universe with the proteome. Methods 2015, 71, 44–57. [Google Scholar] [CrossRef] [PubMed]
- Boobis, A.; Gundert-Remy, U.; Kremers, P.; Macheras, P.; Pelkonen, O. In silico prediction of ADME and pharmacokinetics: Report of an expert meeting organised by COST B15. Eur. J. Pharm. Sci. 2002, 17, 183–193. [Google Scholar] [CrossRef] [PubMed]
- El Fadili, M.; Er-Rajy, M.; Ali Eltayb, W.; Kara, M.; Imtara, H.; Zarougui, S.; Al-Hoshani, N.; Elhallaoui, M. An in-silico investigation based on molecular simulations of novel and potential brain-penetrant GluN2B NMDA receptor antagonists as anti-stroke therapeutic agents. J. Biomol. Struct. Dyn. 2023, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Alameen, A.A.; Abdalla, M.; Alshibl, H.M.; AlOthman, M.R.; Alkhulaifi, M.M.; Mirgany, T.O.; Elsayim, R. In-silico studies of glutathione peroxidase4 activators as candidate for multiple sclerosis management. J. Saudi Chem. Soc. 2022, 26, 101554. [Google Scholar] [CrossRef]
- Zothantluanga, J.H.; Abdalla, M.; Rudrapal, M.; Tian, Q.; Chetia, D.; Li, J. Computational investigations for identification of bioactive molecules from Baccaurearamiflora and Bergeniaciliata as inhibitors of SARS-CoV-2 Mpro. Polycycl. Aromat. Compd. 2023, 43, 2459–2487. [Google Scholar] [CrossRef]
- Yamari, I.; Abchir, O.; Siddique, F.; Zaki, H.; Errougui, A.; Talbi, M.; Bouachrine, M.; ElKouali, M.; Chtita, S. The anticoagulant potential of Lippia Alba extract in inhibiting SARS-CoV-2 Mpro: Density functional calculation, molecular docking analysis, and molecular dynamics simulations. Sci. Afr. 2024, 23, e01986. [Google Scholar] [CrossRef]
- Yamari, I.; Mouhib, A.; Es-Sounni, B.; Nejjari, R.; Mazoir, N.; Bakhouch, M.; Mouzdahir, A.; Benharref, A.; El Kouali, M.; Chtita, S. Oxidative functionalization of triterpenes isolated from Euphorbia resinifera latex: Semisynthesis, ADME-Tox, molecular docking, and molecular dynamics simulations. Chem. Phys. Impact. 2023, 7, 100372. [Google Scholar] [CrossRef]
- Badhani, B.; Kakkar, R. In silico studies on potential MCF-7 inhibitors: A combination of pharmacophore and 3D-QSAR modeling, virtual screening, molecular docking, and pharmacokinetic analysis. J. Biomol. Struct. Dyn. 2017, 35, 1950–1967. [Google Scholar] [CrossRef]
- Roy, K.; Kar, S.; Ambure, P. On a simple approach for determining applicability domain of QSAR models. Chemom. Intell. Lab. Syst. 2015, 145, 22–29. [Google Scholar] [CrossRef]
- Tropsha, A. Best practices for QSAR model development, validation, and exploitation. Mol. Inform. 2010, 29, 476–488. [Google Scholar] [CrossRef]
- Khalil, T.E.; El-Dissouky, A.; Al-Wahaib, D.; Abrar, N.M.; El-Sayed, D.S. Synthesis, characterization, antimicrobial activity, 3D-QSAR, DFT, and molecular docking of some ciprofloxacin derivatives and their copper (II) complexes. Appl. Organomet. Chem. 2020, 34, e5998. [Google Scholar] [CrossRef]
- Gaussian.com. Expanding the Limits of Computational Chemistry. Available online: http://gaussian.com/ (accessed on 20 August 2023).
- Ambarwati, N.S.S.; Azminah, A.; Ahmad, I. Molecular docking, physicochemical and drug-likeness properties of isolated compounds from Garcinia latissimaMiq. on elastase enzyme: In silico analysis. Pharmacogn. J. 2022, 14, 282–288. [Google Scholar] [CrossRef]
- Hyperchem (Molecular Modeling System) Hypercube, Inc., 1115 Nw, 4th Street, Gainesville, Fl 32601, USA, 2007. Available online: http://www.hyperchem.com/ (accessed on 20 August 2023).
- Ononamadu, C.; Ibrahim, A. Molecular docking and prediction of ADME/drug-likeness properties of potentially active antidiabetic compounds isolated from aqueous-methanol extracts of Gymnemasylvestre and Combretummicranthum. BioTechnologia 2021, 102, 85–99. [Google Scholar] [CrossRef] [PubMed]
- Pires, D.E.V.; Blundell, T.L.; Ascher, D.B. pkCSM: Predicting small-molecule pharmacokinetic and toxicity properties using graph-based signatures. J. Med. Chem. 2015, 58, 4066–4072. [Google Scholar] [CrossRef] [PubMed]
- Hazhazi, H.; Melkemi, N.; Salah, T.; Bouachrine, M. DFT-based reactivity and combined QSAR, molecular docking of 1, 2, 4, 5-Tetrazine derivatives as inhibitors of Pim-1 kinase. Heliyon 2019, 5, e02451. [Google Scholar] [CrossRef] [PubMed]
- SPSS Software Packages; SPSS Inc.: Chicago, IL, USA, 2021.
- Olasupo, S.B.; Uzairu, A.; Shallangwa, G.; Uba, S. QSAR modeling, molecular docking and ADMET/pharmacokinetic studies: A chemometrics approach to search for novel inhibitors of norepinephrine transporter as potent antipsychotic drugs. J. Iran. Chem. Soc. 2020, 17, 1953–1966. [Google Scholar] [CrossRef]
- Rücker, C.; Rücker, G.; Meringer, M. y-Randomization and its variants in QSPR/QSAR. J. Chem. Inf. Model. 2007, 47, 2345–2357. [Google Scholar] [CrossRef]
- Roy, K.; Mitra, I. On various metrics used for validation of predictive QSAR models with applications in virtual screening and focused library design. Comb. Chem. High Throughput Screen. 2011, 14, 450–474. [Google Scholar] [CrossRef]
- Ibrahim, Z.Y.U.; Uzairu, A.; Shallangwa, G.; Abechi, S. In-silico design of aryl and aralkyl amine-based triazolopyrimidine derivatives with enhanced activity against resistant Plasmodium falciparum. Chem. Afr. 2021, 4, 137–148. [Google Scholar] [CrossRef]
- Roy, P.P.; Roy, K. On some aspects of variable selection for partial least squares regression models. QSAR Comb. Sci. 2008, 27, 302–313. [Google Scholar] [CrossRef]
- Lemaoui, T.; Darwish, A.S.; Attoui, A.; Hatab, F.A.; Hammoudi, N.E.H.; Benguerba, Y.; Vega, L.F.; Alnashef, I.M. Predicting the density and viscosity of hydrophobic eutectic solvents: Towards the development of sustainable solvents. Green Chem. 2020, 22, 8511–8530. [Google Scholar] [CrossRef]
- Chtita, S.; Belhassan, A.; Aouidate, A.; Belaidi, S.; Bouachrine, M.; Lakhlifi, T. Discovery of potent SARS-CoV-2 inhibitors from approved antiviral drugs via docking and virtual screening. Comb. Chem. High Throughput Screen. 2021, 24, 441–454. [Google Scholar] [CrossRef]
- Bourougaa, L.; Ouassaf, M.; Khan, S.U.; Htar, T.T. Pharmacophore-based virtual screening, molecular docking and molecular dynamics studies for the discovery of novel neuraminidase inhibitors. J. Biomol. Struct. Dyn. 2023, 1–13. [Google Scholar] [CrossRef]
- Haloui, R.; Daoui, O.; Mkhayar, K.; El Yaqoubi, M.; Elkhattabi, S.; Haoudi, A.; Rodi, Y.K.; Ouazzani, F.C.; Chtita, S. 3D-QSAR, drug-likeness, ADMET prediction, and molecular docking studies in silico of novel 5-oxo-1-thioxo-4, 5-dihydro-1H-thiazolo [3, 4-a] quinazoline derivatives as MALT1 protease inhibitors for the treatment of B cell lymphoma. Chem. Pap. 2023, 77, 2255–2274. [Google Scholar] [CrossRef]
- Morris, G.M.; Goodsell, D.S.; Halliday, R.S.; Huey, R.; Hart, W.E.; Belew, R.K.; Olson, A.J. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J. Comput. Chem. 1998, 19, 1639–1662. [Google Scholar] [CrossRef]
- Er-rajy, M.; Imtara, H.; Noman, O.M.; Mothana, R.A.; Abdullah, S.; Zerougui, S.; Elhallaoui, M. QSAR, ADME-Tox, molecular docking and molecular dynamics simulations of novel selective glycine transporter type 1 inhibitors with memory enhancing properties. Heliyon 2023, 9, e13706. [Google Scholar] [CrossRef]
- Luo, J.; Lai, T.; Guo, T.; Chen, F.; Zhang, L.; Ding, W.; Zhang, Y. Synthesis and acaricidal activities of scopoletin phenolic ether derivatives: QSAR, molecular docking study and in silico ADME predictions. Molecules 2018, 23, 995. [Google Scholar] [CrossRef] [PubMed]
- Studio, D. Discovery Studio Life Science Modeling and Simulations; Researchgate: Berlin, Germany, 2008. [Google Scholar]
- Karplus, M.; McCammon, J.A. Molecular dynamics simulations of biomolecules. Nat. Struct. Biol. 2002, 9, 646–652. [Google Scholar] [CrossRef]
- Schrödinger, System, Maestro-Desmond Interoperability Tools, Software. 2021. Available online: https://newsite.schrodinger.com/platform/products/desmond (accessed on 5 February 2024).
- Roos, K.; Wu, C.; Damm, W.; Reboul, M.; Stevenson, J.M.; Lu, C.; Dahlgren, M.K.; Mondal, S.; Chen, W.; Wang, L.; et al. OPLS3e: Extending Force Field Coverage for Drug-Like Small Molecules. J. Chem. Theory Comput. 2019, 15, 1863–1874. [Google Scholar] [CrossRef]
- Mark, P.; Nilsson, L. Structure and Dynamics of the TIP3P, SPC, and SPC/E Water Models at 298 K. J. Phys. Chem. A 2001, 105, 9954–9960. [Google Scholar] [CrossRef]
- Ke, Q.; Gong, X.; Liao, S.; Duan, C.; Li, L. Effects of thermostats/barostats on physical properties of liquids by molecular dynamics simulations. J. Mol. Liq. 2022, 365, 120116. [Google Scholar] [CrossRef]
- Moroy, G.; Martiny, V.Y.; Vayer, P.; Villoutreix, B.O.; Miteva, M.A. Toward in silico structure-based ADMET prediction in drug discovery. Drug Discov. Today 2012, 17, 44–55. [Google Scholar] [CrossRef] [PubMed]
- Bourougaa, L.; Ouassaf, M.; Khan, S.U. Comparative Molecular Field Analysis(CoMFA), Molecular Docking and ADMET Study on Thiazolidine-4-carboxylic acid Derivatives as New Neuraminidase Inhibitors. ActaChimicaSlovenica 2023, 70, 333–344. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef]
- Banerjee, P.; Eckert, A.O.; Schrey, A.K.; Preissner, R. ProTox-II: A webserver for the prediction of toxicity of chemicals. Nucleic Acids Res. 2018, 46, W257–W263. [Google Scholar] [CrossRef] [PubMed]
- Benfenati, E.; Manganaro, A.; Gini, G.C. VEGA-QSAR: AI inside a platformforpredictivetoxicology. PAI@ AI* IA 2013, 1107, 21–28. Available online: http://www.vegahub.eu/portfolio-item/vega-qsar/ (accessed on 15 November 2023).
- Matin, M.M.; Bhattacharjee, S.C.; Chakraborty, P.; Alam, M.S. Synthesis, PASS predication, in vitro antimicrobial evaluation and pharmacokinetic study of novel n-octylglucopyranoside esters. Carbohydr. Res. 2019, 485, 107812. [Google Scholar] [CrossRef]
- Chowdhury, S.A.; Kibria, S.M.; Alam, M.R. DFT Based Pharmacokinetic, Molecular Docking, and ADMET Studies of Some Glucopyranoside Esters. J. Appl. Sci. Process Eng. 2021, 8, 671–683. [Google Scholar] [CrossRef]
- Matin, M.M.; Roshid, M.H.; Bhattacharjee, S.C.; Azad, A.K. PASS predication, antiviral, in vitro Antimicrobial, and ADMET studies of rhamnopyranoside esters. Med. Res. Arch. 2020, 8, 2165. [Google Scholar] [CrossRef]
- Kerassa, A.; Belaidi, S.; Harkati, D.; Lanez, T.; Prasad, O.; Sinha, L. Investigations on molecular structure, electronic properties, NLO properties and comparison of drug-likeness of triazolothiadiazole derivatives by quantum methods and QSAR analysis. Rev. Theo. Sci. 2016, 4, 85–96. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| R2adj | R2cv | SPRESS | SSY | PRESS | PRESS/SSY | PE | 6PE |
|---|---|---|---|---|---|---|---|
| 0.817 | 0.89 | 0.096 | 1.30 | 0.147 | 0.11 | 0.015 | 0.09 |
| Comp | pIC50(Exp) | pIC50(Pred) | Residual |
|---|---|---|---|
| 6a | 4.526 | 4.584 | −0.058 |
| 6b | 4.947 | 4.955 | −0.008 |
| 6c | 4.848 | 4.854 | −0.006 |
| 6d | 5.092 | 5.079 | 0.013 |
| 6e | 5.276 | 5.215 | 0.061 |
| 6f | 5.310 | 5.268 | 0.042 |
| 6g | 5.119 | 5.06 | 0.059 |
| 7a | 4.652 | 4.64 | 0.012 |
| 7b | 5.092 | 5.121 | −0.029 |
| 7c | 5.086 | 5.06 | 0.026 |
| 7d | 4.770 | 4.996 | −0.226 |
| 7e(ref) | 5.337 | 5.239 | 0.098 |
| 7f | 4.910 | 4.908 | 0.002 |
| 7g | 5.000 | 5.047 | −0.047 |
| 7h | 4.987 | 5.037 | −0.05 |
| 7i | 4.917 | 4.901 | 0.016 |
| 7j | 5.102 | 4.998 | 0.104 |
| 7k | 5.013 | 5.14 | −0.127 |
| 7m | 5.180 | 5.076 | 0.104 |
| 12 | 4.914 | 4.98 | −0.066 |
| 13d | 4.644 | 4.601 | 0.043 |
| 13e | 4.686 | 4.65 | 0.036 |
| 14b | 5.194 | 5.124 | 0.07 |
| 14d | 4.921 | 5.021 | −0.1 |
| 14e | 5.018 | 5.064 | −0.046 |
| 14f | 5.143 | 5.129 | 0.014 |
| 14g | 4.533 | 4.47 | 0.063 |
| Model | R | R2 | Q2 | Model | R | R2 | Q2 |
|---|---|---|---|---|---|---|---|
| Original | 0.942 | 0.887 | 0.631 | Original | 0.942 | 0.887 | 0.631 |
| Random 1 | 0.480 | 0.230 | −1.662 | Random 26 | 0.614 | 0.376 | −2.392 |
| Random 2 | 0.425 | 0.181 | −3.135 | Random 27 | 0.654 | 0.428 | −0.496 |
| Random 3 | 0.671 | 0.451 | −0.396 | Random 28 | 0.602 | 0.362 | −0.660 |
| Random 4 | 0.635 | 0.403 | −0.183 | Random 29 | 0.687 | 0.472 | −0.434 |
| Random 5 | 0.595 | 0.354 | −1.660 | Random 30 | 0.546 | 0.299 | −1.066 |
| Random 6 | 0.771 | 0.594 | −0.557 | Random 31 | 0.705 | 0.497 | −0.220 |
| Random 7 | 0.629 | 0.396 | −0.771 | Random 32 | 0.453 | 0.205 | −1.060 |
| Random 8 | 0.565 | 0.320 | −0.734 | Random 33 | 0.598 | 0.358 | −1.243 |
| Random 9 | 0.431 | 0.186 | −0.583 | Random 34 | 0.565 | 0.320 | −1.367 |
| Random 10 | 0.651 | 0.424 | −0.874 | Random 35 | 0.657 | 0.431 | −0.602 |
| Random 11 | 0.719 | 0.517 | −0.429 | Random 36 | 0.563 | 0.317 | −1.031 |
| Random 12 | 0.534 | 0.285 | −1.298 | Random 37 | 0.703 | 0.495 | −0.175 |
| Random 13 | 0.554 | 0.307 | −0.693 | Random 38 | 0.555 | 0.308 | −0.931 |
| Random 14 | 0.764 | 0.584 | −0.294 | Random 39 | 0.604 | 0.365 | −1.955 |
| Random 15 | 0.545 | 0.297 | −1.552 | Random 40 | 0.453 | 0.205 | −2.432 |
| Random 16 | 0.761 | 0.579 | −0.193 | Random 41 | 0.574 | 0.329 | −1.018 |
| Random 17 | 0.454 | 0.206 | −1.148 | Random 42 | 0.497 | 0.247 | −1.088 |
| Random 18 | 0.597 | 0.357 | −0.916 | Random 43 | 0.642 | 0.413 | −0.917 |
| Random 19 | 0.682 | 0.465 | −0.683 | Random 44 | 0.692 | 0.479 | −0.399 |
| Random 20 | 0.676 | 0.457 | −0.448 | Random 45 | 0.405 | 0.164 | −1.100 |
| Random 21 | 0.450 | 0.203 | −1.006 | Random 46 | 0.398 | 0.158 | −1.967 |
| Random 22 | 0.558 | 0.311 | −0.927 | Random 47 | 0.627 | 0.394 | −0.661 |
| Random 23 | 0.678 | 0.460 | −1.264 | Random 48 | 0.642 | 0.412 | −0.715 |
| Random 24 | 0.587 | 0.344 | −0.671 | Random 49 | 0.401 | 0.161 | −0.953 |
| Random 25 | 0.754 | 0.569 | −0.064 | Random 50 | 0.721 | 0.519 | −0.024 |
| Random Models Parameters | |||||||
| Average R | 0.594 | ||||||
| Average R2 | 0.364 | ||||||
| Average Q2 | −0.941 | ||||||
| cRp2 | 0.681 | ||||||
| Compound | Binding Affinity (kcal/mol) | Hydrogen-Binding Interaction | Hydrophobic Interaction | Electrostatic Interaction |
|---|---|---|---|---|
| 11a | −8.18 | ASP48. ASP181. SER216 [1.80–3.59] | ALA217. TYR46. CYS215. VAL49. [3.61–5.06] | ASP48 [3.89] |
| 11b | −8.09 | ASP181. TYR46. [2.88–2.98] | ALA217. TYR46. CYS215. LYS120. [3.12–4.88] | ASP48 [3.59] |
| 11c | −8.30 | ASP48, ASP181, SER216 [1.81–3.59] | ALA217. TYR46. CYS215. VAL49. ARG24.[3.49–5.25] | ASP48 [3.87] |
| 11d | −7.97 | ASP181. TYR20. SER216 [1.81–3.70] | ALA217. TYR46. PHE182. ILE219. [3.19–5.28] | ASP48 [3.48] |
| 11e | −7.41 | ASP181. TYR46. TYR20. [2.87–3.29] | ALA217. TYR46. CYS215. LYS120. PHE182. ILE219. [3.15–5.34] | ASP48 [3.47] |
| 11f | −7.61 | ASP48. SER216 [1.93–3.69] | ALA217. TYR46. CYS215. VAL49. PHE182. ARG221. [3.32–5.29] | ASP48 [3.85] |
| 11g | −7.30 | ASP48 [2.20–3.46] | ALA217. TYR46. CYS215. VAL49. PHE182. ILE219. ARG221. [3.64–5.39] | ASP48 [3.02] |
| 7e(ref) | −7.19 | ASP48. TYR46 [3.16–3.17] | ALA217. ARG221. CYS215. VAL49. | ASP48 [3.03] |
| Compound | MW (g/mol) | Log P (Consensus) | Log S (ESOL) | GI Absorption | Bioavailability Score | Synthetic Accessibility | Lipinski | Pains |
|---|---|---|---|---|---|---|---|---|
| 11a | 455.32 | 2.60 | −4.43 | High | 0.55 | 3.64 | Yes | 0 |
| 11b | 469.35 | 2.94 | −4.65 | High | 0.55 | 3.75 | Yes | 0 |
| 11c | 441.30 | 2.27 | −4.12 | High | 0.55 | 3.56 | Yes | 0 |
| 11d | 471.32 | 1.97 | −4.02 | High | 0.55 | 4.19 | Yes | 0 |
| 11e | 471.32 | 2.11 | −3.89 | High | 0.55 | 3.78 | Yes | 0 |
| 11f | 458.37 | 3.63 | −5.12 | High | 0.55 | 4.61 | Yes | 0 |
| 11g | 456.35 | 3.55 | −5.06 | High | 0.55 | 4.28 | Yes | 0 |
| 7e(ref) | 440.35 | 5.247 | −5.69 | High | 0.55 | 3.81 | Yes | 0 |
| Comp. | Hepatotoxicity | Carcinogenicity | Immunotoxicity | Mutagenicity | Cytotoxicity | Predicted LD50(mg/kg) | Class |
|---|---|---|---|---|---|---|---|
| 11a | Inactive | Inactive | Inactive | Inactive | Inactive | 1180 | 4 |
| 11b | Inactive | Inactive | Inactive | Inactive | Inactive | 1180 | 4 |
| 11c | Inactive | Inactive | Inactive | Inactive | Inactive | 1000 | 4 |
| 11d | Inactive | Inactive | Inactive | Inactive | Inactive | 1190 | 4 |
| 11e | Inactive | Inactive | Inactive | Inactive | Inactive | 1190 | 4 |
| 11f | Inactive | Inactive | Inactive | Inactive | Inactive | 1400 | 4 |
| 11g | Inactive | Inactive | Inactive | Inactive | Inactive | 1000 | 4 |
| Compound | Mutagenicity (Ames Test) | Skin Irritation | Plasma Protein Binding | P-Glycoprotein Activity | Total Body Elimination Half-Life (Hour) |
|---|---|---|---|---|---|
| 11a | No | No | 0.806 | Inactive | 5.192 |
| 11b | No | No | 0.939 | Inactive | 8.289 |
| 11c | No | No | 0.596 | Inactive | 5.522 |
| 11d | No | No | 0.557 | Inactive | 5.335 |
| 11e | No | No | 0.503 | Inactive | 7.975 |
| 11f | No | No | 1.125 | Inactive | 8.051 |
| 11g | No | No | 1.070 | Inactive | 7.202 |
| 7e(ref) | No | No | 1.427 | Inactive | 12.73 |
| Pharmacological Activity | Protein-Tyrosine Phosphatase Beta Inhibitor | Protein-Tyrosine Phosphatase 1B Inhibitor | Protein-Tyrosine Phosphatase Inhibitor | Antidiabetic | Antidiabetic Symptomatic | Antidiabetic (Type 2) | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Compounds | Pa | Pi | Pa | Pi | Pa | Pi | Pa | Pi | Pa | Pi | Pa | Pi | |
| 11a | 0.343 | 0.002 | 0.173 | 0.016 | 0.391 | 0.005 | 0.377 | 0.050 | 0.333 | 0.025 | 0.130 | 0.109 | |
| 11b | 0.368 | 0.002 | 0.181 | 0.015 | 0.411 | 0.005 | 0.426 | 0.037 | 0.393 | 0.014 | 0.137 | 0.100 | |
| 11c | 0.356 | 0.002 | 0.180 | 0.015 | 0.400 | 0.005 | 0.382 | 0.048 | 0.336 | 0.024 | 0.132 | 0.107 | |
| 11d | 0.271 | 0.003 | 0.130 | 0.027 | 0.344 | 0.008 | 0.316 | 0.074 | 0.318 | 0.030 | / | / | |
| 11e | 0.368 | 0.002 | 0.180 | 0.015 | 0.420 | 0.005 | 0.414 | 0.040 | 0.421 | 0.011 | / | / | |
| 11f | 0.290 | 0.003 | 0.181 | 0.015 | 0.346 | 0.008 | 0.372 | 0.052 | 0.299 | 0.040 | 0.178 | 0.069 | |
| 11g | 0.326 | 0.003 | 0.214 | 0.011 | 0.381 | 0.006 | 0.456 | 0.031 | 0.330 | 0.026 | 0.215 | 0.053 | |
| Comp | EHOMO (eV) | ELUMO (eV) | Eg (eV) | ɳ (eV) | (eV−1) | (eV) | ω (eV) |
|---|---|---|---|---|---|---|---|
| 11a | −6.028 | −2.314 | 3.714 | 1.857 | 0.269 | −4.171 | 4.683 |
| 11b | −8.339 | −1.273 | 7.066 | 3.533 | 0.142 | −4.806 | 3.268 |
| 11c | −6.276 | −2.596 | 3.681 | 1.840 | 0.272 | −4.436 | 5.347 |
| 11d | −6.265 | −2.561 | 3.704 | 1.852 | 0.270 | −4.413 | 5.257 |
| 11e | −6.346 | −2.627 | 3.720 | 1.860 | 0.269 | −4.487 | 5.412 |
| 11f | −6.001 | −2.375 | 3.626 | 1.813 | 0.276 | −4.188 | 4.836 |
| 11g | −5.971 | −2.242 | 3.729 | 1.864 | 0.268 | −4.107 | 4.523 |
| 7e(ref) | −6.149 | −2.465 | 3.684 | 1.842 | 0.271 | −4.307 | 5.034 |
 | ||||
|---|---|---|---|---|
| Comp | R1 | R2 | R3 | pIC50 |
| 6a | H | H | H | 4.526 |
| 6b | H | H | Br | 4.947 |
| 6c | CH3 | H | Br | 4.848 |
| 6d | –CH2–CH=CH2 | H | Br | 5.092 |
| 6e | –CH2–CH=C–(CH3)2 | H | Br | 5.276 |
| 6f | –CH2–(CH2)2–CH3 | H | Br | 5.310 |
| 6g | –CH2–C6H5 | H | Br | 5.119 |
| 7a | –CH3 | CH3 | Br | 4.652 |
| 7b | –CH2–C6H5 | CH3 | Br | 5.092 |
| 7c | –CH2–CH=C–(CH3)2 | CH3 | Br | 5.086 |
| 7d | –CH2–CH=C–(CH3)2 | –CH2–CH=CH2 | Br | 4.770 |
| 7e(ref) | –CH2–CH=C–(CH3)2 | –CH–(CH3)2 | Br | 5.337 |
| 7f | –CH2–CH=C–(CH3)2 | –CH2–(CH2)2–CH3 | Br | 4.910 |
| 7g | –CH2–CH=C–(CH3)2 | –CH2–CH=C–(CH3)2 | Br | 5.000 |
| 7h | –CH2–CH=C–(CH3)2 | –CH2–C6H5 | Br | 4.987 |
| 7i | –CH2–(CH2)2–CH3 | CH3 | Br | 4.917 |
| 7j | –CH2–(CH2)2–CH3 | –CH2–CH=CH2 | Br | 5.102 |
| 7k | –CH2–(CH2)2–CH3 | –CH–(CH3)2 | Br | 5.013 |
| 7m | –CH2–(CH2)2–CH3 | –CH2–C6H5 | Br | 5.180 |
| 12 | –CH–O–(CH2)4 (*) | H | –CH2–CH=CH2 | 4.914 |
| 13d | –CH–O–(CH2)4 (*) | –CH2–CH=C–(CH3)2 | –CH2–CH=CH2 | 4.644 |
| 13e | –CH–O–(CH2)4 (*) | –CH2–(CH2)2–CH3 | –CH2–CH=CH2 | 4.686 |
| 14b | H | –CH–(CH3)2 | –CH2–CH=CH2 | 5.194 |
| 14d | H | –CH2–CH=C–(CH3)2 | –CH2–CH=CH2 | 4.921 |
| 14e | H | –CH2–(CH2)2–CH3 | –CH2–CH=CH2 | 5.018 |
| 14f | H | –CH2–C6H5 | –CH2–CH=CH2 | 5.143 |
| 14g | H | H | –CH2–CH=CH2 | 4.533 |
| *: | 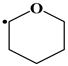 | |||
| Descriptors | Symbol | Class |
|---|---|---|
| Molecular weight | MW | Constitutional |
| Coefficient of partition Octanol/Water | LogP | Physico-chemical |
| Solubility | LogS | |
| Polarizability | Pol | |
| Hydrogen Bond Acceptor | HBA | |
| Hydrogen Bond Donor | HBD | |
| Molar Refractivity | MR | Geometrical |
| Molar Volume | MV | |
| Energy Total | ET | Quantum (Electronic) |
| Energy HOMO | EHOMO | |
| Energy LUMO | ELUMO | |
| Charges | qn | Mulliken Charges |
| Charges | qS | |
| Charges | qC1 | |
| Charges | qC2 | |
| Charges | qC3 | |
| Balaban Index | BIndx | Topological |
| Cluster Count | ClsC | |
| Molecular Topological Index | TIndx |
| Comp | pIC50 | LogP | MW | LogS | MR | MV | POL | EHOMO | ELUMO | ET | HBA | HBD | qS | qN | qC1 | qC2 | qC3 | Clsc | Tindx | Bindx |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 6a | 4.526 | 1.725 | 251.257 | −3.55 | 68.34 | 679.733 | 24.44 | −2.406 | −6.114 | −32,074.5 | 4 | 2 | −0.371 | −0.267 | 0.044 | −0.116 | 0.643 | 17 | 3549 | 106,143 |
| 6b | 4.947 | 2.487 | 330.153 | −4.26 | 75.874 | 735.382 | 27.066 | −2.585 | −6.254 | −102,106 | 4 | 2 | −0.373 | −0.26 | 0.046 | −0.223 | 0.799 | 18 | 3884 | 136,497 |
| 6c | 4.848 | 2.790 | 344.18 | −4.38 | 80.643 | 782.485 | 28.901 | −2.521 | −6.137 | −103,175 | 4 | 1 | 0.412 | −0.262 | 0.083 | −0.079 | 0.596 | 19 | 4627 | 177,387 |
| 6d | 5.092 | 3.346 | 370.218 | −5.04 | 89.806 | 877.383 | 32.379 | −2.653 | −6.337 | −105,282 | 4 | 1 | −0.03 | −0.207 | 0.015 | −0.228 | 0.62 | 21 | 6539 | 295,568 |
| 6e | 5.276 | 4.127 | 398.271 | −5.93 | 99.761 | 962.68 | 36.049 | −2.604 | −6.276 | −107,422 | 4 | 1 | −0.011 | −0.207 | 0.008 | −0.231 | 0.624 | 23 | 8921 | 472,150 |
| 6f | 5.310 | 3.961 | 386.26 | −5.67 | 94.516 | 947.696 | 34.406 | −2.673 | −6.319 | −106,385 | 4 | 1 | −0.17 | −0.238 | 0.01 | −0.18 | 0.589 | 22 | 7722 | 377,714 |
| 6g | 5.119 | 4.361 | 420.277 | −5.92 | 109.394 | 978.128 | 38.561 | −2.607 | −6.272 | −109,464 | 4 | 1 | −0.191 | 0.239 | −0.016 | −0.274 | 0.628 | 25 | 11,391 | 546,426 |
| 7a | 4.652 | 3.132 | 358.207 | −4.38 | 85.54 | 819.806 | 30.736 | −2.428 | −6.060 | −104,245 | 4 | 0 | 0.166 | 0.032 | 0.082 | −0.18 | 0.529 | 20 | 5375 | 224,427 |
| 7b | 5.092 | 4.703 | 434.304 | −5.94 | 114.291 | 1024.088 | 40.396 | −2.512 | −6.199 | −110,534 | 4 | 0 | −0.07 | 0.056 | −0.047 | −0.338 | 0.675 | 26 | 12,739 | 653,163 |
| 7c | 5.086 | 4.469 | 412.298 | −5.94 | 104.658 | 1030.626 | 37.884 | −2.509 | −6.201 | −108,492 | 4 | 0 | −0.07 | 0.056 | −0.015 | −0.295 | 0.542 | 24 | 10,061 | 573,023 |
| 7d | 4.770 | 5.025 | 438.336 | −5.94 | 113.821 | 1083.626 | 41.362 | −2.486 | −6.167 | −110,599 | 4 | 0 | −0.07 | 0.34 | −0.034 | −0.07 | 0.315 | 26 | 12,885 | 843,380 |
| 7e(ref) | 5.337 | 5.247 | 440.352 | −6.77 | 113.824 | 1107.71 | 41.554 | −2.465 | −6.149 | −110,632 | 4 | 0 | −0.231 | 0.412 | −0.031 | 0.104 | 0.162 | 26 | 12,715 | 833,233 |
| 7f | 4.910 | 5.639 | 454.379 | −7.23 | 118.531 | 1151.608 | 43.389 | −2.476 | −6.166 | −111,703 | 4 | 0 | −0.073 | 0.391 | −0.038 | 0.157 | −0.092 | 27 | 14,584 | 1,020,188 |
| 7g | 5.000 | 5.805 | 466.39 | −7.49 | 123.776 | 1190.101 | 45.032 | −2.443 | −6.144 | −112,739 | 4 | 0 | −0.129 | 0.378 | −0.021 | 0.032 | 0.149 | 28 | 16,299 | 1,217,684 |
| 7h | 4.987 | 6.039 | 47.544 | −7.49 | 133.409 | 1195.143 | 47.544 | −2.504 | −6.185 | −114,781 | 4 | 0 | −0.17 | 0.418 | 0.019 | 0.178 | −0.107 | 30 | 19,801 | 1,300,406 |
| 7i | 4.917 | 4.303 | 400.287 | −5.68 | 99.413 | 999.466 | 36.241 | −2.525 | −6.227 | −107,456 | 4 | 0 | −0.067 | 0.057 | 0.002 | −0.289 | 0.554 | 23 | 8758 | 462,362 |
| 7j | 5.102 | 4.859 | 426.325 | −6.35 | 108.576 | 1054.01 | 39.719 | −2.515 | −6.191 | −109,562 | 4 | 0 | −0.107 | 0.322 | −0.012 | −0.01 | 0.279 | 25 | 11,350 | 692,217 |
| 7k | 5.013 | 5.081 | 428.341 | −6.51 | 108.579 | 1077.561 | 39.911 | −2.481 | −6.174 | −109,596 | 4 | 0 | −0.235 | 0.411 | −0.013 | 0.106 | 0.18 | 25 | 11,188 | 683,230 |
| 7m | 5.180 | 5.873 | 476.385 | −7.23 | 128.164 | 1165.816 | 45.901 | −2.517 | −6.033 | −113,744 | 4 | 0 | −0.201 | 0.383 | 0.029 | 0.281 | −0.302 | 29 | 17,754 | 1,097,466 |
| 12 | 4.914 | 3.653 | 375.439 | −5.86 | 104.67 | 1029.891 | 38.791 | −2.272 | −5.889 | −42,615.8 | 5 | 1 | −0.324 | −0.253 | 0.036 | −0.366 | 0.871 | 26 | 12,024 | 614,951 |
| 13d | 4.644 | 5.332 | 443.558 | −7.42 | 128.685 | 1274.358 | 47.774 | −2.127 | −5.772 | −47,932.8 | 5 | 0 | −0.167 | 0.364 | −0.041 | −0.069 | 0.435 | 31 | 20,797 | 1,445,876 |
| 13e | 4.686 | 5.166 | 431.547 | −7.16 | 123.44 | 1242.555 | 46.131 | −2.154 | −5.791 | −46,896.2 | 5 | 0 | −0.118 | 0.368 | −0.046 | 0.104 | 0.183 | 30 | 18,773 | 1,230,606 |
| 14b | 5.194 | 3.574 | 333.402 | −5.49 | 95.931 | 938.266 | 35.258 | −2.229 | −5.880 | −38,451.7 | 4 | 1 | −0.348 | 0.369 | 0.027 | 0.227 | 0.096 | 23 | 8545 | 435,114 |
| 14d | 4.921 | 4.132 | 359.44 | −6.22 | 105.883 | 1015.594 | 38.736 | −2.212 | −5.880 | −40,568.2 | 4 | 1 | −0.209 | 0.338 | −0.02 | 0.157 | 0.033 | 25 | 11,390 | 672,700 |
| 14e | 5.018 | 3.966 | 347.429 | −5.97 | 100.638 | 982.053 | 37.093 | −2.237 | −5.901 | −39,531.6 | 4 | 1 | −0.157 | 0.356 | −0.041 | 0.293 | −0.25 | 24 | 10,034 | 55,093 |
| 14f | 5.143 | 4.366 | 381.446 | −6.22 | 115.516 | 1025.745 | 41.248 | −2.269 | −5.927 | −42,610.1 | 4 | 1 | −0.22 | 0.399 | 0.019 | 0.301 | −0.202 | 27 | 14,174 | 750,061 |
| 14g | 4.533 | 2.453 | 291.321 | −4.66 | 81.868 | 797.075 | 29.753 | −2.363 | −6.008 | −35,251.2 | 4 | 2 | −0.312 | 0.268 | 0.043 | −0.166 | 0.521 | 20 | 5681 | 226,445 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Derki, N.-E.H.; Kerassa, A.; Belaidi, S.; Derki, M.; Yamari, I.; Samadi, A.; Chtita, S. Computer-Aided Strategy on 5-(Substituted benzylidene) Thiazolidine-2,4-Diones to Develop New and Potent PTP1B Inhibitors: QSAR Modeling, Molecular Docking, Molecular Dynamics, PASS Predictions, and DFT Investigations. Molecules 2024, 29, 822. https://doi.org/10.3390/molecules29040822
Derki N-EH, Kerassa A, Belaidi S, Derki M, Yamari I, Samadi A, Chtita S. Computer-Aided Strategy on 5-(Substituted benzylidene) Thiazolidine-2,4-Diones to Develop New and Potent PTP1B Inhibitors: QSAR Modeling, Molecular Docking, Molecular Dynamics, PASS Predictions, and DFT Investigations. Molecules. 2024; 29(4):822. https://doi.org/10.3390/molecules29040822
Chicago/Turabian StyleDerki, Nour-El Houda, Aicha Kerassa, Salah Belaidi, Maroua Derki, Imane Yamari, Abdelouahid Samadi, and Samir Chtita. 2024. "Computer-Aided Strategy on 5-(Substituted benzylidene) Thiazolidine-2,4-Diones to Develop New and Potent PTP1B Inhibitors: QSAR Modeling, Molecular Docking, Molecular Dynamics, PASS Predictions, and DFT Investigations" Molecules 29, no. 4: 822. https://doi.org/10.3390/molecules29040822
APA StyleDerki, N.-E. H., Kerassa, A., Belaidi, S., Derki, M., Yamari, I., Samadi, A., & Chtita, S. (2024). Computer-Aided Strategy on 5-(Substituted benzylidene) Thiazolidine-2,4-Diones to Develop New and Potent PTP1B Inhibitors: QSAR Modeling, Molecular Docking, Molecular Dynamics, PASS Predictions, and DFT Investigations. Molecules, 29(4), 822. https://doi.org/10.3390/molecules29040822