
Machine Learning Methods as a Cost-Effective Alternative to Physics-Based Binding Free Energy Calculations


Abstract
:1. Introduction
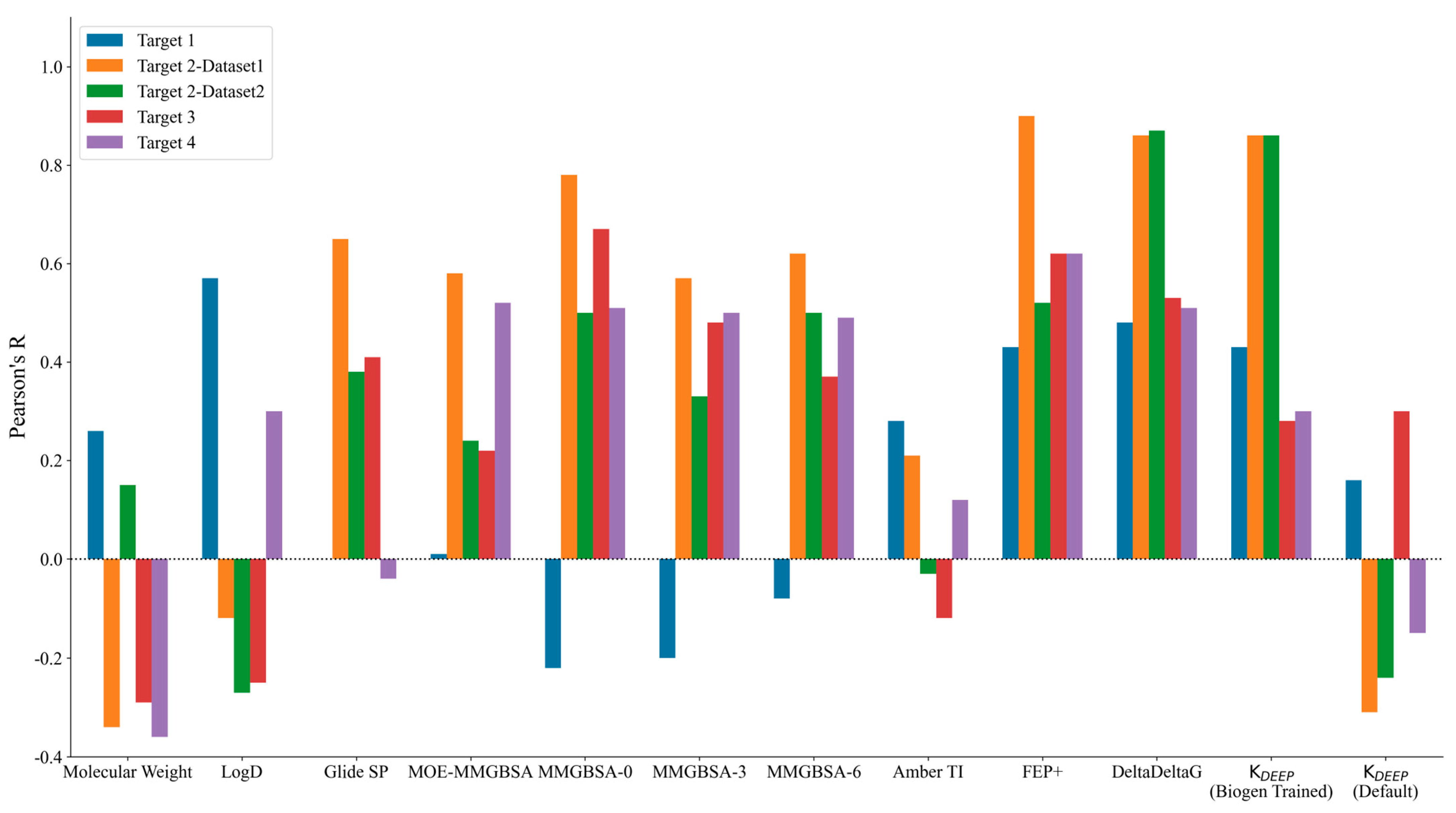
2. Results and Discussion
3. Methods
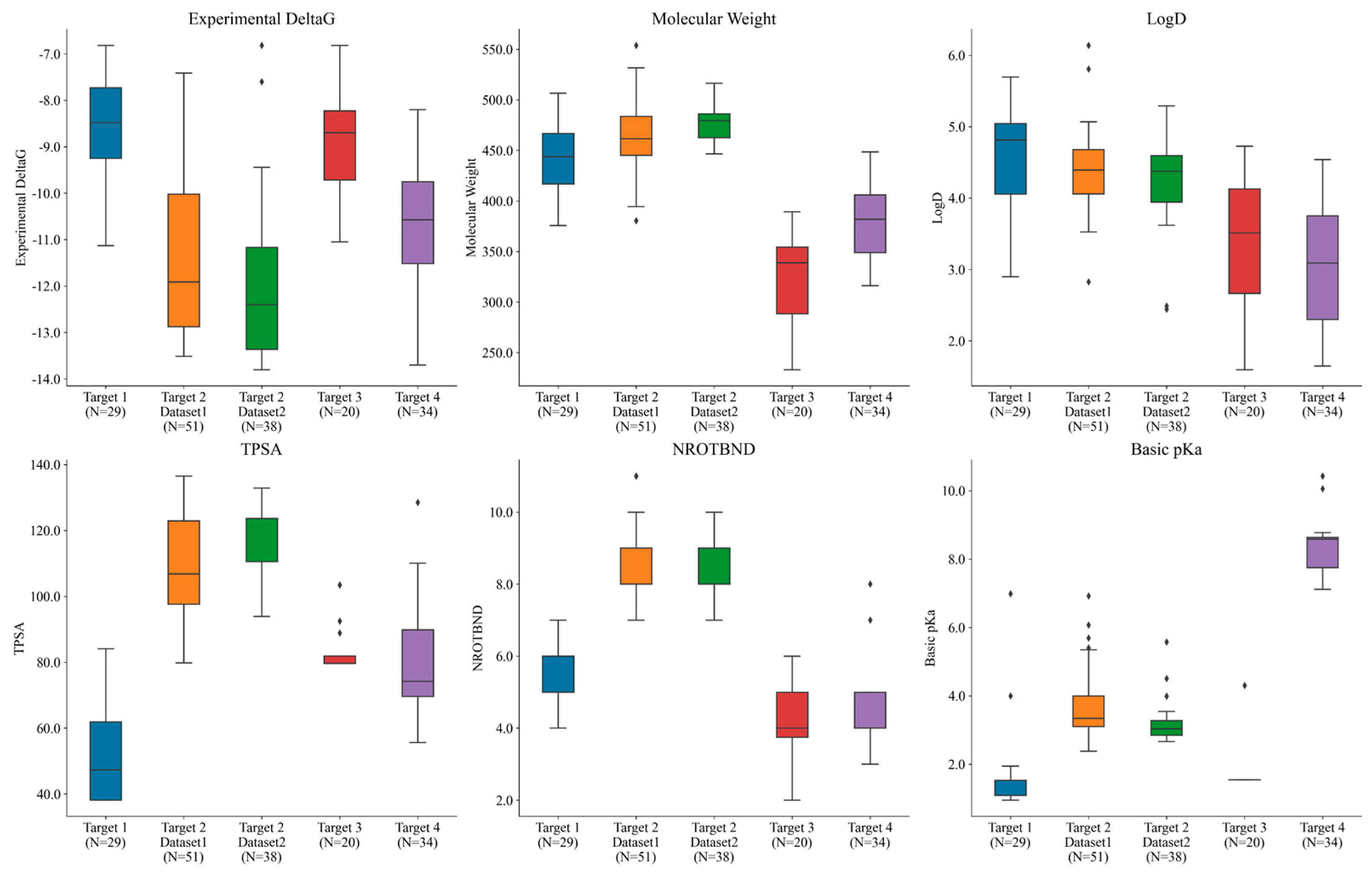
3.1. Datasets
3.2. Computational Details
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Merz, K.M.; Kollman, P.A. Free Energy Perturbation Simulations of the Inhibition of Thermolysin: Prediction of the Free Energy of Binding of a New Inhibitor. J. Am. Chem. Soc. 1989, 111, 5649–5658. [Google Scholar] [CrossRef]
- Wang, L.; Wu, Y.; Deng, Y.; Kim, B.; Pierce, L.; Krilov, G.; Lupyan, D.; Robinson, S.; Dahlgren, M.K.; Greenwood, J.; et al. Accurate and Reliable Prediction of Relative Ligand Binding Potency in Prospective Drug Discovery by Way of a Modern Free-Energy Calculation Protocol and Force Field. J. Am. Chem. Soc. 2015, 137, 2695–2703. [Google Scholar] [CrossRef]
- Mobley, D.L.; Gilson, M.K. Predicting Binding Free Energies: Frontiers and Benchmarks. Annu. Rev. Biophys. 2017, 46, 531–558. [Google Scholar] [CrossRef] [PubMed]
- Mobley, D.L.; Klimovich, P.V. Perspective: Alchemical Free Energy Calculations for Drug Discovery. J. Chem. Phys. 2012, 137, 230901. [Google Scholar] [CrossRef] [PubMed]
- Schindler, C.E.M.; Baumann, H.; Blum, A.; Böse, D.; Buchstaller, H.-P.; Burgdorf, L.; Cappel, D.; Chekler, E.; Czodrowski, P.; Dorsch, D.; et al. Large-Scale Assessment of Binding Free Energy Calculations in Active Drug Discovery Projects. J. Chem. Inf. Model. 2020, 60, 5457–5474. [Google Scholar] [CrossRef] [PubMed]
- Abel, R.; Wang, L.; Mobley, D.L.; Friesner, R.A. A Critical Review of Validation, Blind Testing, and Real- World Use of Alchemical Protein-Ligand Binding Free Energy Calculations. Curr. Top. Med. Chem. 2017, 17, 2577–2585. [Google Scholar] [CrossRef] [PubMed]
- Armacost, K.A.; Riniker, S.; Cournia, Z. Novel Directions in Free Energy Methods and Applications. J. Chem. Inf. Model. 2020, 60, 1–5. [Google Scholar] [CrossRef]
- Song, L.F.; Lee, T.-S.; Zhu, C.; York, D.M.; Merz, K.M., Jr. Using AMBER18 for Relative Free Energy Calculations. J. Chem. Inf. Model. 2019, 59, 3128–3135. [Google Scholar] [CrossRef] [PubMed]
- Ucisik, M.N.; Zheng, Z.; Faver, J.C.; Merz, K.M. Bringing Clarity to the Prediction of Protein–Ligand Binding Free Energies via “Blurring”. J. Chem. Theory Comput. 2014, 10, 1314–1325. [Google Scholar] [CrossRef]
- Cournia, Z.; Allen, B.; Sherman, W. Relative Binding Free Energy Calculations in Drug Discovery: Recent Advances and Practical Considerations. J. Chem. Inf. Model. 2017, 57, 2911–2937. [Google Scholar] [CrossRef]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 1. Method and Assessment of Docking Accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef] [PubMed]
- Jain, A.N. Surflex: Fully Automatic Flexible Molecular Docking Using a Molecular Similarity-Based Search Engine. J. Med. Chem. 2003, 46, 499–511. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization, and Multithreading. J. Comput. Chem. 2009, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Jones, G.; Willett, P.; Glen, R.C.; Leach, A.R.; Taylor, R. Development and Validation of a Genetic Algorithm for Flexible docking11Edited by F. E. Cohen. J. Mol. Biol. 1997, 267, 727–748. [Google Scholar] [CrossRef] [PubMed]
- Rastelli, G.; Del Rio, A.; Degliesposti, G.; Sgobba, M. Fast and Accurate Predictions of Binding Free Energies Using MM-PBSA and MM-GBSA. J. Comput. Chem. 2010, 31, 797–810. [Google Scholar] [CrossRef]
- Kuhn, B.; Kollman, P.A. Binding of a Diverse Set of Ligands to Avidin and Streptavidin: An Accurate Quantitative Prediction of Their Relative Affinities by a Combination of Molecular Mechanics and Continuum Solvent Models. J. Med. Chem. 2000, 43, 3786–3791. [Google Scholar] [CrossRef]
- Kollman, P.A.; Massova, I.; Reyes, C.; Kuhn, B.; Huo, S.; Chong, L.; Lee, M.; Lee, T.; Duan, Y.; Wang, W.; et al. Calculating Structures and Free Energies of Complex Molecules: Combining Molecular Mechanics and Continuum Models. Acc. Chem. Res. 2000, 33, 889–897. [Google Scholar] [CrossRef]
- Li, Y.; Liu, Z.; Wang, R. Test MM-PB/SA on True Conformational Ensembles of Protein−Ligand Complexes. J. Chem. Inf. Model. 2010, 50, 1682–1692. [Google Scholar] [CrossRef]
- Wang, E.; Sun, H.; Wang, J.; Wang, Z.; Liu, H.; Zhang, J.Z.H.; Hou, T. End-Point Binding Free Energy Calculation with MM/PBSA and MM/GBSA: Strategies and Applications in Drug Design. Chem. Rev. 2019, 119, 9478–9508. [Google Scholar] [CrossRef] [PubMed]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA Methods to Estimate Ligand-Binding Affinities. Expert Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Duan, L.; Chen, F.; Liu, H.; Wang, Z.; Pan, P.; Zhu, F.; Zhang, J.Z.H.; Hou, T. Assessing the Performance of MM/PBSA and MM/GBSA Methods. 7. Entropy Effects on the Performance of End-Point Binding Free Energy Calculation Approaches. Phys. Chem. Chem. Phys. 2018, 20, 14450–14460. [Google Scholar] [CrossRef]
- Gaieb, Z.; Liu, S.; Gathiaka, S.; Chiu, M.; Yang, H.; Shao, C.; Feher, V.A.; Walters, W.P.; Kuhn, B.; Rudolph, M.G.; et al. D3R Grand Challenge 2: Blind Prediction of Protein-Ligand Poses, Affinity Rankings, and Relative Binding Free Energies. J. Comput. Aided Mol. Des. 2018, 32, 1–20. [Google Scholar] [CrossRef]
- Goldfeld, D.A.; Murphy, R.; Kim, B.; Wang, L.; Beuming, T.; Abel, R.; Friesner, R.A. Docking and Free Energy Perturbation Studies of Ligand Binding in the Kappa Opioid Receptor. J. Phys. Chem. B 2015, 119, 824–835. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Berne, B.J.; Friesner, R.A. On Achieving High Accuracy and Reliability in the Calculation of Relative Protein–Ligand Binding Affinities. Proc. Natl. Acad. Sci. USA 2012, 109, 1937–1942. [Google Scholar] [CrossRef]
- Ciordia, M.; Pérez-Benito, L.; Delgado, F.; Trabanco, A.A.; Tresadern, G. Application of Free Energy Perturbation for the Design of BACE1 Inhibitors. J. Chem. Inf. Model. 2016, 56, 1856–1871. [Google Scholar] [CrossRef]
- Wan, S.; Bhati, A.P.; Skerratt, S.; Omoto, K.; Shanmugasundaram, V.; Bagal, S.K.; Coveney, P.V. Evaluation and Characterization of Trk Kinase Inhibitors for the Treatment of Pain: Reliable Binding Affinity Predictions from Theory and Computation. J. Chem. Inf. Model. 2017, 57, 897–909. [Google Scholar] [CrossRef]
- Lenselink, E.B.; Louvel, J.; Forti, A.F.; van Veldhoven, J.P.D.; de Vries, H.; Mulder-Krieger, T.; McRobb, F.M.; Negri, A.; Goose, J.; Abel, R.; et al. Predicting Binding Affinities for GPCR Ligands Using Free-Energy Perturbation. ACS Omega 2016, 1, 293–304. [Google Scholar] [CrossRef] [PubMed]
- Heinzelmann, G.; Henriksen, N.M.; Gilson, M.K. Attach-Pull-Release Calculations of Ligand Binding and Conformational Changes on the First BRD4 Bromodomain. J. Chem. Theory Comput. 2017, 13, 3260–3275. [Google Scholar] [CrossRef] [PubMed]
- Keränen, H.; Pérez-Benito, L.; Ciordia, M.; Delgado, F.; Steinbrecher, T.B.; Oehlrich, D.; van Vlijmen, H.W.T.; Trabanco, A.A.; Tresadern, G. Acylguanidine Beta Secretase 1 Inhibitors: A Combined Experimental and Free Energy Perturbation Study. J. Chem. Theory Comput. 2017, 13, 1439–1453. [Google Scholar] [CrossRef]
- Pérez-Benito, L.; Keränen, H.; van Vlijmen, H.; Tresadern, G. Predicting Binding Free Energies of PDE2 Inhibitors. The Difficulties of Protein Conformation. Sci. Rep. 2018, 8, 4883. [Google Scholar] [CrossRef]
- Abel, R.; Wang, L.; Harder, E.D.; Berne, B.J.; Friesner, R.A. Advancing Drug Discovery through Enhanced Free Energy Calculations. Acc. Chem. Res. 2017, 50, 1625–1632. [Google Scholar] [CrossRef] [PubMed]
- Chemical Computing Group ULC. Molecular Operating Environment (MOE); Chemical Computing Group: Montreal, QC, Canada, 2023. [Google Scholar]
- Özen, A.; Perola, E.; Brooijmans, N.; Kim, J. Prospective Application of Free Energy Methods in Drug Discovery Programs. In Free Energy Methods in Drug Discovery: Current State and Future Directions; ACS Symposium Series; American Chemical Society: Washington, DC, USA, 2021; Volume 1397, pp. 127–141. [Google Scholar] [CrossRef]
- Hong, R.S.; Mattei, A.; Sheikh, A.Y.; Bhardwaj, R.M.; Bellucci, M.A.; McDaniel, K.F.; Pierce, M.O.; Sun, G.; Li, S.; Wang, L.; et al. Novel Physics-Based Ensemble Modeling Approach That Utilizes 3D Molecular Conformation and Packing to Access Aqueous Thermodynamic Solubility: A Case Study of Orally Available Bromodomain and Extraterminal Domain Inhibitor Lead Optimization Series. J. Chem. Inf. Model. 2021, 61, 1412–1426. [Google Scholar] [CrossRef] [PubMed]
- Knight, J.L.; Leswing, K.; Bos, P.H.; Wang, L. Impacting Drug Discovery Projects with Large-Scale Enumerations, Machine Learning Strategies, and Free-Energy Predictions. In Free Energy Methods in Drug Discovery: Current State and Future Directions; ACS Symposium Series; American Chemical Society: Washington, DC, USA, 2021; Volume 1397, pp. 205–226. [Google Scholar] [CrossRef]
- Katz, D.; DiMattia, M.A.; Sindhikara, D.; Li, H.; Abraham, N.; Leffler, A.E. Potency- and Selectivity-Enhancing Mutations of Conotoxins for Nicotinic Acetylcholine Receptors Can Be Predicted Using Accurate Free-Energy Calculations. Mar. Drugs 2021, 19, 367. [Google Scholar] [CrossRef] [PubMed]
- Wójcikowski, M.; Ballester, P.J.; Siedlecki, P. Performance of Machine-Learning Scoring Functions in Structure-Based Virtual Screening. Sci. Rep. 2017, 7, 46710. [Google Scholar] [CrossRef]
- Pereira, J.C.; Caffarena, E.R.; dos Santos, C.N. Boosting Docking-Based Virtual Screening with Deep Learning. J. Chem. Inf. Model. 2016, 56, 2495–2506. [Google Scholar] [CrossRef] [PubMed]
- Ragoza, M.; Hochuli, J.; Idrobo, E.; Sunseri, J.; Koes, D.R. Protein-Ligand Scoring with Convolutional Neural Networks. J. Chem. Inf. Model. 2017, 57, 942–957. [Google Scholar] [CrossRef] [PubMed]
- Amini, A.; Shrimpton, P.J.; Muggleton, S.H.; Sternberg, M.J.E. A General Approach for Developing System-Specific Functions to Score Protein–Ligand Docked Complexes Using Support Vector Inductive Logic Programming. Proteins Struct. Funct. Bioinform. 2007, 69, 823–831. [Google Scholar] [CrossRef] [PubMed]
- Zhan, W.; Li, D.; Che, J.; Zhang, L.; Yang, B.; Hu, Y.; Liu, T.; Dong, X. Integrating Docking Scores, Interaction Profiles and Molecular Descriptors to Improve the Accuracy of Molecular Docking: Toward the Discovery of Novel Akt1 Inhibitors. Eur. J. Med. Chem. 2014, 75, 11–20. [Google Scholar] [CrossRef]
- Zilian, D.; Sotriffer, C.A. SFCscoreRF: A Random Forest-Based Scoring Function for Improved Affinity Prediction of Protein–Ligand Complexes. J. Chem. Inf. Model. 2013, 53, 1923–1933. [Google Scholar] [CrossRef]
- Ballester, P.J.; Mitchell, J.B.O. A Machine Learning Approach to Predicting Protein–Ligand Binding Affinity with Applications to Molecular Docking. Bioinformatics 2010, 26, 1169–1175. [Google Scholar] [CrossRef]
- Feinberg, E.N.; Sur, D.; Wu, Z.; Husic, B.E.; Mai, H.; Li, Y.; Sun, S.; Yang, J.; Ramsundar, B.; Pande, V.S. PotentialNet for Molecular Property Prediction. ACS Cent. Sci. 2018, 4, 1520–1530. [Google Scholar] [CrossRef]
- Nguyen, D.D.; Cang, Z.; Wu, K.; Wang, M.; Cao, Y.; Wei, G.-W. Mathematical Deep Learning for Pose and Binding Affinity Prediction and Ranking in D3R Grand Challenges. J. Comput. Aided Mol. Des. 2019, 33, 71–82. [Google Scholar] [CrossRef] [PubMed]
- Gaieb, Z.; Parks, C.D.; Chiu, M.; Yang, H.; Shao, C.; Walters, W.P.; Lambert, M.H.; Nevins, N.; Bembenek, S.D.; Ameriks, M.K.; et al. D3R Grand Challenge 3: Blind Prediction of Protein–Ligand Poses and Affinity Rankings. J. Comput. Aided Mol. Des. 2019, 33, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Ballester, P.J.; Schreyer, A.; Blundell, T.L. Does a More Precise Chemical Description of Protein–Ligand Complexes Lead to More Accurate Prediction of Binding Affinity? J. Chem. Inf. Model. 2014, 54, 944–955. [Google Scholar] [CrossRef] [PubMed]
- Stumpfe, D.; Hu, H.; Bajorath, J. Evolving Concept of Activity Cliffs. ACS Omega 2019, 4, 14360–14368. [Google Scholar] [CrossRef] [PubMed]
- Madhavi Sastry, G.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and Ligand Preparation: Parameters, Protocols, and Influence on Virtual Screening Enrichments. J. Comput. Aided Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, W.L.; Maxwell, D.S.; Tirado-Rives, J. Development and Testing of the OPLS All-Atom Force Field on Conformational Energetics and Properties of Organic Liquids. J. Am. Chem. Soc. 1996, 118, 11225–11236. [Google Scholar] [CrossRef]
- Harder, E.; Damm, W.; Maple, J.; Wu, C.; Reboul, M.; Xiang, J.Y.; Wang, L.; Lupyan, D.; Dahlgren, M.K.; Knight, J.L.; et al. OPLS3: A Force Field Providing Broad Coverage of Drug-like Small Molecules and Proteins. J. Chem. Theory Comput. 2016, 12, 281–296. [Google Scholar] [CrossRef]
- Shivakumar, D.; Williams, J.; Wu, Y.; Damm, W.; Shelley, J.; Sherman, W. Prediction of Absolute Solvation Free Energies Using Molecular Dynamics Free Energy Perturbation and the OPLS Force Field. J. Chem. Theory Comput. 2010, 6, 1509–1519. [Google Scholar] [CrossRef]
- Shelley, J.C.; Cholleti, A.; Frye, L.L.; Greenwood, J.R.; Timlin, M.R.; Uchimaya, M. Epik: A Software Program for pKaprediction and Protonation State Generation for Drug-like Molecules. J. Comput. Aided Mol. Des. 2007, 21, 681–691. [Google Scholar] [CrossRef]
- Greenwood, J.R.; Calkins, D.; Sullivan, A.P.; Shelley, J.C. Towards the Comprehensive, Rapid, and Accurate Prediction of the Favorable Tautomeric States of Drug-like Molecules in Aqueous Solution. J. Comput. Aided Mol. Des. 2010, 24, 591–604. [Google Scholar] [CrossRef]
- Li, J.; Abel, R.; Zhu, K.; Cao, Y.; Zhao, S.; Friesner, R.A. The VSGB 2.0 Model: A next Generation Energy Model for High Resolution Protein Structure Modeling. Proteins 2011, 79, 2794–2812. [Google Scholar] [CrossRef]
- Chen, W.; Cui, D.; Jerome, S.V.; Michino, M.; Lenselink, E.B.; Huggins, D.J.; Beautrait, A.; Vendome, J.; Abel, R.; Friesner, R.A.; et al. Enhancing Hit Discovery in Virtual Screening through Absolute Protein–Ligand Binding Free-Energy Calculations. J. Chem. Inf. Model. 2023, 63, 3171–3185. [Google Scholar] [CrossRef]
- Jiménez, J.; Škalič, M.; Martínez-Rosell, G.; De Fabritiis, G. K DEEP: Protein–Ligand Absolute Binding Affinity Prediction via 3D-Convolutional Neural Networks. J. Chem. Inf. Model. 2018, 58, 287–296. [Google Scholar] [CrossRef] [PubMed]
- Jiménez-Luna, J.; Pérez-Benito, L.; Martínez-Rosell, G.; Sciabola, S.; Torella, R.; Tresadern, G.; De Fabritiis, G. DeltaDelta Neural Networks for Lead Optimization of Small Molecule Potency. Chem. Sci. 2019, 10, 10911–10918. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Liu, Z.; Li, J.; Han, L.; Liu, J.; Zhao, Z.; Wang, R. Comparative Assessment of Scoring Functions on an Updated Benchmark: 1. Compilation of the Test Set. J. Chem. Inf. Model. 2014, 54, 1700–1716. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Train | Test | Max Similarity (µ) | Max Similarity (σ) | |
|---|---|---|---|---|
| Target 1 | 52 | 29 | 0.76 | 0.06 |
| Target 2–Dataset1 | 72 | 51 | 0.77 | 0.07 |
| Target 2–Dataset 2 | 158 | 38 | 0.84 | 0.07 |
| Target 3 | 57 | 20 | 0.83 | 0.11 |
| Target 4 | 195 | 34 | 0.81 | 0.09 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bansal, N.; Wang, Y.; Sciabola, S. Machine Learning Methods as a Cost-Effective Alternative to Physics-Based Binding Free Energy Calculations. Molecules 2024, 29, 830. https://doi.org/10.3390/molecules29040830
Bansal N, Wang Y, Sciabola S. Machine Learning Methods as a Cost-Effective Alternative to Physics-Based Binding Free Energy Calculations. Molecules. 2024; 29(4):830. https://doi.org/10.3390/molecules29040830
Chicago/Turabian StyleBansal, Nupur, Ye Wang, and Simone Sciabola. 2024. "Machine Learning Methods as a Cost-Effective Alternative to Physics-Based Binding Free Energy Calculations" Molecules 29, no. 4: 830. https://doi.org/10.3390/molecules29040830
APA StyleBansal, N., Wang, Y., & Sciabola, S. (2024). Machine Learning Methods as a Cost-Effective Alternative to Physics-Based Binding Free Energy Calculations. Molecules, 29(4), 830. https://doi.org/10.3390/molecules29040830