
Adaptation of a Commercial NAD+ Quantification Kit to Assay the Base-Exchange Activity and Substrate Preferences of SARM1

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
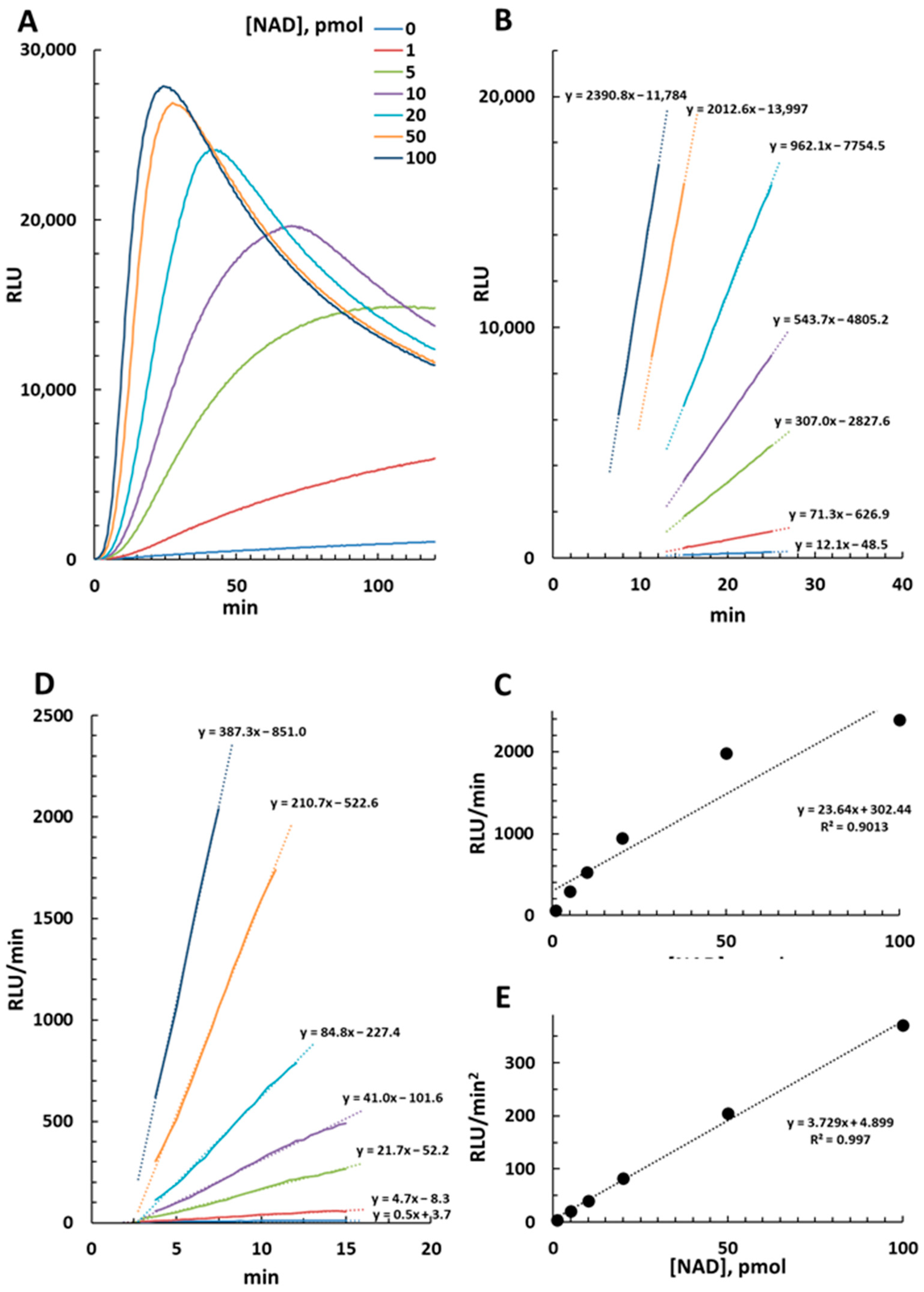
2.1. Luminescence Emission in the NAD/NADH-Glo™ Assay and Derived Calculations
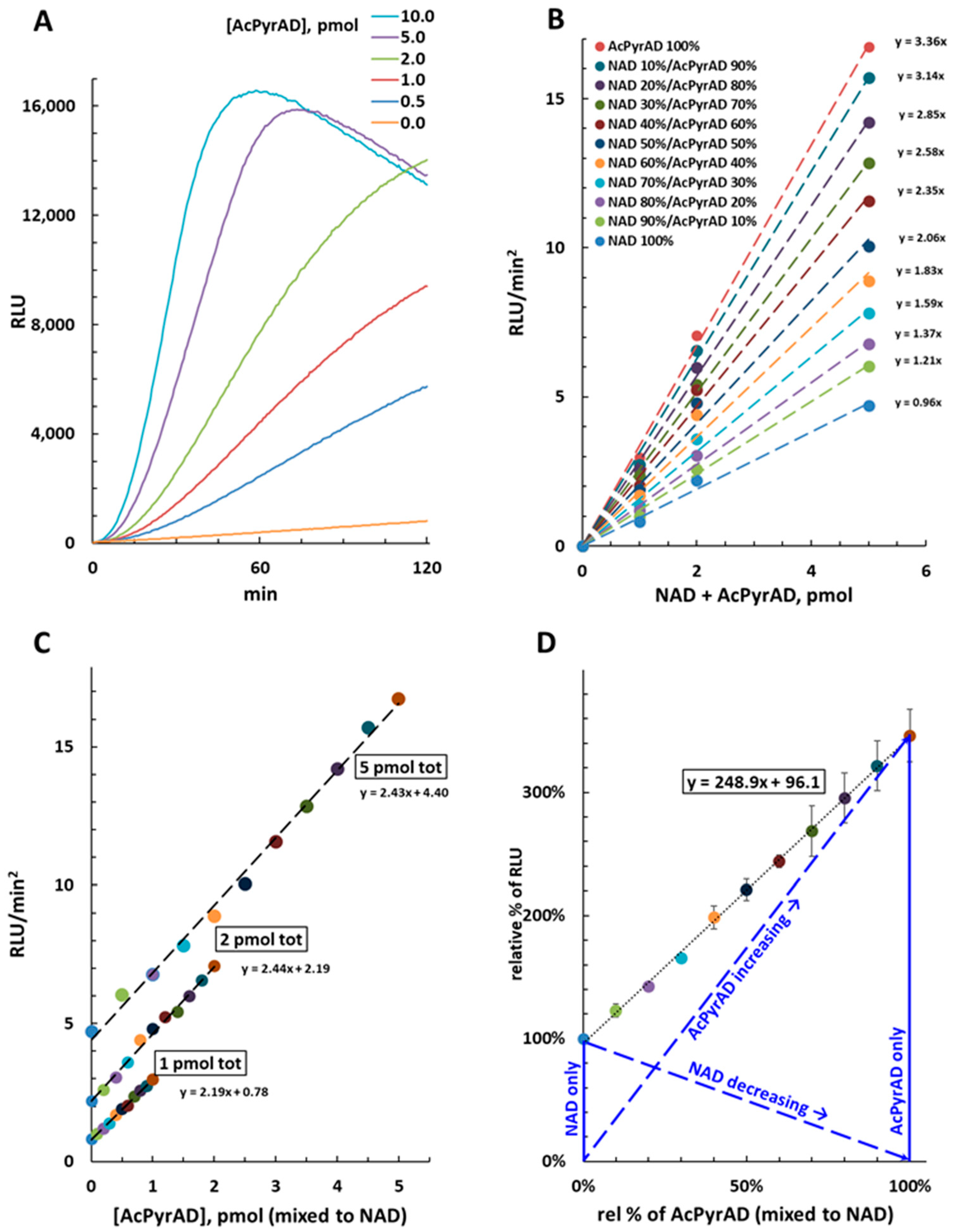
2.2. Cycling Amplification of AcPyrAD in the NAD/NADH-Glo™ Assay
2.3. Quantification of AcPyrAD When Mixed with NAD
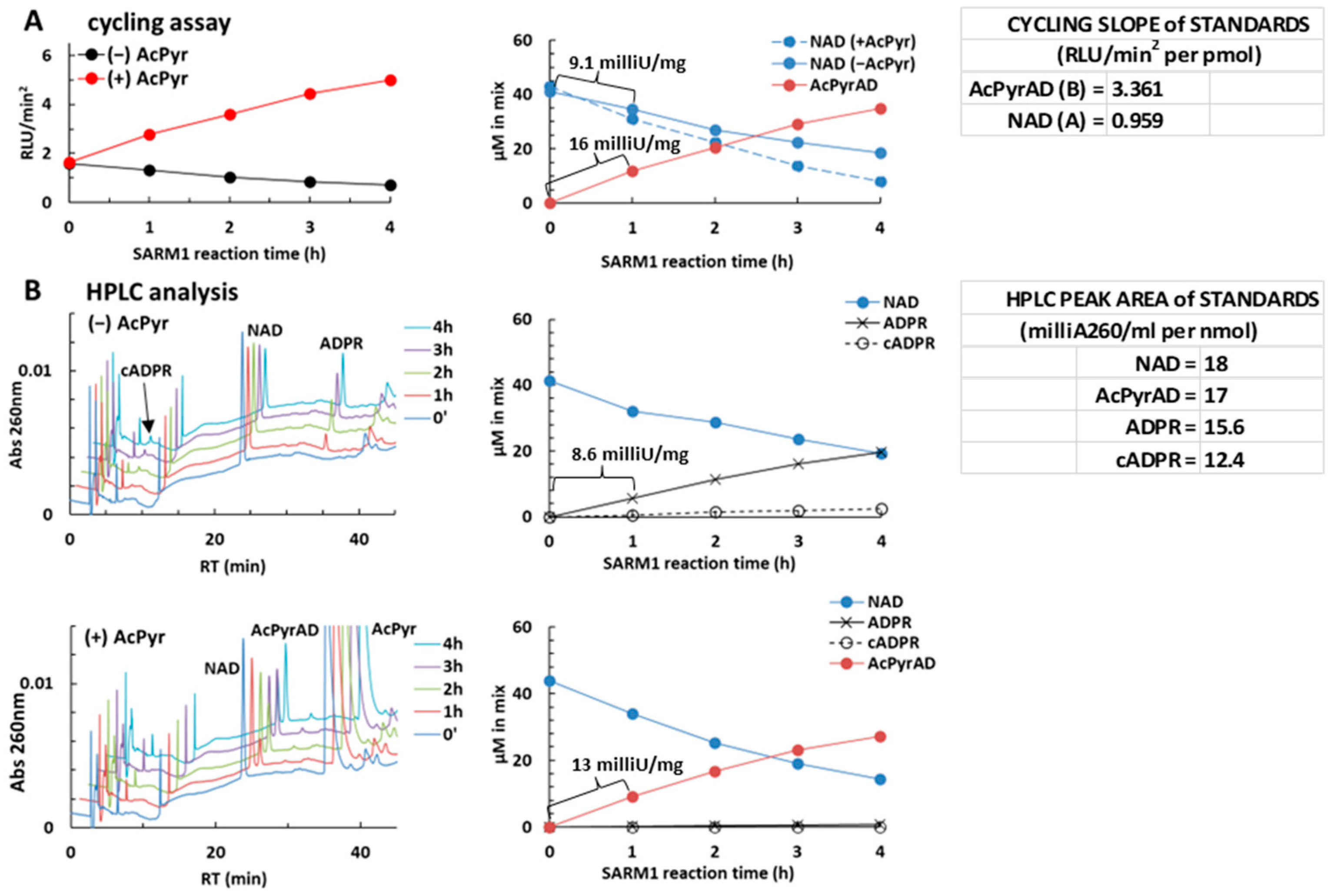
2.4. Using the NAD/NADH-Glo™ Assay to Assay AcPyr Base Exchange on NAD by SARM1
2.5. SARM1 Shows Differing Preferences for Other Free Pyridine Bases When Added in Competition with AcPyr
3. Discussion
4. Methods
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AcPyr | 3-acetylpiridine |
| AcPyrAD | oxidized 3-acetylpiridine adenine dinucleotide used in place of AcPyrAD+ |
| AcPyrADP | 3-acetylpiridine adenine dinucleotide phosphate, used in place of AcPyrADP+ |
| ADPR | adenosine 5′-diphosphoribose |
| ADPRP | adenosine 5′-diphosphoribose phosphate |
| ARM | armadillo/HEAT motif |
| isoQ | isoquinoline |
| Na | nicotinic acid |
| NAD | oxidized nicotinamide adenine dinucleotide used in place of NAD+ |
| NADP | nicotinamide adenine dinucleotide phosphate, used in place of NADP+ |
| NAD(P)ase | NAD+/NADP+-glycohydrolase multifunctional enzyme belonging to EC 3.2.2.6 |
| Nam | nicotinamide |
| NMN | nicotinamide mononucleotide |
| Vacor | pyrinuron (1-(4-nitrophenyl)-3-(pyridin-3-ylmethyl)urea) |
| SARM1 | sterile alpha and TIR motif-containing protein 1 |
| TIR | Toll/interleukin-1 receptor |
References
- Osterloh, J.M.; Yang, J.; Rooney, T.M.; Fox, A.N.; Adalbert, R.; Powell, E.H.; Sheehan, A.E.; Avery, M.A.; Hackett, R.; Logan, M.A.; et al. dSarm/Sarm1 is required for activation of an injury-induced axon death pathway. Science 2012, 337, 481–484. [Google Scholar] [CrossRef]
- Loring, H.S.; Thompson, P.R. Emergence of SARM1 as a Potential Therapeutic Target for Wallerian-type Diseases. Cell Chem. Biol. 2020, 27, 1–13. [Google Scholar] [CrossRef]
- Coleman, M.P.; Hoke, A. Programmed axon degeneration: From mouse to mechanism to medicine. Nat. Rev. Neurosci. 2020, 21, 183–196. [Google Scholar] [CrossRef] [PubMed]
- Essuman, K.; Summers, D.W.; Sasaki, Y.; Mao, X.; DiAntonio, A.; Milbrandt, J. The SARM1 Toll/Interleukin-1 Receptor Domain Possesses Intrinsic NAD(+) Cleavage Activity that Promotes Pathological Axonal Degeneration. Neuron 2017, 93, 1334–1343. [Google Scholar] [CrossRef]
- Angeletti, C.; Amici, A.; Gilley, J.; Loreto, A.; Trapanotto, A.G.; Antoniou, C.; Merlini, E.; Coleman, M.P.; Orsomando, G. SARM1 is a multi-functional NAD(P)ase with prominent base exchange activity, all regulated bymultiple physiologically relevant NAD metabolites. iScience 2022, 25, 103812. [Google Scholar] [CrossRef] [PubMed]
- Loreto, A.; Angeletti, C.; Gu, W.; Osborne, A.; Nieuwenhuis, B.; Gilley, J.; Merlini, E.; Arthur-Farraj, P.; Amici, A.; Luo, Z.; et al. Neurotoxin-mediated potent activation of the axon degeneration regulator SARM1. eLife 2021, 10, e72823. [Google Scholar] [CrossRef]
- Wu, T.; Zhu, J.; Strickland, A.; Ko, K.W.; Sasaki, Y.; Dingwall, C.B.; Yamada, Y.; Figley, M.D.; Mao, X.; Neiner, A.; et al. Neurotoxins subvert the allosteric activation mechanism of SARM1 to induce neuronal loss. Cell Rep. 2021, 37, 109872. [Google Scholar] [CrossRef]
- Di Stefano, M.; Nascimento-Ferreira, I.; Orsomando, G.; Mori, V.; Gilley, J.; Brown, R.; Janeckova, L.; E Vargas, M.; A Worrell, L.; Loreto, A.; et al. A rise in NAD precursor nicotinamide mononucleotide (NMN) after injury promotes axon degeneration. Cell Death Differ. 2015, 22, 731–742. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.Y.; Xie, X.J.; Li, W.H.; Liu, J.; Chen, Z.; Zhang, B.; Li, T.; Li, S.L.; Lu, J.G.; Zhang, L.; et al. A Cell-Permeant Mimetic of NMN Activates SARM1 to Produce Cyclic ADP-Ribose and Induce Non-apoptotic Cell Death. iScience 2019, 15, 452–466. [Google Scholar] [CrossRef]
- Figley, M.; Gu, W.; Nanson, J.D.; Shi, Y.; Sasaki, Y.; Cunnea, K.; Malde, A.K.; Jia, X.; Luo, Z.; Saikot, F.K.; et al. SARM1 is a Metabolic Sensor Activated by an Increased NMN/NAD+ Ratio to Trigger Axon Degeneration. Neuron 2021, 109, 1118–1136. [Google Scholar] [CrossRef]
- Bratkowski, M.; Xie, T.; Thayer, D.A.; Lad, S.; Mathur, P.; Yang, Y.-S.; Danko, G.; Burdett, T.C.; Danao, J.; Cantor, A.; et al. Structural and Mechanistic Regulation of the Pro-degenerative NAD Hydrolase SARM1. Cell Rep. 2020, 32, 107999. [Google Scholar] [CrossRef]
- Jiang, Y.; Liu, T.; Lee, C.-H.; Chang, Q.; Yang, J.; Zhang, Z. The NAD(+)-mediated self-inhibition mechanism of pro-neurodegenerative SARM1. Nature 2020, 588, 658–663. [Google Scholar] [CrossRef]
- Li, W.H.; Huang, K.; Cai, Y.; Wang, Q.W.; Zhu, W.J.; Hou, Y.N.; Wang, S.; Cao, S.; Zhao, Z.Y.; Xie, X.J.; et al. Permeant fluorescent probes visualize the activation of SARM1 and uncover an anti-neurodegenerative drug candidate. eLife 2021, 10, e67381. [Google Scholar] [CrossRef]
- Huang, K.; Zhu, W.J.; Li, W.H.; Lee, H.C.; Zhao, Y.J.; Lee, C.-S. Base-Exchange Enabling the Visualization of SARM1 Activities in Sciatic Nerve-Injured Mice. ACS Sens. 2023, 8, 767–773. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Kerry, P.S.; Nanson, J.D.; Bosanac, T.; Sasaki, Y.; Krauss, R.; Saikot, F.K.; Adams, S.E.; Mosaiab, T.; Masic, V.; et al. Structural basis of SARM1 activation, substrate recognition, and inhibition by small molecules. Mol. Cell 2022, 82, 1643–1659.e10. [Google Scholar] [CrossRef]
- Guse, A.H. Enzymology of Ca2+-Mobilizing Second Messengers Derived from NAD: From NAD Glycohydrolases to (Dual) NADPH Oxidases. Cells 2023, 12, 675. [Google Scholar] [CrossRef] [PubMed]
- Icso, J.D.; Barasa, L.; Thompson, P.R. SARM1, an Enzyme Involved in Axon Degeneration, Catalyzes Multiple Activities through a Ternary Complex Mechanism. Biochemistry 2023, 62, 2065–2078. [Google Scholar] [CrossRef]
- Sverkeli, L.J.; Hayat, F.; Migaud, M.E.; Ziegler, M. Enzymatic and Chemical Syntheses of Vacor Analogs of Nicotinamide Riboside, NMN and NAD. Biomolecules 2021, 11, 1044. [Google Scholar] [CrossRef]
- Gomi, K.; Kajiyama, N. Oxyluciferin, a luminescence product of firefly luciferase, is enzymatically regenerated into luciferin. J. Biol. Chem. 2001, 276, 36508–36513. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.Y.; Liu, Y.J. Luciferin Regeneration in Firefly Bioluminescence via Proton-Transfer-Facilitated Hydrolysis, Condensation and Chiral Inversion. ChemPhysChem 2019, 20, 1719–1727. [Google Scholar] [CrossRef]
- Hughes, R.O.; Bosanac, T.; Mao, X.; Engber, T.M.; DiAntonio, A.; Milbrandt, J.; Devraj, R.; Krauss, R. Small Molecule SARM1 Inhibitors Recapitulate the SARM1. Cell Rep. 2021, 34, 108588. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cirilli, I.; Amici, A.; Gilley, J.; Coleman, M.P.; Orsomando, G. Adaptation of a Commercial NAD+ Quantification Kit to Assay the Base-Exchange Activity and Substrate Preferences of SARM1. Molecules 2024, 29, 847. https://doi.org/10.3390/molecules29040847
Cirilli I, Amici A, Gilley J, Coleman MP, Orsomando G. Adaptation of a Commercial NAD+ Quantification Kit to Assay the Base-Exchange Activity and Substrate Preferences of SARM1. Molecules. 2024; 29(4):847. https://doi.org/10.3390/molecules29040847
Chicago/Turabian StyleCirilli, Ilenia, Adolfo Amici, Jonathan Gilley, Michael P. Coleman, and Giuseppe Orsomando. 2024. "Adaptation of a Commercial NAD+ Quantification Kit to Assay the Base-Exchange Activity and Substrate Preferences of SARM1" Molecules 29, no. 4: 847. https://doi.org/10.3390/molecules29040847
APA StyleCirilli, I., Amici, A., Gilley, J., Coleman, M. P., & Orsomando, G. (2024). Adaptation of a Commercial NAD+ Quantification Kit to Assay the Base-Exchange Activity and Substrate Preferences of SARM1. Molecules, 29(4), 847. https://doi.org/10.3390/molecules29040847