
Electrografting of Phenyl Phosphate Layers onto Glassy Carbon for Tuning Catalytic Activity toward the Hydrogen Evolution Reaction



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
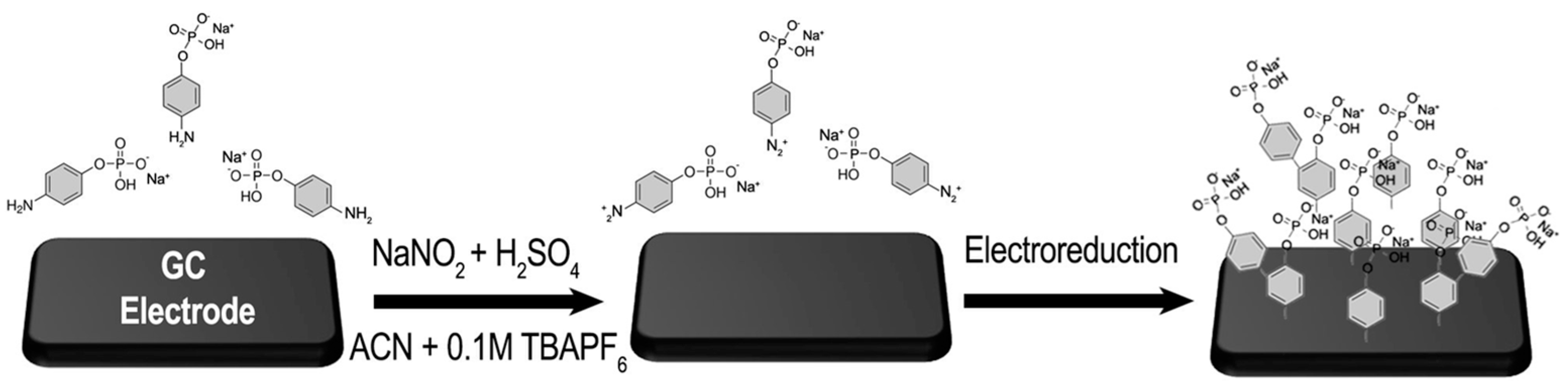
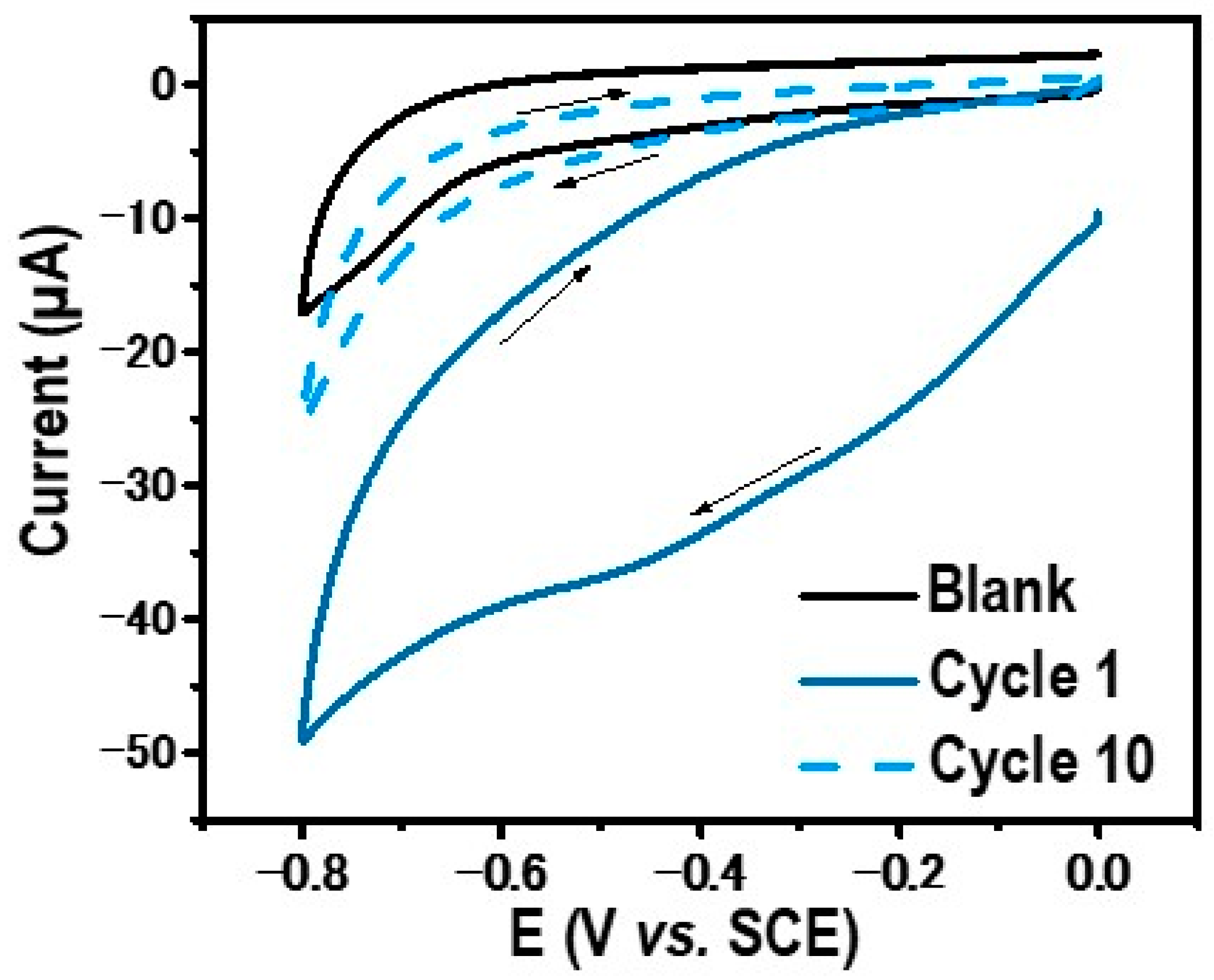
2.1. Electrografting of 4-Diazoniumphenyl Phosphate onto Glassy Carbon
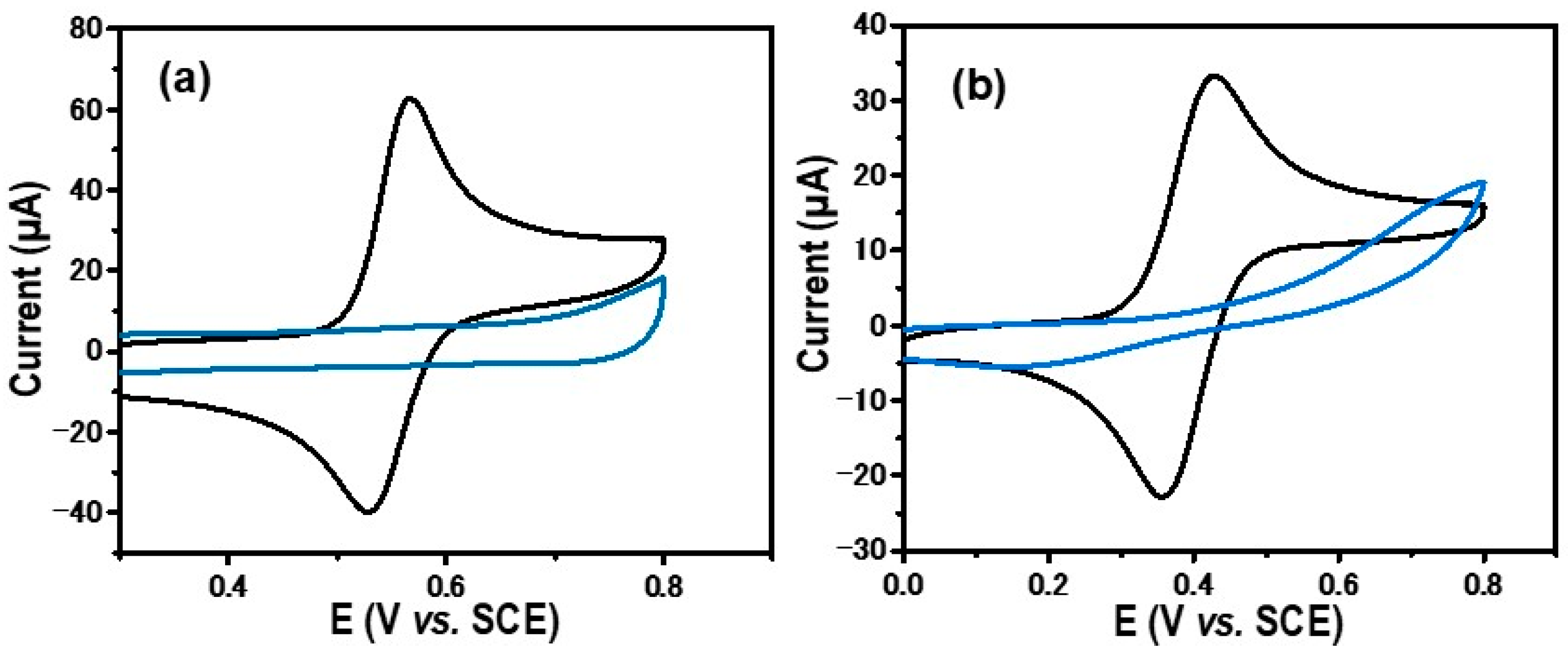
2.2. Electrodeposition of Palladium and Cobalt Nanoparticles on GC/Ph-PO4
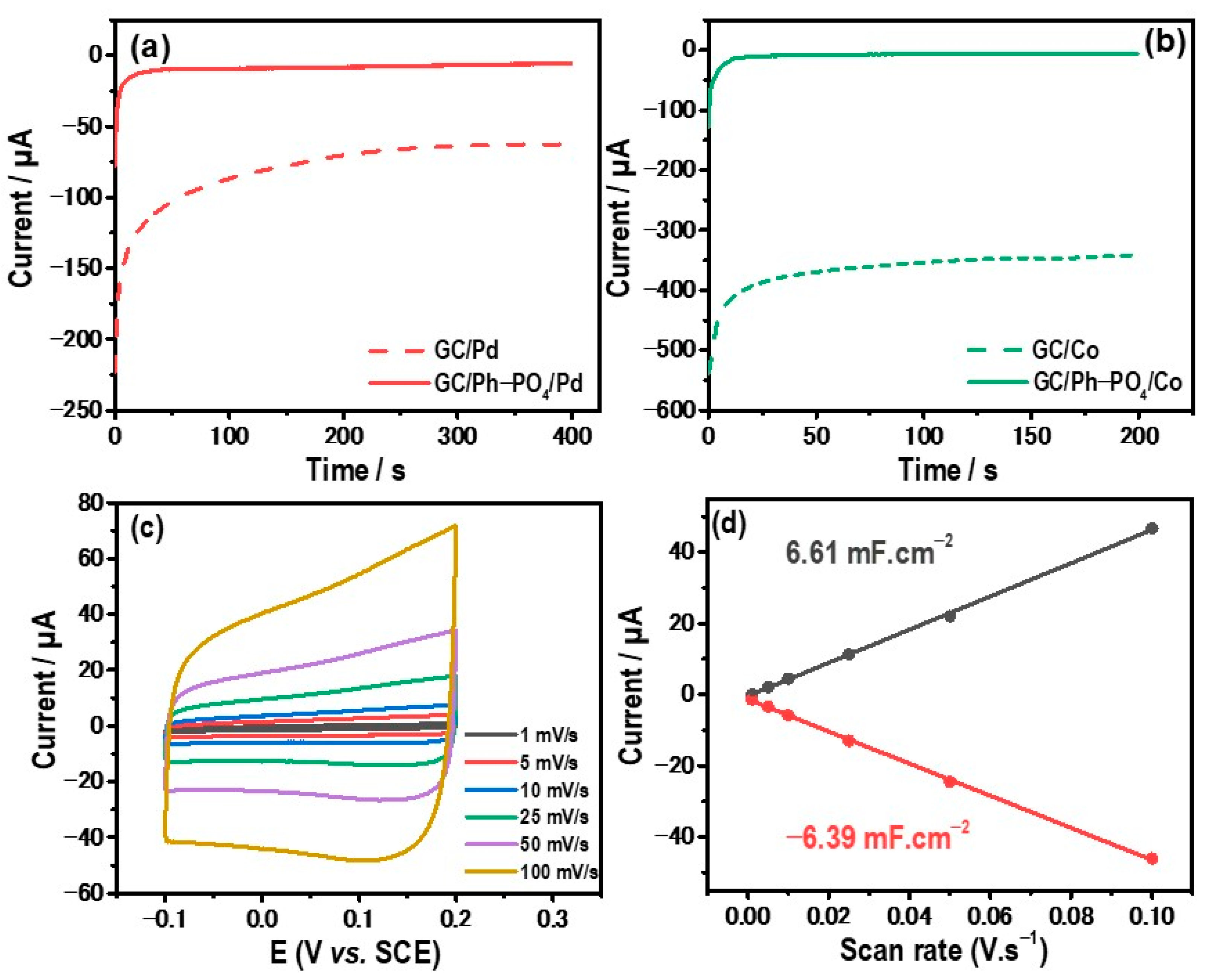
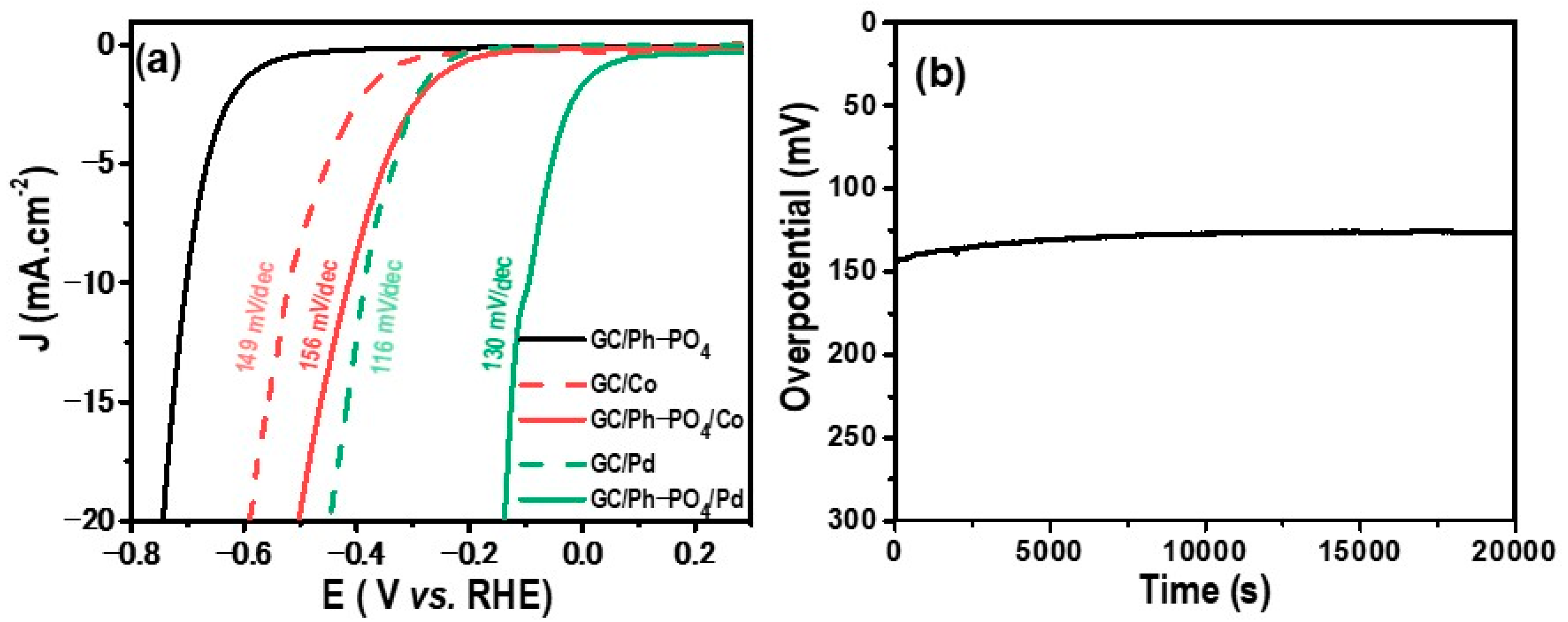
2.3. Electrochemical Activity
3. Materials and Methods
3.1. Materials
3.1.1. Chemicals
3.1.2. Surface Characterization
3.1.3. Electrochemical Measurements
3.2. Methods
3.2.1. GC Surface Functionalization
3.2.2. Metals Electrodeposition on Functionalized GC Electrodes
- C is the charge (C) integrated from the chronoamperometric curve;
- M is the molar mass of the metal;
- Ce− is the charge of a single electron (1.6 × 10−19 C);
- ne− is the number of electrons involved in the reduction (i.e., 2);
- NA is the Avogadro number (6.022 × 1023 mol−1);
- A is the surface of the electrode (0.0707 cm2).
3.2.3. Electrochemical Activity Investigations
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mozetič, M. Surface modification to improve properties of materials. Materials 2019, 12, 441. [Google Scholar] [CrossRef]
- Smith, R.K.; Lewis, P.A.; Weiss, P.S. Patterning self-assembled monolayers. Prog. Surf. Sci. 2004, 75, 1–68. [Google Scholar] [CrossRef]
- Pinson, J.; Podvorica, F. Attachment of organic layers to conductive or semiconductive surfaces by reduction of diazonium salts. Chem. Soc. Rev. 2005, 34, 429–439. [Google Scholar] [CrossRef]
- Orqusha, N. Grafting of the gold surface by heterocyclic moieties derived through electrochemical oxidation of amino triazole—An experimental and “ab initio” study. RSC Adv. 2022, 12, 23017–23025. [Google Scholar] [CrossRef]
- Hetemi, D.; Noël, N.; Pinson, J. Grafting of diazonium salts on surfaces: Application to biosensors. Biosensors 2020, 10, 4. [Google Scholar] [CrossRef] [PubMed]
- Bélanger, D.; Pinson, J. Electrografting: A powerful method for surface modification. Chem. Soc. Rev. 2011, 40, 3995. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Huez, C.; Nguyen, Q.V.; Bellynck, S.; Decorse, P.; Martin, P.; Lacroix, J.C. Nanometer-thick bilayers by stepwise electrochemical reduction of diazonium compounds for molecular junctions. ACS Appl. Nano Mater. 2021, 4, 13861–13870. [Google Scholar] [CrossRef]
- Menanteau, T.; Dias, M.; Levillain, E.; Downard, A.J.; Breton, T. Electrografting via diazonium chemistry: The key role of the aryl substituent in the layer growth mechanism. J. Phys. Chem. C 2016, 120, 4423–4429. [Google Scholar] [CrossRef]
- Baranton, S.; Belanger, D. Electrochemical derivatization of carbon surface by reduction of in situ generated diazonium cations. J. Phys. Chem. B 2005, 109, 24401–24410. [Google Scholar] [CrossRef] [PubMed]
- Baranton, S.; Bélanger, D. In situ generation of diazonium cations in organic electrolyte for electrochemical modification of electrode surface. Electrochim. Acta 2008, 53, 6961–6967. [Google Scholar] [CrossRef]
- Shul, G.; Parent, R.; Mosqueda, H.A.; Bélanger, D. Localized in situ generation of diazonium cations by electrocatalytic formation of a diazotization reagent. ACS Appl. Mater. Interfaces 2013, 5, 1468–1473. [Google Scholar] [CrossRef]
- Smith, W. The role of fuel cells in energy storage. J. Power Sources 2000, 86, 74–83. [Google Scholar] [CrossRef]
- Tang, J.; Xu, X.; Tang, T.; Zhong, Y.; Shao, Z. Perovskite-based electrocatalysts for cost-effective ultrahigh-current-density water splitting in anion exchange membrane electrolyzer cell. Small Methods 2022, 6, 2201099. [Google Scholar] [CrossRef]
- Santos, D.M.F.; Šljukić, B. Advanced materials for electrochemical energy conversion and storage devices. Materials 2021, 14, 7711. [Google Scholar] [CrossRef]
- Espegren, K.; Damman, S.; Pisciella, P.; Graabak, I.; Tomasgard, A. The role of hydrogen in the transition from a petroleum economy to a low-carbon society. Int. J. Hydrogen Energy 2021, 46, 23125–23138. [Google Scholar] [CrossRef]
- Sun, H.; Xu, X.; Kim, H.; Shao, Z.; Jung, W.C. Advanced electrocatalysts with unusual active sites for electrochemical water splitting. InfoMat 2023, 6, e12494. [Google Scholar] [CrossRef]
- Li, M.; Yin, W.; Pan, J.; Zhu, Y.; Sun, N.; Zhang, X.; Wan, Y.; Luo, Z.; Yi, L.; Wang, L. Hydrogen spillover as a promising strategy for boosting heterogeneous catalysis and hydrogen storage. Chem. Eng. J. 2023, 471, 144691. [Google Scholar] [CrossRef]
- Kumar, S.; Kaur, R.; Sharma, S. Recent reports on hydrogen evolution reactions and catalysis. Results Chem. 2022, 4, 100613. [Google Scholar] [CrossRef]
- Abdelghafar, F.; Xu, X.; Jiang, S.P.; Shao, Z. Designing single-atom catalysts toward improved alkaline hydrogen evolution reaction. Mater. Rep. Energy 2022, 2, 100144. [Google Scholar] [CrossRef]
- Smołka, S.; Krukiewicz, K. Catalyst Design through Grafting of Diazonium Salts—A Critical Review on Catalyst Stability. Int. J. Mol. Sci. 2023, 24, 12575. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.-Z.; Bencherif, S.; Pham-Truong, T.-N.; Ghilane, J. Immobilization of molecule-based ionic liquids: A promising approach to improve electrocatalyst performance towards the hydrogen evolution reaction. New J. Chem. 2022, 46, 454–464. [Google Scholar] [CrossRef]
- Pham-Truong, T.-N.; Mebarki, O.; Ranjan, C.; Randriamahazaka, H.; Ghilane, J. Electrochemical growth of metallic nanoparticles onto immobilized polymer brush ionic liquid as a hybrid electrocatalyst for the hydrogen evolution reaction. ACS Appl. Mater. Interfaces 2019, 11, 38265–38275. [Google Scholar] [CrossRef]
- Shukla, C.A.; Kulkarni, A.A.; Ranade, V.V. Selectivity engineering of the diazotization reaction in a continuous flow reactor. React. Chem. Eng. 2016, 1, 387–396. [Google Scholar] [CrossRef]
- Phal, P.; Shimizu, K.; Mwanza, D.; Mashazi, P.; Shchukarev, A.; Tesfalidet, S. Electrografting of 4-carboxybenzenediazonium on glassy carbon electrode: The effect of concentration on the formation of mono and multilayers. Molecules 2020, 25, 4575. [Google Scholar] [CrossRef] [PubMed]
- Kurapati, N.; Pathirathna, P.; Ziegler, C.J.; Amemiya, S. Adsorption and electron-transfer mechanisms of ferrocene carboxylates and sulfonates at highly oriented pyrolytic graphite. ChemElectroChem 2019, 6, 5651–5660. [Google Scholar] [CrossRef]
- Eblagon, K.M.; Arenillas, A.; Malaika, A.; Pereira, M.F.R.; Figueiredo, J.L. The influence of the surface chemistry of phosphorylated carbon xerogel catalysts on the production of HMF from fructose in water. Fuel 2023, 334, 126610. [Google Scholar] [CrossRef]
- Mattiuzzi, A.; Jabin, I.; Mangeney, C.; Roux, C.; Reinaud, O.; Santos, L.; Bergamini, J.-F.; Hapiot, P.; Lagrost, C. Electrografting of calix[4]arenediazonium salts to form versatile robust platforms for spatially controlled surface functionalization. Nat. Commun. 2012, 3, 1130. [Google Scholar] [CrossRef]
- Wu, X.; Gong, K.; Zhao, G.; Lou, W.; Wang, X.; Liu, W. Mechanical synthesis of chemically bonded phosphorus—Graphene hybrid as high-temperature lubricating oil additive. RSC Adv. 2018, 8, 4595–4603. [Google Scholar] [CrossRef] [PubMed]
- Asnavandi, M.; Suryanto, B.H.R.; Zhao, C. Controlled electrodeposition of nanostructured Pd thin films from protic ionic liquids for electrocatalytic oxygen reduction reactions. RSC Adv. 2015, 5, 74017–74023. [Google Scholar] [CrossRef]
- Fayette, M.; Bertocci, U.; Stafford, G.R. In situ stress measurements during cobalt electrodeposition on (111)-textured Au. J. Electrochem. Soc. 2016, 163, D146. [Google Scholar] [CrossRef]
- Kibis, L.S.; Simanenko, A.A.; Stadnichenko, A.I.; Zaikovskii, V.I.; Boronin, A.I. Probing of Pd4+ species in a PdOx-CeO2 system by X-ray photoelectron spectroscopy. J. Phys. Chem. C 2021, 125, 20845–20854. [Google Scholar] [CrossRef]
- Martinez, E.Y.; Zhu, K.; Li, C.W. Influence of the defect stability on n-type conductivity in electron-doped α- and β-Co(OH)2 nanosheets. Inorg. Chem. 2021, 60, 6950–6956. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Liu, H.; Martens, W.N.; Frost, R.L. Synthesis and characterization of Cobalt hydroxide, Cobalt oxyhydroxide, and Cobalt oxide nanodiscs. J. Phys. Chem. C 2010, 114, 111–119. [Google Scholar] [CrossRef]
- Tourneur, J.; Lagrost, C.; Fabre, B. Robust and bifunctional electrodeposited NiCoCr ternary alloy for alkaline water electrolysis. Adv. Energy Sustain. Res. 2024, 5, 2300133. [Google Scholar] [CrossRef]
- Li, H.B.; Yu, M.H.; Lu, X.H.; Liu, P.; Liang, Y.; Xiao, J.; Tong, X.Y.; Yang, G.W. Amorphous cobalt hydroxide with superior pseudocapacitive performance. ACS Appl. Mater. Interfaces 2014, 6, 745–749. [Google Scholar] [CrossRef]
- Bao, F.; Kemppainen, E.; Dorbandt, I.; Bors, R.; Xi, F.; Schlatmann, R.; van de Krol, R.; Calnan, S. Understanding the hydrogen evolution reaction kinetics of electrodeposited nickel-molybdenum in acidic, near-neutral, and alkaline conditions. ChemElectroChem 2021, 8, 195–208. [Google Scholar] [CrossRef]
- Monteiro, M.C.O.; Goyal, A.; Moerland, P.; Koper, M.T.M. Understanding cation trends for hydrogen evolution on platinum and gold electrodes in alkaline media. ACS Catal. 2021, 11, 14328–14335. [Google Scholar] [CrossRef]
- Zhang, C.; Qu, P.; Zhou, M.; Qian, L.; Bai, T.; Jin, J.; Xin, B. Ionic liquids as promisingly multi-functional participants for electrocatalyst of water splitting: A review. Molecules 2023, 28, 3051. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, S.; Peter, S.C. An overview on Pd-based electrocatalysts for the hydrogen evolution reaction. Inorg. Chem. Front. 2018, 5, 2060–2080. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Atyf, Z.; Lenne, Q.; Ghilane, J. Electrografting of Phenyl Phosphate Layers onto Glassy Carbon for Tuning Catalytic Activity toward the Hydrogen Evolution Reaction. Molecules 2024, 29, 835. https://doi.org/10.3390/molecules29040835
Atyf Z, Lenne Q, Ghilane J. Electrografting of Phenyl Phosphate Layers onto Glassy Carbon for Tuning Catalytic Activity toward the Hydrogen Evolution Reaction. Molecules. 2024; 29(4):835. https://doi.org/10.3390/molecules29040835
Chicago/Turabian StyleAtyf, Zaynab, Quentin Lenne, and Jalal Ghilane. 2024. "Electrografting of Phenyl Phosphate Layers onto Glassy Carbon for Tuning Catalytic Activity toward the Hydrogen Evolution Reaction" Molecules 29, no. 4: 835. https://doi.org/10.3390/molecules29040835
APA StyleAtyf, Z., Lenne, Q., & Ghilane, J. (2024). Electrografting of Phenyl Phosphate Layers onto Glassy Carbon for Tuning Catalytic Activity toward the Hydrogen Evolution Reaction. Molecules, 29(4), 835. https://doi.org/10.3390/molecules29040835