
Enhanced Selectivity in 4-Quinolone Formation: A Dual-Base System for Palladium-Catalyzed Carbonylative Cyclization with Fe(CO)5

 , ,
, ,
Abstract
:
1. Introduction
2. Results and Discussion
3. Experimental
3.1. Subsection General Information
3.2. General Method for the Synthesis of 4-Quinolones
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Velema, W.A.; van der Berg, J.P.; Hansen, M.J.; Szymanski, W.; Driessen, A.J.M.; Feringa, B.L. Optical Control of Antibacterial Activity. Nat. Chem. 2013, 5, 924–928. [Google Scholar] [CrossRef]
- Pescatori, L.; Métifiot, M.; Chung, S.; Masoaka, T.; Cuzzucoli Crucitti, G.; Messore, A.; Pupo, G.; Madia, V.N.; Saccoliti, F.; Scipione, L.; et al. N-Substituted Quinolinonyl Diketo Acid Derivatives as HIV Integrase Strand Transfer Inhibitors and Their Activity against RNase H Function of Reverse Transcriptase. J. Med. Chem. 2015, 58, 4610–4623. [Google Scholar] [CrossRef] [PubMed]
- Shen, C.; Wang, A.; Xu, J.; An, Z.; Loh, K.Y.; Zhang, P.; Liu, X. Recent Advances in the Catalytic Synthesis of 4-Quinolones. Chem 2019, 5, 1059–1107. [Google Scholar] [CrossRef]
- Wu, X.-F.; Neumann, H.; Beller, M. Synthesis of Heterocycles via Palladium-Catalyzed Carbonylations. Chem. Rev. 2012, 113, 1–35. [Google Scholar] [CrossRef] [PubMed]
- Torii, S.; Okumoto, H.; Xu, L.H. Palladium-catalyzed Carbonylation to Form 2-Substituted 1,4-Dihydro-4-oxo-quinoline. Tetrahedron Lett. 1991, 32, 237–240. [Google Scholar] [CrossRef]
- Haddad, N.; Tan, J.; Farina, V. Convergent Synthesis of the Quinolone Substructure of BILN 2061 via Carbonylative Sonogashira Coupling/Cyclization. J. Org. Chem. 2006, 71, 5031–5034. [Google Scholar] [CrossRef]
- Genelot, M.; Bendjeriou, A.; Dufaud, V.; Djakovitch, L. Optimised Procedures for the One-pot selective Syntheses of Indoxyls and 4-Quinolones by a Carbonylative Sonogashira/Cyclisation Sequence. Appl. Catal. A-Gen. 2009, 369, 125–132. [Google Scholar] [CrossRef]
- Kalinin, V.N.; Shostakovsky, M.V.; Ponomaryov, A.B. A New Route to 2-Aryl-4-quinolones via Palladium-catalyzed Carbonylative Coupling of O-iodoanilines with Terminal Arylacetylenes. Tetrahedron Lett. 1992, 33, 373–376. [Google Scholar] [CrossRef]
- Genelot, M.; Dufaud, V.; Djakovitch, L. Heterogeneous Metallo-Organocatalysis for the Selective One-Pot Synthesis of 2-Benzylidene-indoxyl and 2-phenyl-4-quinolone. Tetrahedron 2011, 67, 976–981. [Google Scholar] [CrossRef]
- Wu, J.; Zhou, Y.; Wu, T.; Zhou, Y.; Chiang, C.-W.; Lei, A. From Ketones, Amines, and Carbon Monoxide to 4-Quinolones: Palladium-Catalyzed Oxidative Carbonylation. Org. Lett. 2017, 19, 6432–6435. [Google Scholar] [CrossRef]
- Roderique, J.D.; Josef, C.S.; Feldman, M.J.; Spiess, B.D. A Modern Literature Review of Carbon Monoxide Poisoning Theories, Therapies, and potential targets for therapy advancement. Toxicology 2015, 334, 45–58. [Google Scholar] [CrossRef]
- Åkerbladh, L.; Nordeman, P.; Wejdemar, M.; Odell, L.R.; Larhed, M. Synthesis of 4-Quinolones via a Carbonylative Sonogashira Cross-Coupling Using Molybdenum Hexacarbonyl as a CO Source. J. Org. Chem. 2015, 80, 1464–1471. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, P.; Nandi, A.K.; Das, S. Carbonylative Sonogashira Annulation Sequence: One-pot Synthesis of 4-Quinolone and 4H-chromen-4-one Derivatives. Tetrahedron Lett. 2018, 59, 2025–2029. [Google Scholar] [CrossRef]
- Wang, J.-S.; Li, C.; Ying, J.; Xu, T.; Lu, W.; Li, C.-Y.; Wu, X.-F. Supported Palladium-catalyzed Carbonylative Cyclization of 2-Bromonitrobenzenes and Alkynes to Access Quinolin-4(1H)-ones. J. Catal. 2022, 408, 81–87. [Google Scholar] [CrossRef]
- Fortes, A.D.; Parker, S.F. Structure and Spectroscopy of Iron Pentacarbonyl, Fe(CO)5. J. Am. Chem. Soc. 2022, 144, 17376–17386. [Google Scholar] [CrossRef] [PubMed]
- Mond, L.; Langer, C. On Iron Carbonyls. J. Chem. Soc. Trans. 1891, 59, 1090–1093. [Google Scholar] [CrossRef]
- Zhu, Z.; Wang, Z.; Jian, Y.; Sun, H.; Zhang, G.; Lynam, J.M.; McElroy, C.R.; Burden, T.J.; Inight, R.L.; Fairlamb, I.J.S.; et al. Pd-Catalysed Carbonylative Suzuki–Miyaura Cross-couplings using Fe(CO)5 under Mild Conditions: Generation of a Highly Active, Recyclable and Scalable ‘Pd–Fe’ Nanocatalyst. Green Chem. 2021, 23, 920–926. [Google Scholar] [CrossRef]
- Babjak, M.; Markovič, M.; Kandríková, B.; Gracza, T. Homogeneous Cyclocarbonylation of Alkenols with Iron Pentacarbonyl. Synthesis 2014, 46, 809–816. [Google Scholar] [CrossRef]
- Babjak, M.; Caletková, O.; Ďurišová, D.; Gracza, T. Iron Pentacarbonyl in Alkoxy- and Aminocarbonylation of Aromatic Halides. Synlett 2014, 25, 2579–2584. [Google Scholar] [CrossRef]
- Baxter, A.; Chambers, M.; Edfeldt, F.; Edman, K.; Freeman, A.; Johansson, C.; King, S.; Morley, A.; Petersen, J.; Rawlins, P.; et al. Non-covalent Inhibitors of Rhinovirus 3C Protease. Bioorg. Med. Chem. Lett. 2011, 21, 777–780. [Google Scholar] [CrossRef]
- Kim, M.; Jung, I.; Lee, J.; Na, J.; Kim, W.; Kim, Y.-O.; Park, Y.-D.; Lee, J. Pseudane-VII Isolated from Pseudoalteromonas sp. M2 Ameliorates LPS-Induced Inflammatory Response In Vitro and In Vivo. Mar. Drugs 2017, 15, 336. [Google Scholar] [CrossRef]
- Wang, J.Y.; Strom, A.E.; Hartwig, J.F. Mechanistic Studies of Palladium-Catalyzed Aminocarbonylation of Aryl Chlorides with Carbon Monoxide and Ammonia. J. Am. Chem. Soc. 2018, 140, 7979–7993. [Google Scholar] [CrossRef]
- Zhang, J.; Lan, T.; Lu, Y. Translating in Vitro Diagnostics from Centralized Laboratories to Point-of-Care Locations using Commercially-Available Handheld Meters. Trends Analyt. Chem. 2020, 124, 115782–115797. [Google Scholar] [CrossRef]
- Xu, S.; Sun, H.; Zhuang, M.; Zheng, S.; Jian, Y.; Zhang, W.; Gao, Z. Divergent Synthesis of Flavones and Aurones via Base-controlled Regioselective Palladium Catalyzed Carbonylative Cyclization. Mol. Catal. 2018, 452, 264–270. [Google Scholar] [CrossRef]
- Romek, A.; Opatz, T. Microwave-Assisted Synthesis of Polysubstituted 4-Quinolones from Deprotonated α-Aminonitriles. Eur. J. Org. Chem. 2010, 2010, 5841–5849. [Google Scholar] [CrossRef]
- Xu, X.; Sun, R.; Zhang, S.; Zhang, X.; Yi, W. Divergent Synthesis of Quinolones and Dihydroepindolidiones via Cu(I)-Catalyzed Cyclization of Anilines with Alkynes. Org. Lett. 2018, 20, 1893–1897. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.; Lin, J.-P.; Song, L.-R.; Long, Y.-Q. Direct Synthesis of 2-Aryl-4-quinolones via Transition-Metal-Free Intramolecular Oxidative C(sp3)–H/C(sp3)–H Coupling. Org. Lett. 2015, 17, 1268–1271. [Google Scholar] [CrossRef]
- Seppänen, O.; Muuronen, M.; Helaja, J. Gold-Catalyzed Conversion of Aryl- and Alkyl-Substituted 1-(o-Aminophenyl)-2-propyn-1-ones to the Corresponding 2-Substituted 4-Quinolones. Eur. J. Org. Chem. 2014, 2014, 4044–4052. [Google Scholar] [CrossRef]
- Jones, C.P.; Anderson, K.W.; Buchwald, S.L. Sequential Cu-Catalyzed Amidation-Base-Mediated CampsCyclization: A Two-Step Synthesis of 2-Aryl-4-quinolones fromo-Halophenones. J. Org. Chem. 2007, 72, 7968–7973. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Nerella, S.; Pabbaraja, S.; Mehta, G. Access to 2-Alkyl/Aryl-4-(1H)-Quinolones via Orthogonal “NH3” Insertion into o-Haloaryl Ynones: Total Synthesis of Bioactive Pseudanes, Graveoline, Graveolinine, and Waltherione F. Org. Lett. 2020, 22, 1575–1579. [Google Scholar] [CrossRef] [PubMed]
- Ward, T.R.; Turunen, B.J.; Haack, T.; Neuenswander, B.; Shadrick, W.; Georg, G.I. Synthesis of a Quinolone Library from Ynones. Tetrahedron Lett. 2009, 50, 6494–6497. [Google Scholar] [CrossRef] [PubMed]
- Ferretti, F.; Fouad, M.A.; Abbo, C.; Ragaini, F. Effective Synthesis of 4-Quinolones by Reductive Cyclization of 2′-Nitrochalcones Using Formic Acid as a CO Surrogate. Molecules 2023, 28, 5424–5438. [Google Scholar] [CrossRef] [PubMed]


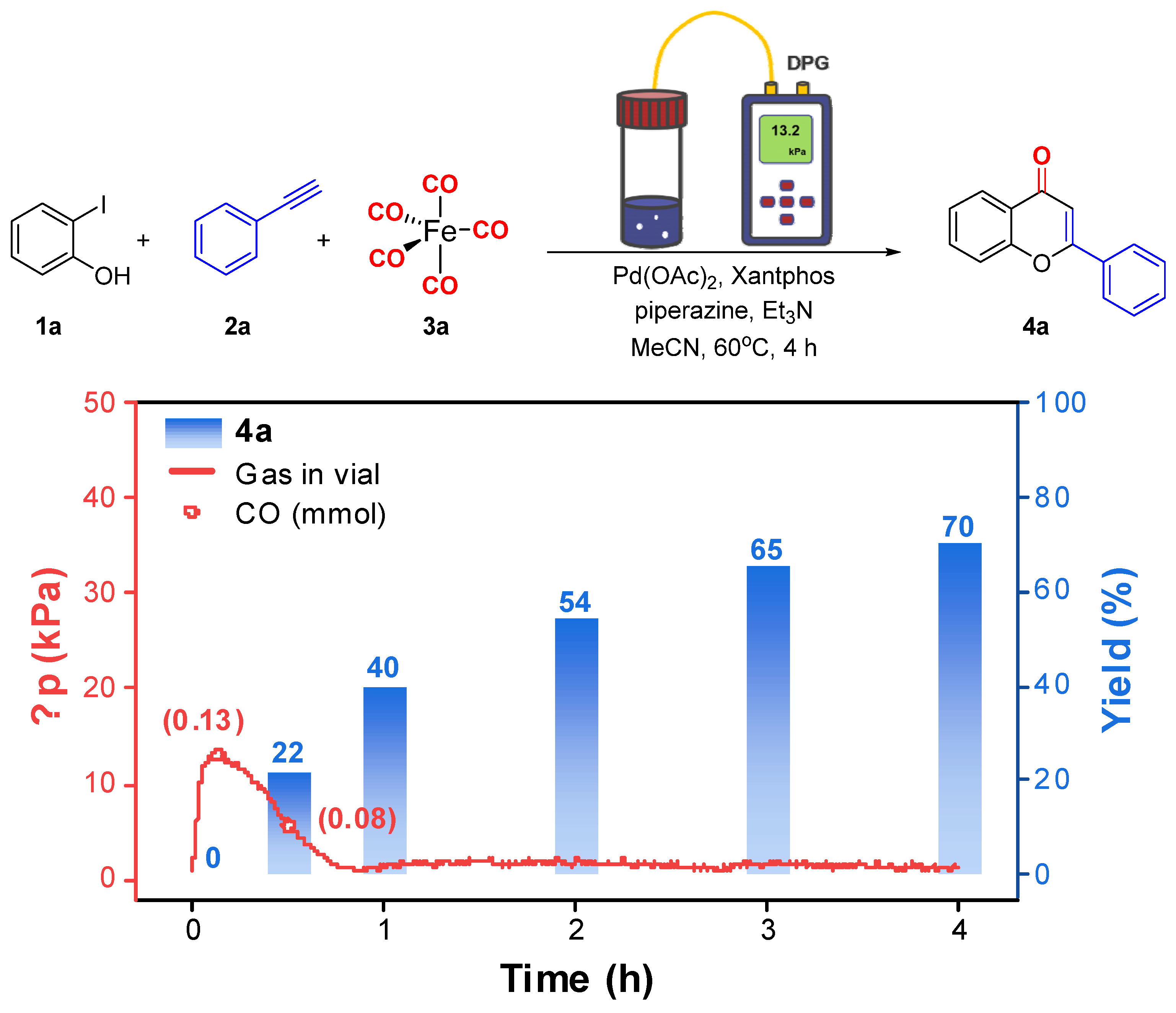
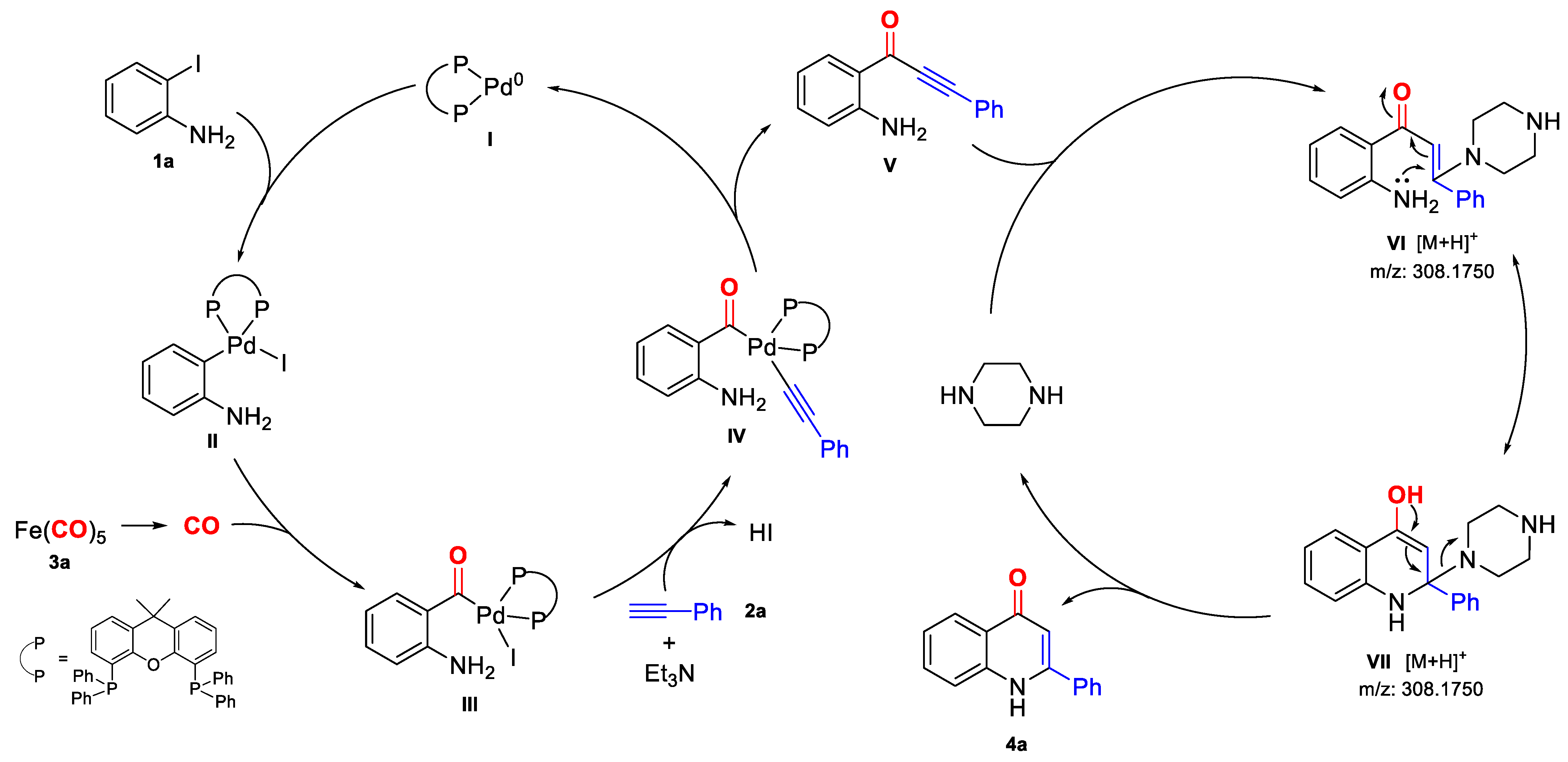

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
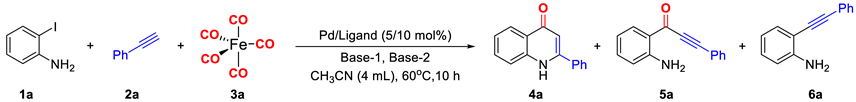 | |||||||
|---|---|---|---|---|---|---|---|
| Entry a | Pd | Ligand | Base-1 (3 Equiv.) | Base-2 (1 Equiv.) | Yield (%) b | ||
| 4a | 5a | 6a | |||||
| 1 | Pd(OAc)2 | - | Piperazine | - | 57 | - | 32 |
| 2 | Pd(OAc)2 | - | K2CO3 | - | 41 | - | 15 |
| 3 | Pd(OAc)2 | - | K3PO4 | - | 29 | - | 20 |
| 4 | Pd(OAc)2 | - | Et3N | - | 53 | 3 | 4 |
| 5 | Pd(OAc)2 | - | TMEDA | - | 25 | 12 | 3 |
| 6 | Pd(OAc)2 | - | Piperazine | Et3N | 70 | - | 21 |
| 7 | Pd(OAc)2 | - | Et3N | Piperazine | 85 | - | 10 |
| 8 | Pd2(dba)3 | - | Et3N | Piperazine | 63 | 8 | 21 |
| 9 | Pd(OAc)2 | PPh3 | Et3N | Piperazine | 66 | 8 | 26 |
| 10 | Pd(OAc)2 | dppp | Et3N | Piperazine | 88 | 7 | 5 |
| 11 | Pd(OAc)2 | Xantphos | Et3N | Piperazine | 97 | - | 3 |
| 12 c | Pd(OAc)2 | Xantphos | Et3N | Piperazine | 90 | - | 10 |
| 13 d | Pd(OAc)2 | Xantphos | Et3N | Piperazine | 78 | - | 22 |
| Entry | CO/CORM | Conditions | 4a-Yield (%) |
|---|---|---|---|
| 1 [5] | CO (20 bar) | PdCl2(PPh3)2, Et2NH, 120 °C, 6 h | 90 |
| 2 [7] | CO (5 bar) | PdCl2(dppp), toluene, Et3N, 80 °C, 6 h | 62 |
| b. Et2NH, 1 h | |||
| 3 [12] | Mo(CO)6 (2 equiv.) | Pd2(dba)3, dppf, Et2NH, MW 120 °C, 20 min | 85 |
| 4 [12] | Mo(CO)6 (1.5 equiv.) | Pd(OAc)2, [HP(tBu)3]BF4, MeCN, Et3N, r.t., 16 h | 84 |
| Et2NH, 5 h | |||
| 5 | Fe(CO)5 (0.25 equiv.) | Pd(OAc)2, Xantphos, piperazine, Et3N, CH3CN, 60 °C 10 h | 91 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guo, M.; Wu, D.; Yang, H.; Zhang, X.; Xue, D.-X.; Zhang, W. Enhanced Selectivity in 4-Quinolone Formation: A Dual-Base System for Palladium-Catalyzed Carbonylative Cyclization with Fe(CO)5. Molecules 2024, 29, 850. https://doi.org/10.3390/molecules29040850
Guo M, Wu D, Yang H, Zhang X, Xue D-X, Zhang W. Enhanced Selectivity in 4-Quinolone Formation: A Dual-Base System for Palladium-Catalyzed Carbonylative Cyclization with Fe(CO)5. Molecules. 2024; 29(4):850. https://doi.org/10.3390/molecules29040850
Chicago/Turabian StyleGuo, Meng, Dou Wu, Hongyu Yang, Xiao Zhang, Dong-Xu Xue, and Weiqiang Zhang. 2024. "Enhanced Selectivity in 4-Quinolone Formation: A Dual-Base System for Palladium-Catalyzed Carbonylative Cyclization with Fe(CO)5" Molecules 29, no. 4: 850. https://doi.org/10.3390/molecules29040850