Microwave-Mediated, Catalyst-Free Synthesis of 1,2,4-Triazolo[1,5-a]pyridines from Enaminonitriles

 , and
, and
Abstract
:
1. Introduction
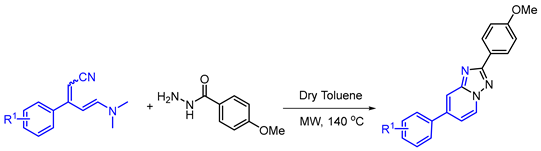
2. Results and Discussion
3. Materials and Methods
3.1. General Information
3.2. Typical Procedure for the Preparation of Enaminonitriles [55]
3.3. Typical Procedure for the Preparation of 1,2,4-triazolo[1,5-a]Pyridines in Microwave Conditions
3.4. Typical Procedure for the Preparation of 1,2,4-triazolo[1,5-a]Pyridines in Reflux Conditions
3.5. Procedure for the Scale-Up Reaction
3.6. Procedure for the Reaction with Radical Scavenger
3.7. Procedure for the Suzuki–Miyaura Coupling Reaction
3.8. Procedure for the Sonogashira Coupling Reaction
3.9. Anlytical Data of Synthezised Compounds
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Maes, B.W. Microwave-Assisted Synthesis of Heterocycles; Eycken, E., Kappe, C.O., Eds.; Springer: Berlin/Heidelberg, Germany, 2006; p. 155. [Google Scholar]
- Henary, M.; Kananda, C.; Rotolo, L.; Savino, B.; Owens, E.A.; Cravotto, G. Benefits and applications of microwave-assisted synthesis of nitrogen containing heterocycles in medicinal chemistry. RSC Adv. 2020, 10, 14170–14197. [Google Scholar] [CrossRef]
- Mohite, P.; Nahar, D.; Pawara, R.; Alqahtani, T.; Eldin, S.M.; Mukherje, N.; Rahman Mohammad Said Al-Tawaha, A.; Iqbal, R.; Bawazeer, S.; Ali, I. Triazolopyridine, a leitmotif of synthetic methods and pharmacological attributes: An extensive review. Arab. J. Chem. 2023, 16, 105181. [Google Scholar] [CrossRef]
- Jones, G. The chemistry of the triazolopyridines: An update. In Advances in Heterocyclic Chemistry; Academic Press: Cambridge, MA, USA, 2002; Volume 83, pp. 1–70. [Google Scholar]
- Vorob’ev, A.Y. Methods of synthesis of [1,2,4]triazolo[1,5-а]pyridines (microreview). Chem. Heterocycl. Compd. 2019, 55, 695–697. [Google Scholar] [CrossRef]
- Fischer, G. Chapter One—Recent advances in 1,2,4-triazolo[1,5-a]pyrimidine chemistry. In Advances in Heterocyclic Chemistry; Scriven, E.F.V., Ramsden, C.A., Eds.; Academic Press: Cambridge, MA, USA, 2019; Volume 128, pp. 1–101. [Google Scholar]
- Nakajima, R.; Oono, H.; Sugiyama, S.; Matsueda, Y.; Ida, T.; Kakuda, S.; Hirata, J.; Baba, A.; Makino, A.; Matsuyama, R.; et al. Discovery of [1,2,4]Triazolo[1,5-a]pyridine Derivatives as Potent and Orally Bioavailable RORγt Inverse Agonists. ACS Med. Chem. Lett. 2020, 11, 528–534. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, S.; Ayscough, A.; Barker, G.R.; Canning, H.E.; Davenport, R.; Downham, R.; Harrison, D.; Jenkins, K.; Kinsella, N.; Livermore, D.G.; et al. 1,2,4-Triazolo-[1,5-a]pyridine HIF Prolylhydroxylase Domain-1 (PHD-1) Inhibitors With a Novel Monodentate Binding Interaction. J. Med. Chem. 2017, 60, 5663–5672. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Mao, W.; Su, Y.; Zheng, X.; Qian, W.; Shen, M.; Shan, N.; Li, Y.; Wang, D.; Wu, S.; et al. Identification of TUL01101: A Novel Potent and Selective JAK1 Inhibitor for the Treatment of Rheumatoid Arthritis. J. Med. Chem. 2022, 65, 16716–16740. [Google Scholar] [CrossRef] [PubMed]
- Siu, M.; Pastor, R.; Liu, W.; Barrett, K.; Berry, M.; Blair, W.S.; Chang, C.; Chen, J.Z.; Eigenbrot, C.; Ghilardi, N.; et al. 2-Amino-[1,2,4]triazolo[1,5-a]pyridines as JAK2 inhibitors. Bioorg. Med. Chem. Lett. 2013, 23, 5014–5021. [Google Scholar] [CrossRef] [PubMed]
- Englert, H.; Mania, D.; Wettlaufer, D.; Klaus, E. Arylcarbonylaminoalkyl-dihydro-oxo-pyridines, Their Production and Their Use. U.S. Patent 5,360,808, 1 November 1994. [Google Scholar]
- Edmondson, S.D.; Mastracchio, A.; Mathvink, R.J.; He, J.; Harper, B.; Park, Y.-J.; Beconi, M.; Di Salvo, J.; Eiermann, G.J.; He, H.; et al. (2S,3S)-3-Amino-4-(3,3-difluoropyrrolidin-1-yl)-N,N-dimethyl-4-oxo-2-(4-[1,2,4]triazolo[1,5-a]- pyridin-6-ylphenyl)butanamide: A Selective α-Amino Amide Dipeptidyl Peptidase IV Inhibitor for the Treatment of Type 2 Diabetes. J. Med. Chem. 2006, 49, 3614–3627. [Google Scholar] [CrossRef] [PubMed]
- Blake, J.; Lyssikatos, J.P.; Marlow, A.L.; Seo, J.; Wallace, E.; Yang, H.W. Heterocyclic Inhibitors of MEK And Methods of Use Thereof. International Patent Application No. WO2005051301, 18 May 2006. [Google Scholar]
- Song, W.; Shi, L.; Gao, L.; Hu, P.; Mu, H.; Xia, Z.; Huang, J.; Su, J. [1,2,4]Triazolo[1,5-a]pyridine as Building Blocks for Universal Host Materials for High-Performance Red, Green, Blue and White Phosphorescent Organic Light-Emitting Devices. ACS Appl. Mater. Interfaces 2018, 10, 5714–5722. [Google Scholar] [CrossRef] [PubMed]
- Ueda, S.; Nagasawa, H. Facile Synthesis of 1,2,4-Triazoles via a Copper-Catalyzed Tandem Addition−Oxidative Cyclization. J. Am. Chem. Soc. 2009, 131, 15080–15081. [Google Scholar] [CrossRef] [PubMed]
- Grenda, V.J.; Jones, R.E.; Gal, G.; Sletzinger, M. Novel Preparation of Benzimidazoles from N-Arylamidines. New Synthesis of Thiabendazole1. J. Org. Chem. 1965, 30, 259–261. [Google Scholar] [CrossRef]
- Potts, K.T.; Burton, H.R.; Bhattacharyya, J. 1,2,4-Triazoles. XIII. Derivatives of the s-Triazolo[1,5-a]pyridine Ring System1a. J. Org. Chem. 1966, 31, 260–265. [Google Scholar] [CrossRef]
- Raval, J.P.; Desai, K.R. Synthesis and antimicrobial activity of new triazolopyridinyl phenothiazines. ARKIVOC 2005, 2005, 21–28. [Google Scholar] [CrossRef]
- Meng, X.; Yu, C.; Zhao, P. An efficient and recyclable heterogeneous catalytic system for the synthesis of 1,2,4-triazoles using air as the oxidant. RSC Adv. 2014, 4, 8612–8616. [Google Scholar] [CrossRef]
- Zheng, Z.; Ma, S.; Tang, L.; Zhang-Negrerie, D.; Du, Y.; Zhao, K. PhI(OCOCF3)2-Mediated Intramolecular Oxidative N–N Bond Formation: Metal-Free Synthesis of 1,2,4-Triazolo[1,5-a]pyridines. J. Org. Chem. 2014, 79, 4687–4693. [Google Scholar] [CrossRef] [PubMed]
- Song, L.; Tian, X.; Lv, Z.; Li, E.; Wu, J.; Liu, Y.; Yu, W.; Chang, J. I2/KI-Mediated Oxidative N–N Bond Formation for the Synthesis of 1,5-Fused 1,2,4-Triazoles from N-Aryl Amidines. J. Org. Chem. 2015, 80, 7219–7225. [Google Scholar] [CrossRef]
- Li, Y.; Ye, Z.; Chen, N.; Chen, Z.; Zhang, F. Intramolecular electrochemical dehydrogenative N–N bond formation for the synthesis of 1,2,4-triazolo[1,5-a]pyridines. Green Chem. 2019, 21, 4035–4039. [Google Scholar] [CrossRef]
- Bartels, B.; Bolas, C.G.; Cueni, P.; Fantasia, S.; Gaeng, N.; Trita, A.S. Cu-Catalyzed Aerobic Oxidative Cyclization of Guanidylpyridines and Derivatives. J. Org. Chem. 2015, 80, 1249–1257. [Google Scholar] [CrossRef]
- Khatua, H.; Das, S.K.; Roy, S.; Chattopadhyay, B. Dual Reactivity of 1,2,3,4-Tetrazole: Manganese-Catalyzed Click Reaction and Denitrogenative Annulation. Angew. Chem. Int. Ed. 2021, 60, 304–312. [Google Scholar] [CrossRef]
- Zhu, B.; Li, W.; Chen, H.; Wu, M.; Hu, J.; Cao, H.; Liu, X. Mechanochemical Synthesis of 1,2,4-Triazoles via a [3+2] Cycloaddition of Azinium-N-Imines and Nitriles. Adv. Synth. Catal. 2022, 364, 2911–2915. [Google Scholar] [CrossRef]
- Li, Z.; Qiu, K.; Yang, X.; Zhou, W.; Cai, Q. Base-Promoted Tandem SNAr/Boulton–Katritzky Rearrangement: Access to [1,2,4]Triazolo[1,5-a]pyridines. Org. Lett. 2022, 24, 2989–2992. [Google Scholar] [CrossRef]
- Xiong, H.; Li, Z.; Tang, H.; He, L.; Zhou, W. Tandem C–N coupling/Boulton–Katritzky rearrangement reactions of 3-aminoisoxazoles or 1,2,4-oxadiazol-3-amines with 2-pyridyl trifluoromethanesulfonate: A rapid access to [1,2,4]triazolo[1,5-a]pyridines. Org. Chem. Front. 2022, 9, 3527–3531. [Google Scholar] [CrossRef]
- Liu, X.; Li, W.; Jiang, W.; Lu, H.; Liu, J.; Lin, Y.; Cao, H. Cu(II)-Catalyzed C–H Amidation/Cyclization of Azomethine Imines with Dioxazolones via Acyl Nitrenes: A Direct Access to Diverse 1,2,4-Triazole Derivatives. Org. Lett. 2022, 24, 613–618. [Google Scholar] [CrossRef]
- Kujur, S.; Verma, S.; Kumar, A.; Sharma, R.; Pathak, D.D. A green polyol approach for the synthesis of Cu2O NPs adhered on graphene oxide: A robust and efficient catalyst for 1,2,4-triazole and imidazo[1,2-a]pyridine synthesis. New J. Chem. 2022, 46, 8094–8104. [Google Scholar] [CrossRef]
- Vorob′ev, A.Y.; Borodkin, G.I.; Andreev, R.V.; Shubin, V.G. 1,3-Dipolar cycloaddition of cyanopyridines to heterocyclic N-imines: Experimental and theoretical study. Chem. Heterocycl. Compd. 2021, 57, 284–291. [Google Scholar] [CrossRef]
- Hassen, M.B.; Masmoudi, F.; Zribi, L.; Trigui, M.; Ismaili, L.; Marco-Contelles, J.; Chabchoub, F. Regioselective Synthesis of Novel [1,2,4]Triazolo[1,5-a]pyridine Derivatives. ChemistrySelect 2021, 6, 945–950. [Google Scholar] [CrossRef]
- Ibrahim, H.M.; Behbehani, H.; Ahmed Arafa, W.A. A facile, practical and metal-free microwave-assisted protocol for mono- and bis-[1,2,4]triazolo[1,5-a]pyridines synthesis utilizing 1-amino-2-imino-pyridine derivatives as versatile precursors. RSC Adv. 2020, 10, 15554–15572. [Google Scholar] [CrossRef] [PubMed]
- Xia, J.; Huang, X.; Cai, M. Heterogeneous Copper(I)-Catalyzed Cascade Addition–Oxidative Cyclization of Nitriles with 2-Aminopyridines or Amidines: Efficient and Practical Synthesis of 1,2,4-Triazoles. Synthesis 2019, 51, 2014–2022. [Google Scholar] [CrossRef]
- Starosotnikov, A.M.; Bastrakov, M.A.; Knyazev, D.A.; Fedyanin, I.V.; Kachala, V.V.; Dalinger, I.L. Synthesis of N-Bridged 6,8-Dinitrotriazolo[1,5-a]pyridines. ChemistrySelect 2019, 4, 1510–1515. [Google Scholar] [CrossRef]
- Nagaraju, T.; Krishna, P.R.; Sridhar, B.; Mangarao, N. PhI(OAc)2-Mediated Regioselective Synthesis of 5-Guanidino-1,2,4-thiadiazoles and 1,2,4-Triazolo[1,5-a]pyridines via Oxidative N–S and N–N Bond Formation. Synthesis 2019, 51, 3600–3610. [Google Scholar] [CrossRef]
- Bhatt, A.; Singh, R.K.; Sarma, B.K.; Kant, R. Trichloroisocyanuric acid-mediated synthesis of 1,5-fused 1,2,4-triazoles from N-heteroaryl benzamidines via intramolecular oxidative N–N bond formation. Tetrahedron Lett. 2019, 60, 151026. [Google Scholar] [CrossRef]
- Bhatt, A.; Singh, R.K.; Kant, R.; Sarma, B.K. A Convenient Synthesis of 1,5-Fused 1,2,4-Triazoles from N-Arylamidines via Chloramine-T Mediated Intramolecular Oxidative N–N Bond Formation. Synthesis 2019, 51, 3883–3890. [Google Scholar] [CrossRef]
- Ayothiraman, R.; Bandaru, D.; Paranthaman, R.; Fenster, M.; Eastgate, M.D.; Vaidyanathan, R. T3P-Mediated N–N Cyclization for the Synthesis of 1,2,4-Triazolo[1,5-a]pyridines. Org. Process Res. Dev. 2019, 23, 2510–2515. [Google Scholar] [CrossRef]
- Lv, J.; He, Z.; Zhang, J.; Guo, Y.; Han, Z.; Bao, X. One-pot synthesis of [1,2,4]Triazolo[1,5-a]pyridines from azines and benzylidenemalononitriles via copper-catalyzed tandem cyclization. Tetrahedron 2018, 74, 3996–4004. [Google Scholar] [CrossRef]
- Hamdouchi, C.; Maiti, P.; Warshawsky, A.M.; DeBaillie, A.C.; Otto, K.A.; Wilbur, K.L.; Kahl, S.D.; Patel Lewis, A.; Cardona, G.R.; Zink, R.W.; et al. Discovery of LY3104607: A Potent and Selective G Protein-Coupled Receptor 40 (GPR40) Agonist with Optimized Pharmacokinetic Properties to Support Once Daily Oral Treatment in Patients with Type 2 Diabetes Mellitus. J. Med. Chem. 2018, 61, 934–945. [Google Scholar] [CrossRef]
- Moloney, H.; Magnus, N.A.; Buser, J.Y.; Embry, M.C. Cyclization of Methyl-Coumalate-Derived Methyl 1-Benzamido-6-oxo-1,6-dihydropyridine-3-carboxylates: Assembly of the [1,2,4]Triazolo[1,5-a]pyridine Ring System. J. Org. Chem. 2017, 82, 6279–6288. [Google Scholar] [CrossRef]
- Mammoliti, O.; Quinton, E.M.; Loones, K.T.J.; Nguyen, A.T.; Wouters, J.; Van Lommen, G. Direct access to 1-aryl-5-amino-1,2,4-triazoles and [1,2,4]triazolo[1,5-a]pyridines by two new single-step reactions from 1,3,4-thiadiazol-2-amines. Tetrahedron 2013, 69, 1669–1680. [Google Scholar] [CrossRef]
- Zhang, G.; Hu, Y. Synthesis and antitumor activities of 2-(substituted)phenyl-1,2,4-triazolo[1,5-a]pyridines. J. Heterocycl. Chem. 2007, 44, 919–922. [Google Scholar] [CrossRef]
- Huntsman, E.; Balsells, J. New Method for the General Synthesis of [1,2,4]Triazolo[1,5-a]pyridines. Eur. J. Org. Chem. 2005, 2005, 3761–3765. [Google Scholar] [CrossRef]
- Guba, W.; Nettekoven, M.; Püllmann, B.; Riemer, C.; Schmitt, S. Comparison of inhibitory activity of isomeric triazolopyridine derivatives towards adenosine receptor subtypes or do similar structures reveal similar bioactivities? Bioorg. Med. Chem. Lett. 2004, 14, 3307–3312. [Google Scholar] [CrossRef]
- Brodbeck, B.; Püllmann, B.; Schmitt, S.; Nettekoven, M. Parallel iterative solution-phase synthesis of 5-amino-1-aryl-[1,2,4]triazolo[1,5-a]pyridine-7-carboxylic acid amide derivatives. Tetrahedron Lett. 2003, 44, 1675–1678. [Google Scholar] [CrossRef]
- Kakehi, A.; Ito, S.; Uchiyama, K.; Konno, Y.; Kondo, K. Thermolysis and photolysis of various N-imidoyliminopyridinium ylides. J. Org. Chem. 1977, 42, 443–448. [Google Scholar] [CrossRef]
- Kakehi, A.; Ito, S.; Uchiyama, K.; Konno, Y. Synthesis and Characterization of 1-imidoyliminopyridinium n-ylides. Chem. Lett. 1976, 5, 413–414. [Google Scholar] [CrossRef]
- Fairoosa, J.; Saranya, S.; Radhika, S.; Anilkumar, G. Recent Advances in Microwave Assisted Multicomponent Reactions. ChemistrySelect 2020, 5, 5180–5197. [Google Scholar] [CrossRef]
- Kruithof, A.; Ruijter, E.; Orru, R.V.A. Microwave-Assisted Multicomponent Synthesis of Heterocycles. Curr. Org. Chem. 2011, 15, 204–236. [Google Scholar] [CrossRef]
- Roberts, B.A.; Strauss, C.R. Toward Rapid, “Green”, Predictable Microwave-Assisted Synthesis. Acc. Chem. Res. 2005, 38, 653–661. [Google Scholar] [CrossRef]
- Rakshit, A.; Dhara, H.N.; Sahoo, A.K.; Patel, B.K. The Renaissance of Organo Nitriles in Organic Synthesis. Chem. Asian J. 2022, 17, e202200792. [Google Scholar] [CrossRef] [PubMed]
- Cui, S.Q.; Liao, W.W. Recent Advances in Transition-Metal-Catalyzed C-H Addition to Nitriles. Synthesis 2022, 54, 33–48. [Google Scholar]
- Xia, Y.; Jiang, H.; Wu, W. Recent Advances in Chemical Modifications of Nitriles. Eur. J. Org. Chem. 2021, 2021, 6658–6669. [Google Scholar] [CrossRef]
- Sim, J.; Viji, M.; Rhee, J.; Jo, H.; Cho, S.J.; Park, Y.; Seo, S.Y.; Jung, K.Y.; Lee, H.; Jung, J.K. γ-Functionalization of α,β-Unsaturated Nitriles under Mild Conditions: Versatile Synthesis of 4-Aryl-2-Bromopyridines. Adv. Synth. Catal. 2019, 361, 5458–5465. [Google Scholar] [CrossRef]
- Jung, C.; Li, S.; Lee, K.; Viji, M.; Lee, H.; Hyun, S.; Lee, K.; Kang, Y.K.; Chaudhary, C.L.; Jung, J.K. Reagent-free intramolecular hydrofunctionalization: A regioselective 6-endo-dig cyclization of o-alkynoylphenols. Green Chem. 2022, 24, 2376–2384. [Google Scholar] [CrossRef]
- Viji, M.; Vishwanath, M.; Sim, J.; Park, Y.; Jung, C.; Lee, S.; Lee, H.; Lee, K.; Jung, J.K. α-Hydroxy acid as an aldehyde surrogate: Metal-free synthesis of pyrrolo[1,2-a]quinoxalines, quinazolinones, and other N-heterocycles via decarboxylative oxidative annulation reaction. RSC Adv. 2020, 10, 37202–37208. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Sim, J.; Jo, H.; Viji, M.; Srinu, L.; Lee, K.; Lee, H.; Manjunatha, V.; Jung, J.K. Transition metal-free synthesis of quinazolinones using dimethyl sulfoxide as a synthon. Org. Biomol. Chem. 2019, 17, 8067–8070. [Google Scholar] [CrossRef] [PubMed]
- Viji, M.; Sim, J.; Li, S.; Lee, H.; Oh, K.; Jung, J.K. Organocatalytic and Regiodivergent Mannich Reaction of Ketones with Benzoxazinones. Adv. Synth. Catal. 2018, 360, 4464–4469. [Google Scholar] [CrossRef]
- Sim, J.; Jo, H.; Viji, M.; Choi, M.; Jung, J.A.; Lee, H.; Jung, J.K. Rapid, Operationally Simple, and Metal-free NBS Mediated One-pot Synthesis of 1,2-Naphthoquinone from 2-Naphthol. Adv. Synth. Catal. 2018, 360, 852–858. [Google Scholar] [CrossRef]
- Choi, M.; Viji, M.; Kim, D.; Lee, Y.H.; Sim, J.; Kwak, Y.S.; Lee, K.; Lee, H.; Jung, J.K. Metal-free approach for the ʟ-proline mediated synthesis of nitrones from nitrosobenzene. Tetrahedron 2018, 74, 4182–4187. [Google Scholar] [CrossRef]
- Oh, H.; Kim, H.; Kim, I. Regioselective Access to 1,2,4-Triazole-Fused N-Heterocycle, Pyrrolo[1,2-a][1,2,4]triazolo[5,1-c]pyrazine, via Double Dehydrative Cyclizations. J. Org. Chem. 2023, 88, 11748–11761. [Google Scholar] [CrossRef]
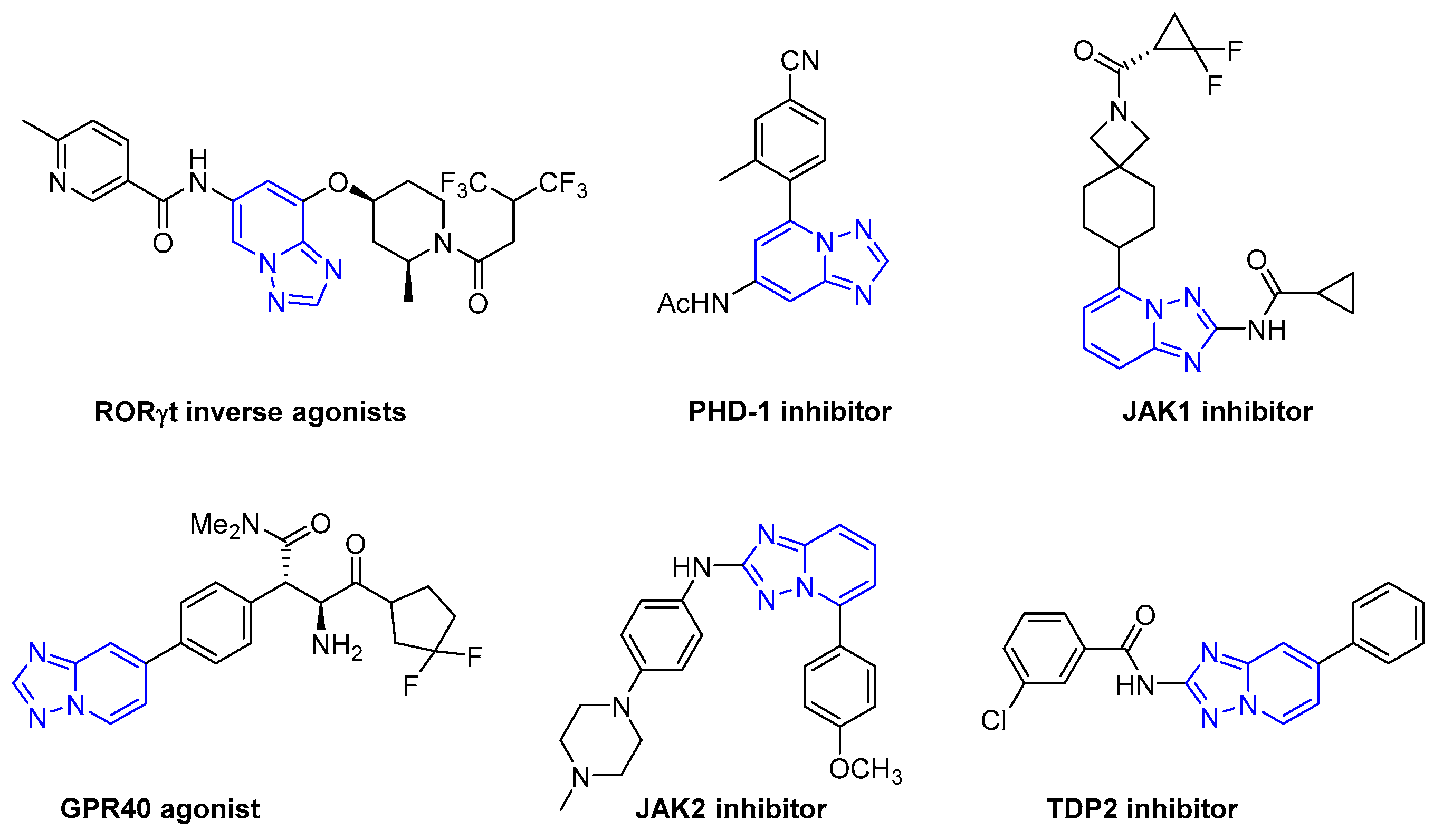
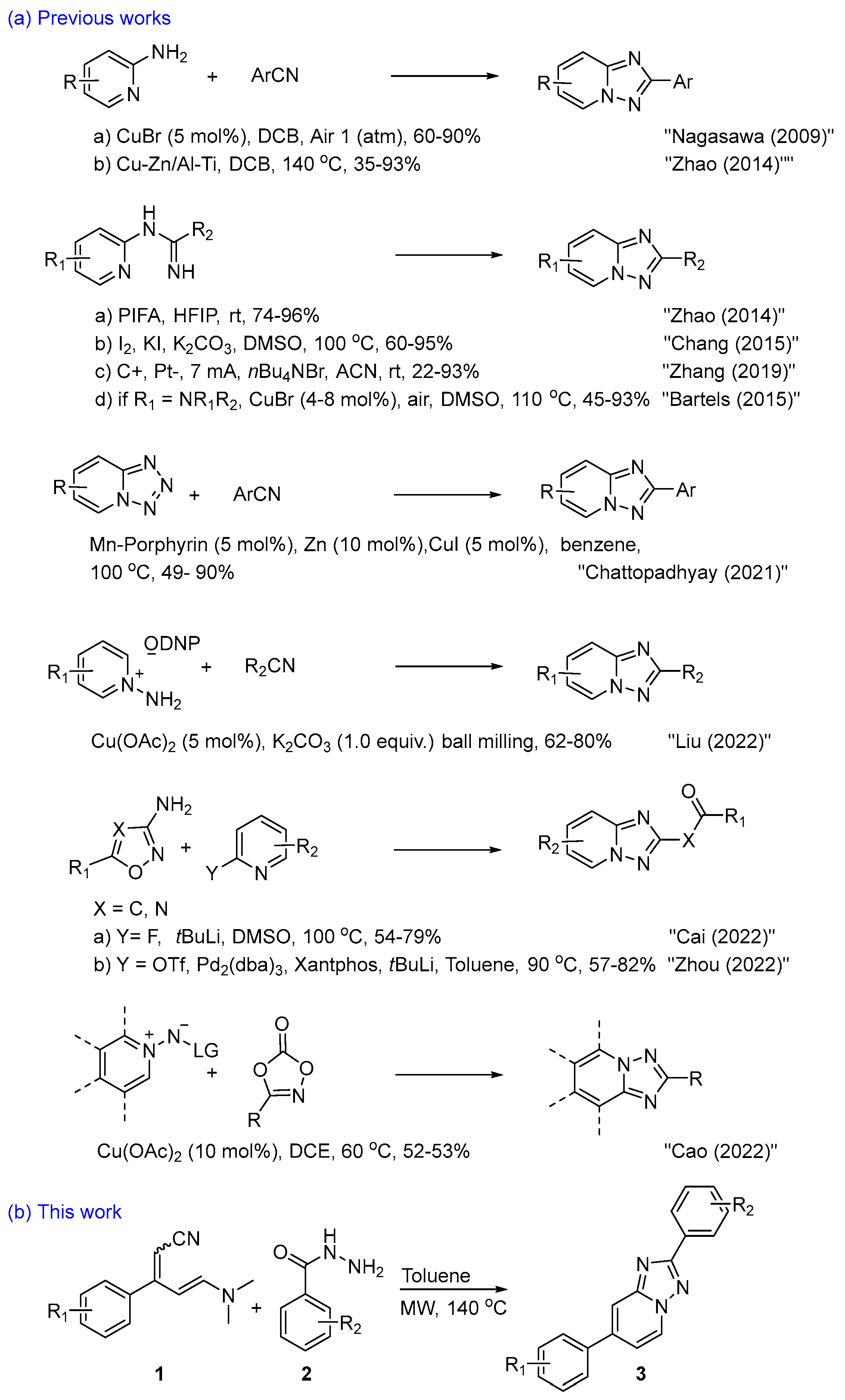
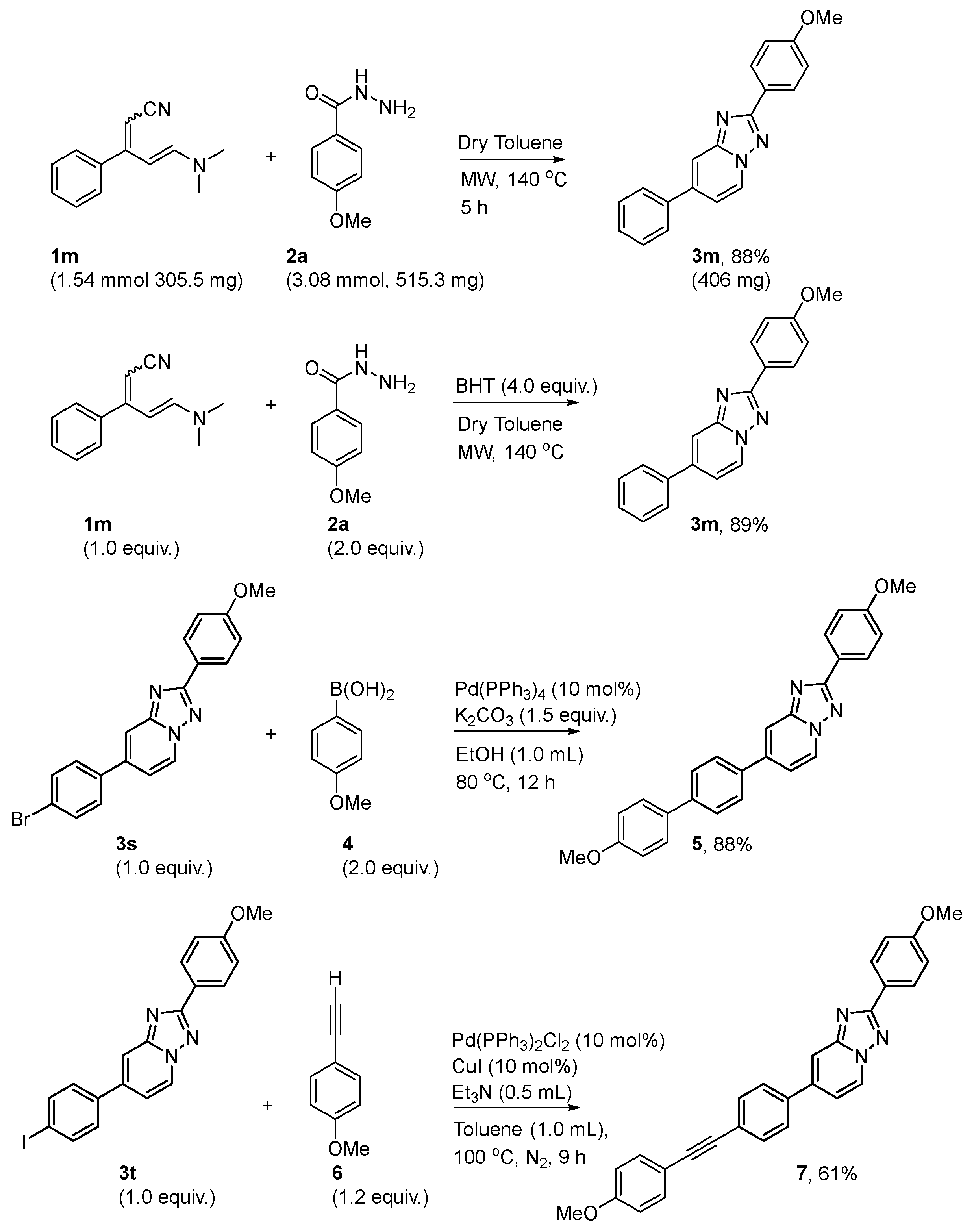


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Entry | Solvent | Additive | Time (h) | Yield (%) b |
|---|---|---|---|---|
| 1 | Toluene | 24 | 83 | |
| 2 | Solvents c | 24 | NR d | |
| 3 | DMF | 24 | 20 | |
| 4 | ACN | 24 | 17 | |
| 5 | Pyridine | 24 | 76 | |
| 6 | Xylene | 24 | 69 | |
| 7 | Chlorobenzene | 24 | 79 | |
| 8 | Dry Toluene | 12 | 86 | |
| 9 e | Dry Toluene | 3Å MS | 5 | 89 |
| 10 f | Dry Toluene | 13 | 27 | |
| 11 g | Dry Toluene | 26 | 51 | |
| 12 h | Dry Toluene | TFA | 2 | 79 |
| 13 h | Dry Toluene | p-TSA | 1 | 72 |
| 14 h | Dry Toluene | MsOH | 0.5 | 50 |
| 15 h | Dry Toluene | Bases i | 2 | NR |
| 16 j,k | Dry Toluene | 3 | 89 | |
| 17 l,k | Dry Toluene | 6 | 28 | |
| 18 m,k | Dry toluene | 6 | 62 | |
| 19 n,k | Dry Toluene | 90 min | 81 | |
| 20 o,k | Dry Toluene | 40 min | 76 | |
| 21 p,k | TBME | 3 | 69 |
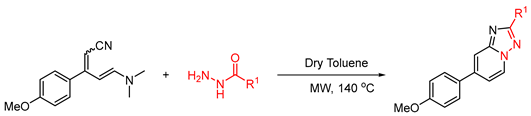
| Entry No | Compound Code | R1 | Time (h) | Yield (%) | Under Reflux Conditions b |
|---|---|---|---|---|---|
| 1 | 3a | phenyl | 3.5 | 83 | 8 h, 85% |
| 2 | 3b | 4-methoxyphenyl | 3 | 89 | 6 h, 89% |
| 3 | 3c | 4-tolyl | 3 | 82 | |
| 4 | 3d | 4-(trifluoromethyl)phenyl | 3 | 74 | |
| 5 | 3e | 4-nitrophenyl | 3.5 | 24 | 8 h, 24% |
| 6 | 3f | 4-chlorophenyl | 3 | 41 | 6 h, 42% |
| 7 | 3g | 4-bromophenyl | 3 | 43 | |
| 8 | 3h | 3-pyridinyl | 3 | 77 | |
| 9 | 3i | 2-thiophenyl | 2 | 94 | 5 h, 85% |
| 10 | 3j | 2-furanyl | 2 | 73 | |
| 11 | 3k | methyl | 7 | 67 | 12 h, 66% |
| 12 | 3l | heptyl | 6 | 46 |
| Entry No | Compound Code | R1 | Time (h) | Yield (%) | Under Reflux Conditions b |
|---|---|---|---|---|---|
| 13 | 3m | H | 3 | 89 | |
| 14 | 3n | 4-n-Pr | 4.5 | 80 | 7 h, 84% |
| 15 | 3o | 4-SMe | 3 | 71 | 5 h, 54% |
| 16 | 3p | 2-NO2 | 7 | 48 | 25 h, 47% |
| 17 | 3q | 4-F | 5 | 91 | |
| 18 | 3r | 4-Cl | 5 | 94 | |
| 19 | 3s | 4-Br | 6 | 90 | |
| 20 | 3t | 4-I | 7 | 71 | 12 h, 76% |
| 21 | 3u | 2,5-dichloro | 7 | 67 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, K.; Kim, Y.-A.; Jung, C.; Sim, J.; Rajasekar, S.; Kwak, J.-H.; Viji, M.; Jung, J.-K. Microwave-Mediated, Catalyst-Free Synthesis of 1,2,4-Triazolo[1,5-a]pyridines from Enaminonitriles. Molecules 2024, 29, 894. https://doi.org/10.3390/molecules29040894
Lee K, Kim Y-A, Jung C, Sim J, Rajasekar S, Kwak J-H, Viji M, Jung J-K. Microwave-Mediated, Catalyst-Free Synthesis of 1,2,4-Triazolo[1,5-a]pyridines from Enaminonitriles. Molecules. 2024; 29(4):894. https://doi.org/10.3390/molecules29040894
Chicago/Turabian StyleLee, Kwanghee, Young-Ah Kim, Chanhyun Jung, Jaeuk Sim, Shanmugam Rajasekar, Jae-Hwan Kwak, Mayavan Viji, and Jae-Kyung Jung. 2024. "Microwave-Mediated, Catalyst-Free Synthesis of 1,2,4-Triazolo[1,5-a]pyridines from Enaminonitriles" Molecules 29, no. 4: 894. https://doi.org/10.3390/molecules29040894
APA StyleLee, K., Kim, Y.-A., Jung, C., Sim, J., Rajasekar, S., Kwak, J.-H., Viji, M., & Jung, J.-K. (2024). Microwave-Mediated, Catalyst-Free Synthesis of 1,2,4-Triazolo[1,5-a]pyridines from Enaminonitriles. Molecules, 29(4), 894. https://doi.org/10.3390/molecules29040894