

Electronic Structures of Radical-Pair-Forming Cofactors in a Heliobacterial Reaction Center

Abstract
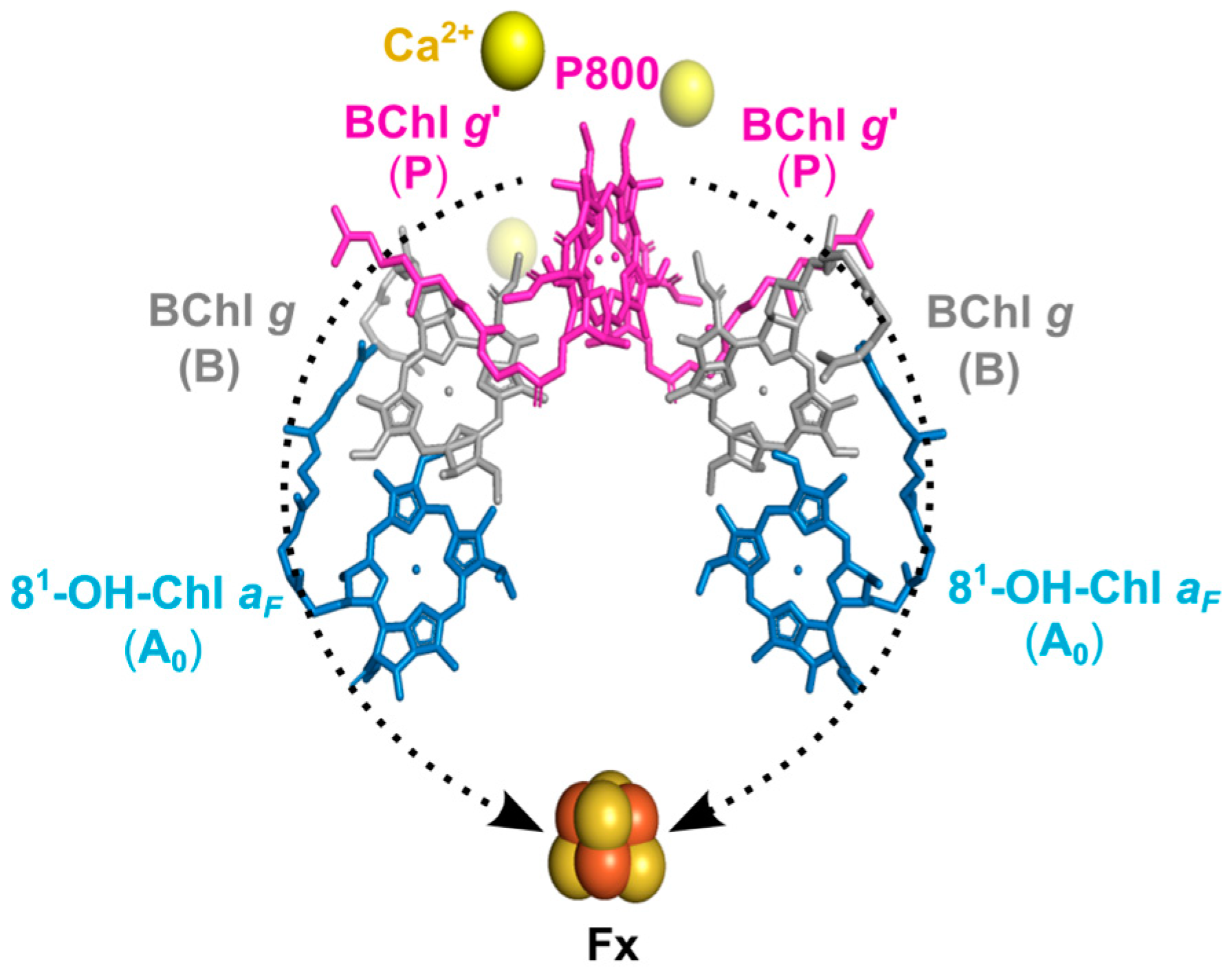
1. Introduction
2. Results
2.1. Isolation of Membrane Fragments
2.2. Magnetic-Field Dependency
2.3. 15N Chemical Shift Assignment
2.4. The Effect of 13C Isotope Enrichment
2.5. 13C Chemical Shift Assignment Based on One-Dimensional MAS NMR Spectra
2.6. 13C Chemical Shift Assignment Based on Two-Dimensional MAS NMR Spectra
2.7. Long-Range Transfer of Nuclear Hyperpolarization
2.8. The Relation of Chemical Shifts and Redox Potential
2.9. Electron Spin Density Distribution
3. Materials and Methods
3.1. Sample Preparation
3.2. NMR Measurement
3.2.1. NMR Set Up
3.2.2. One-Dimensional 13C and 15N Photo-CIDNP MAS NMR
3.2.3. Two-Dimensional 13C-13C Photo-CIDNP DARR MAS NMR under Continuous Illumination
3.3. DFT Calculation
4. Conclusions
- These RCs are homodimeric, i.e., only two cofactors appear in the NMR spectra.
- The lifetime of the SCRP is similar in both spin-states, singlet, and triplet. That implies that the DD mechanism is negligible, and the number of spin-dynamical mechanisms is reduced to two: the DR and TSM.
- All positive 13C signals originate from the donor caused by the dominance of the DR over the TSM (except for methine carbons). The TSM mechanism causes negative signals originating from the acceptor cofactor.
- In the 15N photo-CIDNP MAS NMR spectra, all signals are negative since both DR and TSM produce negative intensities, implying that, due to the negative gyromagnetic ratio of 15N spin sorting during the coherent singlet–triplet interconversion, this changes the sign.
- The field dependence of DR and TSM provides a simple tool to optimize the observation of either the donor or acceptor cofactor.
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ALA | δ-Aminolevulinic acid |
| BChl | Bacteriochlorophyll |
| BPhe | Bacteriopheophytin |
| Chl | Chlorophyll |
| CIDNP | Chemically induced dynamic nuclear polarization |
| DARR | Dipolar-assisted rotational resonance |
| DD | Differential decay |
| DFT | Density functional theory |
| DR | Differential relaxation |
| ET | Electron transfer |
| H. chlorum | Heliobacterium chlorum |
| Hb. mobilis | Heliobacillus mobilis |
| H. modesticaldum | Heliobacterium modesticaldum |
| MAS NMR | Magic-angle spinning nuclear magnetic resonance |
| OCSD | Overall chemical shift difference |
| Phe | Pheophytin |
| PSI | Photosystem I |
| PSII | Photosystem II |
| RC | Reaction center |
| r.f. | Radio-frequency |
| R. sphaeroides | Rhodobacter sphaeroides |
| SACS | Sum of the aromatic chemical shifts |
| SCRPs | Spin-correlated radical pairs |
| SWf-TPPM | Sweep-frequency two-pulse phase pulse modulation |
| TSM | Three-spin mixing |
| WT | Wild type |
References
- Wasielewski, M.R.; Niemczyk, M.P.; Svec, W.A.; Pewitt, E.B. High-Quantum-Yield Long-Lived Charge Separation in a Photosynthetic Reaction Center Model. J. Am. Chem. Soc. 1985, 107, 5562–5563. [Google Scholar] [CrossRef]
- Amoruso, G.; Liu, J.; Polak, D.W.; Tiwari, K.; Jones, M.R.; Oliver, T.A.A. High-Efficiency Excitation Energy Transfer in Biohybrid Quantum Dot-Bacterial Reaction Center Nanoconjugates. J. Phys. Chem. Lett. 2021, 12, 5448–5455. [Google Scholar] [CrossRef] [PubMed]
- Gorka, M.; Baldansuren, A.; Malnati, A.; Gruszecki, E.; Golbeck, J.H.; Lakshmi, K.V. Shedding Light on Primary Donors in Photosynthetic Reaction Centers. Front. Microbiol. 2021, 12, 735666. [Google Scholar] [CrossRef] [PubMed]
- Gest, H.; Favinger, J.L. Heliobacterium chlorum, an Anoxygenic Brownish-Green Photosynthetic Bacterium Containing a “New” Form of Bacteriochlorophyll. Arch. Microbiol. 1983, 136, 11–16. [Google Scholar] [CrossRef]
- Gest, H. Discovery of the Heliobacteria. Photosynth. Res. 1994, 41, 17–21. [Google Scholar] [CrossRef] [PubMed]
- Michalski, T.J.; Hunt, J.E.; Bowman, M.K.; Smith, U.; Bardeen, K.; Gest, H.; Norris, J.R.; Katz, J.J. Bacteriopheophytin g: Properties and some speculations on a possible primary role for bacteriochlorophylls b and g in the biosynthesis of chlorophylls. Proc. Natl. Acad. Sci. USA 1987, 84, 2570–2574. [Google Scholar] [CrossRef] [PubMed]
- Brockmann, H.; Lipinski, A. Bacteriochlorophyll g. A new bacteriochlorophyll from Heliobacterium chiorum. Arch. Microbiol. 1983, 136, 17–19. [Google Scholar] [CrossRef]
- Beer-Romero, P.; Gest, H. Heliobacillus mobilis, a Peritrichously Flagellated Anoxyphototroph Containing Bacteriochlorophyll g. FEMS Microbiol. Lett. 1987, 41, 109–114. [Google Scholar] [CrossRef]
- Kimble, L.K.; Mandelco, L.; Woese, C.R.; Madigan, M.T. Heliobacterium modesticaldum, sp. Nov., a Thermophilic Heliobacterium of Hot Springs and Volcanic Soils. Arch. Microbiol. 1995, 163, 259–267. [Google Scholar] [CrossRef]
- Trost, J.T.; Blankenship, R.E. Isolation of a Photoactive Photosynthetic Reaction Center-Core Antenna Complex from Heliobacillus mobilis. Biochemistry 1989, 28, 9898–9904. [Google Scholar] [CrossRef]
- Kobayashi, M.; van de Meent, E.J.; Erkelens, C.; Amesz, J.; Ikegami, I.; Watanabe, T. Bacteriochlorophyll g Epimer as a Possible Reaction Center Component of Heliobacteria. Biochim. Biophys. Acta-Bioenerg. 1991, 1057, 89–96. [Google Scholar] [CrossRef]
- Mizoguchi, T.; Oh-oka, H.; Hitoshi, T. Determination of stereochemistry of bacteriochlorophyll gF and 81-hydroxy-chlorophyll aF from Heliobacterium modesticaldum. Photochem. Photobiol. 2005, 81, 666–673. [Google Scholar] [CrossRef] [PubMed]
- van de Meent, E.J.; Kobayashi, M.; Erkelens, C.; van Veelen, P.A.; Amesz, J.; Watanabe, T. Identification of 81-Hydroxychlorophyll a as a Functional Reaction Center Pigment in Heliobacteria. Biochim. Biophys. Acta-Bioenerg. 1991, 1058, 356–362. [Google Scholar] [CrossRef]
- Miyamoto, R.; Iwaki, M.; Mino, H.; Harada, J.; Itoh, S.; Oh-Oka, H. ESR Signal of the Iron-Sulfur Center FX and Its Function in the Homodimeric Reaction Center of Heliobacterium modesticaldum. Biochemistry 2006, 45, 6306–6316. [Google Scholar] [CrossRef] [PubMed]
- Liebl, U.; Mockensturm-Wilson, M.; Trost, J.T.; Brune, D.C.; Blankenship, R.E.; Vermaas, W. Single Core Polypeptide in the Reaction Center of the Photosynthetic Bacterium Heliobacillus Mobilis: Structural Implications and Relations to Other Photosystems. Proc. Natl. Acad. Sci. USA 1993, 90, 7124–7128. [Google Scholar] [CrossRef]
- Prince, R.C.; Gest, H.; Blankenship, R.E. Thermodynamic Properties of the Photochemical Reaction Center of Heliobacterium chlorum. Biochim. Biophys. Acta (BBA)-Bioenerg. 1985, 810, 377–384. [Google Scholar] [CrossRef]
- Van Noort, P.I.; Aartsma, T.J.; Amesz, J. Energy Transfer and Primary Charge Separation in Heliobacteria by Picosecond Transient Absorption Spectroscopy. Springer Ser. Chem. Phys. 1993, 1140, 549–551. [Google Scholar] [CrossRef]
- Lin, S.; Chiou, H.C.; Kleinherenbrink, F.A.; Blankenship, R.E. Time-Resolved Spectroscopy of Energy and Electron Transfer Processes in the Photosynthetic Bacterium Heliobacillus mobilis. Biophys. J. 1994, 66, 437–445. [Google Scholar] [CrossRef]
- Fischer, M.R. Photosynthetic Electron Transfer in Heliobacterium chlorum Studied by EPR Spectroscopy. Biochim. Biophys. Acta-Bioenerg. 1990, 1015, 471–481. [Google Scholar] [CrossRef]
- Nuijs, A.M.; van Dorssen, R.J.; Duysens, L.N.M.; Amesz, J. Excited States and Primary Photochemical Reactions in the Photosynthetic Bacterium Heliobacterium chlorum. Proc. Nati. Acad. Sci. USA 1985, 82, 6865–6868. [Google Scholar] [CrossRef] [PubMed]
- Gisriel, C.; Sarrou, I.; Ferlez, B.; Golbeck, J.H.; Redding, K.E.; Fromme, R. Structure of a Symmetric Photosynthetic Reaction Center-Photosystem. Science 2017, 357, 1021–1025. [Google Scholar] [CrossRef]
- Song, Y.; Sechrist, R.; Nguyen, H.H.; Johnson, W.; Abramavicius, D.; Redding, K.E.; Ogilvie, J.P. Excitonic Structure and Charge Separation in the Heliobacterial Reaction Center Probed by Multispectral Multidimensional Spectroscopy. Nat. Commun. 2021, 12, 2801. [Google Scholar] [CrossRef]
- Oh-oka, H. Type 1 Reaction Center of Photosynthetic Heliobacteria. Photochem. Photobiol. 2007, 83, 177–186. [Google Scholar] [CrossRef] [PubMed]
- Sarrou, I.; Khan, Z.; Cowgill, J.; Lin, S.; Brune, D.; Romberger, S.; Golbeck, J.H.; Redding, K.E. Purification of the Photosynthetic Reaction Center from Heliobacterium modesticaldum. Photosynth. Res. 2012, 111, 291–302. [Google Scholar] [CrossRef]
- Hiraishi, A. Occurrence of menaquinone as the sole isoprenoid quinone in the photosynthetic bacterium Heliobacterium chlorum. Arch. Microbiol. 1989, 151, 378–379. [Google Scholar] [CrossRef]
- Kashey, T.S.; Luu, D.D.; Cowgill, J.C.; Baker, P.L.; Redding, K.E. Light-Driven Quinone Reduction in Heliobacterial Membranes. Photosynth. Res. 2018, 138, 1–9. [Google Scholar] [CrossRef]
- Lin, S.; Chiou, H.C.; Blankenship, R.E. Secondary Electron Transfer Processes in Membranes of Heliobacillus Mobilis. Biochemistry 1995, 34, 12761–12767. [Google Scholar] [CrossRef]
- Van Der Est, A.; Hager-Braun, C.; Leibl, W.; Hauska, G.; Stehlik, D. Transient Electron Paramagnetic Resonance Spectroscopy on Green-Sulfur Bacteria and Heliobacteria at Two Microwave Frequencies. Biochim. Biophys. Acta-Bioenerg. 1998, 1409, 87–98. [Google Scholar] [CrossRef] [PubMed]
- Brettel, K.; Leibl, W.; Liebl, U. Electron Transfer in the Heliobacterial Reaction Center: Evidence against a Quinone-Type Electron Acceptor Functioning Analogous to A1 in Photosystem I. Biochim. Biophys. Acta-Bioenerg. 1998, 1363, 175–181. [Google Scholar] [CrossRef]
- Kleinherenbrink, F.A.M.; Ikegami, I.; Hiraishi, A.; Otte, S.C.M.; Amesz, J. Electron Transfer in Menaquinone-Depleted Membranes of Heliobacterium chlorum. Biochim. Biophys. Acta-Bioenerg. 1993, 1142, 69–73. [Google Scholar] [CrossRef]
- Chauvet, A.; Sarrou, J.; Lin, S.; Romberger, S.P.; Golbeck, J.H.; Savikhin, S.; Redding, K.E. Temporal and Spectral Characterization of the Photosynthetic Reaction Center from Heliobacterium modesticaldum. Photosynth. Res. 2013, 116, 1–9. [Google Scholar] [CrossRef]
- Miyamoto, R.; Mino, H.; Kondo, T.; Itoh, S.; Oh-oka, H. An Electron Spin-Polarized Signal of the P800+A1(Q)− State in the Homodimeric Reaction Center Core Complex of Heliobacterium modesticaldum. Biochemistry 2008, 47, 4386–4393. [Google Scholar] [CrossRef] [PubMed]
- Kondo, T.; Itoh, S.; Matsuoka, M.; Azai, C.; Oh-Oka, H. Menaquinone as the Secondary Electron Acceptor in the Type I Homodimeric Photosynthetic Reaction Center of Heliobacterium modesticaldum. J. Phys. Chem. B 2015, 119, 8480–8489. [Google Scholar] [CrossRef] [PubMed]
- Kondo, T.; Matsuoka, M.; Azai, C.; Kobayashi, M.; Itoh, S.; Oh-Oka, H. Light-Induced Electron Spin-Polarized (ESP) EPR Signal of the P800+ Menaquinone- Radical Pair State in Oriented Membranes of Heliobacterium modesticaldum: Role/Location of Menaquinone in the Homodimeric Type I Reaction Center. J. Phys. Chem. B 2018, 122, 2536–2543. [Google Scholar] [CrossRef]
- Orf, G.S.; Gisriel, C.; Redding, K.E. Evolution of Photosynthetic Reaction Centers: Insights from the Structure of the Heliobacterial Reaction Center. Photosynth. Res. 2018, 138, 11–37. [Google Scholar] [CrossRef] [PubMed]
- Imada, Y.; Nakamura, H.; Takano, Y. Density Functional Study of Porphyrin Distortion Effects on Redox Potential of Heme. J. Comput. Chem. 2018, 39, 143–150. [Google Scholar] [CrossRef] [PubMed]
- De Wijn, R.; Van Gorkom, H.J. The Rate of Charge Recombination in Photosystem II. Biochim. Biophys. Acta-Bioenerg. 2002, 1553, 302–308. [Google Scholar] [CrossRef]
- Ishikita, H.; Saenger, W.; Biesiadka, J.; Loll, B.; Knapp, E.W. How Photosynthetic Reaction Centers Control Oxidation Power in Chlorophyll Pairs P680, P700, and P870. Proc. Natl. Acad. Sci. USA 2006, 103, 9855–9860. [Google Scholar] [CrossRef]
- Kanda, T.; Ishikita, H. Energetic Diversity in the Electron-Transfer Pathways of Type I Photosynthetic Reaction Centers. Biochemistry 2023, 62, 934–941. [Google Scholar] [CrossRef]
- Ferlez, B.; Cowgill, J.; Dong, W.; Gisriel, C.; Lin, S.; Flores, M.; Walters, K.; Cetnar, D.; Redding, K.E.; Golbeck, J.H. Thermodynamics of the Electron Acceptors in Heliobacterium modesticaldum: An Exemplar of an Early Homodimeric Type I Photosynthetic Reaction Center. Biochemistry 2016, 55, 2358–2370. [Google Scholar] [CrossRef]
- Kleinherenbrink, F.A.M.; Aartsma, T.J.; Amesz, J. Charge Separation and Formation of Bacteriochlorophyll Triplets in Heliobacterium chlorum. Biochim. Biophys. Acta-Bioenerg. 1991, 1057, 346–352. [Google Scholar] [CrossRef]
- Smit, H.W.J.; Amesz, J.; van der Hoeven, M.F.R. Electron Transport and Triplet Formation in Membranes of the Photosynthetic Bacterium Heliobacterium chlorum. Biochim. Biophys. Acta-Bioenerg. 1987, 893, 232–240. [Google Scholar] [CrossRef]
- Ferlez, B.; Agostini, A.; Carbonera, D.; Golbeck, J.H.; Van Der Est, A. Triplet Charge Recombination in Heliobacterial Reaction Centers Does Not Produce a Spin-Polarized EPR Spectrum. Z. Phys. Chem. 2017, 231, 593–607. [Google Scholar] [CrossRef]
- Zysmilich, M.G.; McDermott, A. Photochemically Induced Dynamic Nuclear Polarization in the Solid-State 15N Spectra of Reaction Centers from Photosynthetic Bacteria Rhodobacter spbaeroides R-26. J. Am. Chem. Soc. 1994, 116, 8362–8363. [Google Scholar] [CrossRef]
- Hore, P.J.; Winder, S.L.; Roberts, C.H.; Dobson, C.M. Stopped-Flow Photo-CIDNP Observation of Protein Folding. J. Am. Chem. Soc. 1997, 119, 5049–5050. [Google Scholar] [CrossRef]
- Hore, J.; Broadhurst, R.W. Photo-CIDNP of Biopolymers. Prog. Nucl. Magn. Reson. Spectrosc. 1993, 25, 345–402. [Google Scholar] [CrossRef]
- Jeschke, G.; Matysik, J. A Reassessment of the Origin of Photochemically Induced Dynamic Nuclear Polarization Effects in Solids. Chem. Phys. 2003, 294, 239–255. [Google Scholar] [CrossRef]
- Matysik, J.; Diller, A.; Roy, E.; Alia, A. The Solid-State Photo-CIDNP Effect. Photosynth. Res. 2009, 102, 427–435. [Google Scholar] [CrossRef][Green Version]
- Sai Sankar Gupta, K.B.; Alia, A.; De Groot, H.J.M.; Matysik, J. Symmetry Break of Special Pair: Photochemically Induced Dynamic Nuclear Polarization NMR Confirms Control by Nonaromatic Substituents. J. Am. Chem. Soc. 2013, 135, 10382–10387. [Google Scholar] [CrossRef]
- Prakash, S.; Alia; Gast, P.; De Groot, H.J.M.; Jeschke, G.; Matysik, J. Magnetic Field Dependence of Photo-CIDNP MAS NMR on Photosynthetic Reaction Centers of Rhodobacter sphaeroides WT. J. Am. Chem. Soc. 2005, 127, 14290–14298. [Google Scholar] [CrossRef]
- Prakash, S.; Alia, A.; Gast, P.; De Groot, H.J.M.; Matysik, J.; Jeschke, G. Photo-CIDNP MAS NMR in Intact Cells of Rhodobacter Sphaeroides R26: Molecular and Atomic Resolution at Nanomolar Concentration. J. Am. Chem. Soc. 2006, 128, 12794–12799. [Google Scholar] [CrossRef]
- Daviso, E.; Prakash, S.; Alia, A.; Gast, P.; Jeschke, G.; Matysik, J. Nanosecond-Flash 15N Photo-CIDNP MAS NMR on Reaction Centers of Rhodobacter sphaeroides R26. Appl. Magn. Reson. 2010, 37, 49–63. [Google Scholar] [CrossRef][Green Version]
- Janssen, G.J.; Roy, E.; Matysik, J.; Alia, A. 15N Photo-CIDNP MAS NMR To Reveal Functional Heterogeneity in Electron Donor of Different Plant Organisms. Appl. Magn. Reson. 2012, 42, 57–67. [Google Scholar] [CrossRef]
- Janssen, G.J.; Eschenbach, P.; Kurle, P.; Bode, B.E.; Neugebauer, J.; de Groot, H.J.M.; Matysik, J.; Alia, A. Analysis of the Electronic Structure of the Primary Electron Donor of Photosystem I of Spirodela oligorrhiza by Photochemically Induced Dynamic Nuclear Polarization (Photo-CIDNP) Solid-State Nuclear Magnetic Resonance (NMR). Magn. Reson. 2020, 1, 261–274. [Google Scholar] [CrossRef]
- Zill, J.C.; Kansy, M.; Goss, R.; Köhler, L.; Alia, A.; Wilhelm, C.; Matysik, J. Photo-CIDNP in the Reaction Center of the Diatom Cyclotella meneghiniana Observed by 13C MAS NMR the Solid-State Photo-CIDNP Effect in a New Kingdom of the Tree of Life. Z. Phys. Chem. 2017, 231, 347–367. [Google Scholar] [CrossRef]
- Roy, E.; Rohmer, T.; Gast, P.; Jeschke, G.; Alia, A.; Matysik, J. Characterization of the Primary Radical Pair in Reaction Centers of Heliobacillus mobilis by 13C Photo-CIDNP MAS NMR. Biochemistry 2008, 47, 4629–4635. [Google Scholar] [CrossRef]
- Thamarath, S.S.; Alia, A.; Daviso, E.; Mance, D.; Golbeck, J.H.; Matysik, J. Whole Cell Nuclear Magnetic Resonance Characterization of Two Photochemically Active States of the Photosynthetic Reaction Center in Heliobacteria. Biochemistry 2012, 51, 5763–5773. [Google Scholar] [CrossRef]
- Thamarath, S.S.; Alia, A.; Roy, E.; Sai Sankar Gupta, K.B.; Golbeck, J.H.; Matysik, J. The Field-Dependence of the Solid-State Photo-CIDNP Effect in Two States of Heliobacterial Reaction Centers. Photosynth. Res. 2013, 117, 461–469. [Google Scholar] [CrossRef]
- Richter, G.; Weber, S.; Römisch, W.; Bacher, A.; Fischer, M.; Eisenreich, W. Photochemically Induced Dynamic Nuclear Polarization in a C450A Mutant of the LOV2 Domain of the Avena Sativa Blue-Light Receptor Phototropin. J. Am. Chem. Soc. 2005, 127, 17245–17252. [Google Scholar] [CrossRef] [PubMed]
- Kothe, G.; Lukaschek, M.; Link, G.; Kacprzak, S.; Illarionov, B.; Fischer, M.; Eisenreich, W.; Bacher, A.; Weber, S. Detecting a New Source for Photochemically Induced Dynamic Nuclear Polarization in the LOV2 Domain of Phototropin by Magnetic-Field Dependent 13C NMR Spectroscopy. J. Phys. Chem. B 2014, 118, 11622–11632. [Google Scholar] [CrossRef] [PubMed]
- Thamarath, S.S.; Heberle, J.; Hore, P.J.; Kottke, T.; Matysik, J. Solid-State Photo-CIDNP Effect Observed in Phototropin LOV1-C57S by 13C Magic-Angle Spinning NMR Spectroscopy. J. Am. Chem. Soc. 2010, 132, 15542–15543. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Kiryutin, A.S.; Yurkovskaya, A.V.; Sosnovsky, D.V.; Sagdeev, R.Z.; Bannister, S.; Kottke, T.; Kar, R.K.; Schapiro, I.; Ivanov, K.L.; et al. Nuclear Spin-Hyperpolarization Generated in a Flavoprotein under Illumination: Experimental Field-Dependence and Theoretical Level Crossing Analysis. Sci. Rep. 2019, 9, 18436. [Google Scholar] [CrossRef]
- Ding, Y.; Kiryutin, A.S.; Zhao, Z.; Xu, Q.Z.; Zhao, K.H.; Kurle, P.; Bannister, S.; Kottke, T.; Sagdeev, R.Z.; Ivanov, K.L.; et al. Tailored Flavoproteins Acting as Light-Driven Spin Machines Pump Nuclear Hyperpolarization. Sci. Rep. 2020, 10, 18658. [Google Scholar] [CrossRef]
- Jeschke, G. Electron-Electron-Nuclear Three-Spin Mixing in Spin-Correlated Radical Pairs. J. Chem. Phys. 1997, 106, 10072–10086. [Google Scholar] [CrossRef]
- Mcdermott, A.; Zysmilich, M.G.; Polenová, T. Solid State NMR Studies of Photoinduced Polarization in Photosynthetic Reaction Centers: Mechanism and Simulations. Solid State Nucl. Magn. Reson. 1998, 11, 21–47. [Google Scholar] [CrossRef]
- Polenova, T.; McDermott, A.E. A Coherent Mixing Mechanism Explains the Photoinduced Nuclear Polarization in Photosynthetic Reaction Centers. J. Phys. Chem. B 1999, 103, 535–548. [Google Scholar] [CrossRef]
- Redding, K.E.; Sarrou, I.; Rappaport, F.; Santabarbara, S.; Lin, S.; Reifschneider, K.T. Modulation of the Fluorescence Yield in Heliobacterial Cells by Induction of Charge Recombination in the Photosynthetic Reaction Center. Photosynth. Res. 2014, 120, 221–235. [Google Scholar] [CrossRef]
- Camara-Artigas, A.; Brune, D.; Allen, J.P. Interactions between Lipids and Bacterial Reaction Centers Determined by Protein Crystallography. Proc. Natl. Acad. Sci. USA 2002, 99, 11055–11060. [Google Scholar] [CrossRef]
- Thamarath, S.S.; Bode, B.E.; Prakash, S.; Sai Sankar Gupta, K.B.; Alia, A.; Jeschke, G.; Matysik, J. Electron Spin Density Distribution in the Special Pair Triplet of Rhodobacter sphaeroides R26 Revealed by Magnetic Field Dependence of the Solid-State Photo-CIDNP Effect. J. Am. Chem. Soc. 2012, 134, 5921–5930. [Google Scholar] [CrossRef] [PubMed]
- Matysik, J.; Ding, Y.; Kim, Y.; Kurle, P.; Yurkovskaya, A.; Ivanov, K.; Alia, A. Photo-CIDNP in Solid State. Appl. Magn. Reson. 2022, 53, 521–537. [Google Scholar] [CrossRef] [PubMed]
- Valanne, N.; Aro, E.-M. Incorporation of 5-Aminolevulinic Acid in the Chlorophyll-Protein Complexes of the Moss Ceratodon Purpureus. Physiol. Plant. 1976, 37, 218–222. [Google Scholar] [CrossRef] [PubMed]
- Bhaya, D.; Castelfranco, P.A. Chlorophyll Biosynthesis and Assembly into Chlorophyll-Protein Complexes in Isolated Developing Chloroplasts. Proc. Natl. Acad. Sci. USA 1985, 82, 5370–5374. [Google Scholar] [CrossRef] [PubMed]
- Diller, A.; Roy, E.; Gast, P.; Van Gorkom, H.J.; De Groot, H.J.M.; Glaubitz, C.; Jeschke, G.; Matysik, J.; Alia, A. 15N Photochemically Induced Dynamic Nuclear Polarization Magic-Angle Spinning NMR Analysis of the Electron Donor of Photosystem II. Proc. Natl. Acad. Sci. USA 2007, 104, 12767–12771. [Google Scholar] [CrossRef] [PubMed]
- Paul, S.; Roy, U.; Böckers, M.; Neugebauer, J.; Alia, A.; Matysik, J. 15N Photo-CIDNP MAS NMR Analysis of a Bacterial Photosynthetic Reaction Center of Rhodobacter sphaeroides Wildtype. J. Chem. Phys. 2019, 151, 195101. [Google Scholar] [CrossRef]
- Prakash, S.; Alia, A.; Gast, P.; De Groot, H.J.M.; Jeschke, G.; Matysik, J. 13C Chemical Shift Map of the Active Cofactors in Photosynthetic Reaction Centers of Rhodobacter sphaeroides Revealed by Photo-CIDNP MAS NMR. Biochemistry 2007, 46, 8953–8960. [Google Scholar] [CrossRef] [PubMed]
- Najdanova, M.; Gräsing, D.; Alia, A.; Matysik, J. Analysis of the Electronic Structure of the Special Pair of a Bacterial Photosynthetic Reaction Center by 13C Photochemically Induced Dynamic Nuclear Polarization Magic-Angle Spinning NMR Using a Double-Quantum Axis. Photochem. Photobiol. 2018, 94, 69–80. [Google Scholar] [CrossRef]
- Qiu, N.W.; Jiang, D.C.; Wang, X.S.; Wang, B.S.; Zhou, F. Advances in the Members and Biosynthesis of Chlorophyll Family. Photosynthetica 2019, 57, 974–984. [Google Scholar] [CrossRef]
- Tsukatani, Y.; Yamamoto, H.; Mizoguchi, T.; Fujita, Y.; Tamiaki, H. Completion of Biosynthetic Pathways for Bacteriochlorophyll g in Heliobacterium modesticaldum: The C8-Ethylidene Group Formation. Biochim. Biophys. Acta-Bioenerg. 2013, 1827, 1200–1204. [Google Scholar] [CrossRef]
- Boender, G.J. The Stacking of Chlorophylls in the Chlorosomal Antennae of Green Bacteria. Ph.D. Thesis, University of Leiden, Leiden, The Netherlands, 1996. [Google Scholar]
- Matysik, J.; Alia, A.; Gast, P.; Lugtenburg, J.; Hoff, A.J.; de Groot, H.J.M. Perspectives on Solid State NMR in Biology; Kiihne, S.R., de Groot, H.J.M., Eds.; Kluwer Academic: Dordrecht, The Netherlands, 2001; pp. 215–225. [Google Scholar]
- Pandit, A.; Buda, F.; Van Gammeren, A.J.; Ganapathy, S.; De Groot, H.J.M. Selective Chemical Shift Assignment of Bacteriochlorophyll α in Uniformly [13C-15N]-Labeled Light-Harvesting 1 Complexes by Solid-State NMR in Ultrahigh Magnetic Field. J. Phys. Chem. B 2010, 114, 6207–6215. [Google Scholar] [CrossRef]
- Daviso, E.; Prakash, S.; Alia, A.; Gast, P.; Neugebauer, J.; Jeschke, G.; Matysik, J. The Electronic Structure of the Primary Electron Donor of Reaction Centers of Purple Bacteria at Atomic Resolution as Observed by Photo-CIDNP 13C NMR. Proc. Natl. Acad. Sci. USA 2009, 106, 22281–22286. [Google Scholar] [CrossRef]
- Takegoshi, K. Homonuclear Shift-Correlation Experiment in Solids. In Modern Magnetic Resonance; Webb, G.A., Ed.; Springer: Dordrecht, The Netherlands, 2008. [Google Scholar] [CrossRef]
- Takegoshi, K.; Nakamura, S.; Terao, T. 13C-1H Dipolar-Driven 13C-13C Recoupling without 13C rf Irradiation in Nuclear Magnetic Resonance of Rotating Solids. J. Chem. Phys. 2003, 118, 2325–2341. [Google Scholar] [CrossRef]
- Veshtort, M.; Griffin, R.G. Proton-Driven Spin Diffusion in Rotating Solids via Reversible and Irreversible Quantum Dynamics. J. Chem. Phys. 2011, 135, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Huster, D.; Yao, X.; Hong, M. Membrane Protein Topology Probed by 1H Spin Diffusion from Lipids Using Solid-State NMR Spectroscopy. J. Am. Chem. Soc. 2002, 124, 874–883. [Google Scholar] [CrossRef] [PubMed]
- Addison, B.; Stengel, D.; Bharadwaj, V.S.; Happs, R.M.; Doeppke, C.; Wang, T.; Bomble, Y.J.; Holland, G.P.; Harman-Ware, A.E. Selective One-Dimensional 13C-13C Spin-Diffusion Solid-State Nuclear Magnetic Resonance Methods to Probe Spatial Arrangements in Biopolymers Including Plant Cell Walls, Peptides, and Spider Silk. J. Phys. Chem. B 2020, 124, 9870–9883. [Google Scholar] [CrossRef] [PubMed]
- Hesse, M.; Meier, H.; Zeeh, B. Spektroskopische Methoden in der Organischen Chemie, 7th ed.; Thieme: Stuttgart, Germany, 2005; p. 165. [Google Scholar]
- Sai Sankar Gupta, K.B.; Daviso, E.; Jeschke, G.; Alia, A.; Ernst, M.; Matysik, J. Spectral Editing through Laser-Flash Excitation in Two-Dimensional Photo-CIDNP MAS NMR Experiments. J. Magn. Reson. 2014, 246, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Sai Sankar Gupta, K.B.; Alia, A.; Buda, F.; De Groot, H.J.M.; Matysik, J. Bacteriopheophytin a in the Active Branch of the Reaction Center of Rhodobacter sphaeroides Is Not Disturbed by the Protein Matrix as Shown by 13C Photo-CIDNP MAS NMR. J. Phys. Chem. B 2013, 117, 3287–3297. [Google Scholar] [CrossRef] [PubMed]
- Gräsing, D. Photo-CIDNP MAS NMR—Generation, Transfer and Application of Nuclear Hyperpolarization. Ph.D. Thesis, Universität Leipzig, Leipzig, Germany, 2019. [Google Scholar]
- Janssen, G.J.; Bielytskyi, P.; Artiukhin, D.G.; Neugebauer, J.; de Groot, H.J.M.; Matysik, J.; Alia, A. Photochemically Induced Dynamic Nuclear Polarization NMR on Photosystem II: Donor Cofactor Observed in Entire Plant. Sci. Rep. 2018, 8, 17853. [Google Scholar] [CrossRef] [PubMed]
- Egorova-Zachernyuk, T.A.; Van Rossum, B.; Boender, G.J.; Franken, E.; Ashurst, J.; Raap, J.; Gast, P.; Hoff, A.J.; Oschkinat, H.; De Groot, H.J.M. Characterization of Pheophytin Ground States in Rhodobacter sphaeroides R26 Photosynthetic Reaction Centers from Multispin Pheophytin Enrichment and 2-D 13C MAS NMR Dipolar Correlation Spectroscopy. Biochemistry 1997, 36, 7513–7519. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Murchison, H.A.; Nagarajan, V.; Parson, W.W.; Allen, J.P.; Williams, J.C. Specific Alteration of the Oxidation Potential of the Electron Donor in Reaction Centers from Rhodobacter sphaeroides. Proc. Natl. Acad. Sci. USA 1994, 91, 10265–10269. [Google Scholar] [CrossRef]
- Spiedel, D.; Jones, M.R.; Robert, B. Tuning of the Redox Potential of the Primary Electron Donor in Reaction Centres of Purple Bacteria: Effects of Amino Acid Polarity and Position. FEBS Lett. 2002, 527, 171–175. [Google Scholar] [CrossRef]
- Allen, J.P.; Williams, J.C. Relationship between the Oxidation Potential of the Bacteriochlorophyll Dimer and Electron Transfer in Photosynthetic Reaction Centers. J. Bioenerg. Biomembr. 1995, 27, 275–283. [Google Scholar] [CrossRef]
- Cogdell, R.J.; Valentine, J. Bacterial Photosynthesis. Photochem. Photobiol. 1983, 38, 769–772. [Google Scholar] [CrossRef]
- Allakhverdiev, S.I.; Tomo, T.; Shimada, Y.; Kindo, H.; Nagao, R.; Klimov, V.V.; Mimuro, M. Redox Potential of Pheophytin a in Photosystem II of Two Cyanobacteria Having the Different Special Pair Chlorophylls. Proc. Natl. Acad. Sci. USA 2010, 107, 3924–3929. [Google Scholar] [CrossRef] [PubMed]
- Rutherford, A.W.; Mullet, J.E.; Crofts, A.R. Measurement of the Midpoint Potential of the Pheophytin Acceptor of Photosystem II. FEBS Lett. 1981, 123, 235–237. [Google Scholar] [CrossRef]
- Diers, J.R.; Kirmaier, C.; Taniguchi, M.; Lindsey, J.S.; Bocian, D.F.; Holten, D. A Perspective on the Redox Properties of Tetrapyrrole Macrocycles. Phys. Chem. Chem. Phys. 2021, 23, 19130–19140. [Google Scholar] [CrossRef]
- Alric, J.; Cuni, A.; Maki, H.; Nagashima, K.V.P.; Verméglio, A.; Rappaport, F. Electrostatic Interaction between Redox Cofactors in Photosynthetic Reaction Centers. J. Biol. Chem. 2004, 279, 47849–47855. [Google Scholar] [CrossRef] [PubMed]
- Parson, W.W.; Chu, Z.T.; Warshel, A. Electrostatic Control of Charge Separation in Bacterial Photosynthesis. Biochim. Biophys. Acta-Bioenerg. 1990, 1017, 251–272. [Google Scholar] [CrossRef]
- Mandal, M.; Kawashima, K.; Saito, K.; Ishikita, H. Redox Potential of the Oxygen-Evolving Complex in the Electron Transfer Cascade of Photosystem II. J. Phys. Chem. Lett. 2020, 11, 249–255. [Google Scholar] [CrossRef] [PubMed]
- Matysik, J.; Alia, A.; Gast, P.; Van Gorkom, H.J.; Hoff, A.J.; De Groot, H.J.M. Photochemically Induced Nuclear Spin Polarization in Reaction Centers of Photosystem II Observed by 13C-Solid-State NMR Reveals a Strongly Asymmetric Electronic Structure of the P•+680 Primary Donor Chlorophyll. Proc. Natl. Acad. Sci. USA 2000, 97, 9865–9870. [Google Scholar] [CrossRef] [PubMed]
- Najdanova, M.; Janssen, G.J.; De Groot, H.J.M.; Matysik, J.; Alia, A. Analysis of Electron Donors in Photosystems in Oxygenic Photosynthesis by Photo-CIDNP MAS NMR. J. Photochem. Photobiol. B Biol. 2015, 152, 261–271. [Google Scholar] [CrossRef]
- van de Meent, E.J.; Kleinherenbrink, F.A.M.; Amesz, J. Purification and Properties of an Antenna-Reaction Center Complex from Heliobacteria. Biochim. Biophys. Acta-Bioenerg. 1990, 1015, 223–230. [Google Scholar] [CrossRef]
- Matysik, J.; Alia, A.; Hollander, J.G.; Egorova-Zachernyuk, T.; Gast, P.; De Groot, H.J.M. A Set-up to Study Photochemically Induced Dynamic Nclear Polarization in Photosynthetic Reaction Centrer by Solid-State NMR. Indian J. Biochem. Biophys. 2000, 37, 418–423. [Google Scholar]
- Fischer, M.R.; Hoff, A.J.; de Groot, H.J.M.; Raap, J.; Lugtenburg, J. 13C Magic Angle Spinning NMR Study of the Light-Induced and Temperature-Dependent Changes in Rhodobacter Sphaeroides R26 Reaction Centers Enriched in [4′-13C] Tyrosinet. Biochemistry 1992, 31, 11038–11049. [Google Scholar] [CrossRef]
- Thakur, R.S.; Kurur, N.D.; Madhu, P.K. Swept-Frequency Two-Pulse Phase Modulation for Heteronuclear Dipolar Decoupling in Solid-State NMR. Chem. Phys. Lett. 2006, 426, 459–463. [Google Scholar] [CrossRef]
- Marion, D.; Ikura, M.; Tschudin, R.; Bax, A. Rapid Recording of 2D NMR Spectra without Phase Cycling. Application To The Study of Hydrogen Exchange in Proteins. J. Magn. Reson. 1989, 85, 393–399. [Google Scholar] [CrossRef]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeersch, T.; Zurek, E.; Hutchison, G.R. Avogadro: An advanced semantic chemical editor, visualization, and analysis platform. J. Cheminform. 2012, 4, 17. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Brandenburg, J.G.; Bannwarth, C.; Hansen, A. Consistent structures and interactions by density functional theory with small atomic orbital basis sets. J. Chem. Phys. 2015, 143, 054107. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Adamo, C.; Barone, V. Toward reliable density functional methods without adjustable parameters: The PBE0 model. J. Chem. Phys. 1999, 110, 6158–6170. [Google Scholar] [CrossRef]
- Jensen, F. Segmented Contracted Basis Sets Optimized for Nuclear Magnetic Shielding. J. Chem. Theory Comput. 2015, 11, 132–138. [Google Scholar] [CrossRef] [PubMed]
- Forsyth, D.A.; Sebag, A.B. Computed 13C NMR Chemical Shifts via Empirically Scaled GIAO Shieldings and Molecular Mechanics Geometries. Conformation and Configuration from 13C Shifts. J. Am. Chem. Soc. 1997, 119, 9483–9494. [Google Scholar] [CrossRef]
- Mulder, F.A.A.; Filatov, M. NMR chemical shift data and ab initio shielding calculations: Emerging tools for protein structure determination. Chem. Soc. Rev. 2010, 39, 578–590. [Google Scholar] [CrossRef] [PubMed]
- Staroverov, V.N.; Scuseria, G.E.; Tao, J.; Perdew, J.P. Comparative assessment of a new nonempirical density functional: Molecules and hydrogen-bonded complexes. J. Chem. Phys. 2003, 119, 12129–12137. [Google Scholar] [CrossRef]
- Tao, J.; Perdew, J.P. Climbing the Density Functional Ladder: Nonempirical Meta–Generalized Gradient Approximation Designed for Molecules and Solids. Phys. Rev. Lett. 2003, 91, 146401. [Google Scholar] [CrossRef] [PubMed]
- Jakobsen, P.; Jensen, F. Probing basis set requirements for calculating hyperfine coupling constants. J. Chem. Phys. 2019, 151, 174107. [Google Scholar] [CrossRef]
- Neese, F. Software update: The ORCA program system, version 4.0, WIREs. Comput. Mol. Sci. 2018, 8, e1327. [Google Scholar] [CrossRef]
- Neese, F. Software update: The ORCA program system—Version 5.0, WIREs. Comput. Mol. Sci. 2022, 12, e1606. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Assignment | Electron Donor, BChl g′ | Electron Acceptor, 81-OH-Chl aF | ||||
|---|---|---|---|---|---|---|
| Expt. | Shield. | Fitted. | Expt. | Shield. | Fitted. | |
| N21 | 189.0 (E) | 55.3 | 190.1 | 189.0 (E) | 51.0 | 190.4 |
| N22 | 259.6 (E) | −18.1 | 261.2 | 216.1 (E) | 26.6 | 216.8 |
| N23 | 193.2 (E) | 53.2 | 192.2 | 193.2 (E) | 50.2 | 191.3 |
| N24 | 253.2 (E) | −8.1 | 251.5 | 249.3 (E) | −3.3 | 249.1 |
| C131 | 187.4 (A) | −14.6 | 188.6 | 190.8 (E) | −14.5 | 189.8 |
| C19 | 169.6 (A) | 9.9 | 164.9 | 172.0 (E) | 3.9 | 171.6 |
| C6 | 166.1 (A) | 11.4 | 163.4 | 153.8 (E) | 28.2 | 147.8 |
| C14 | 162.3 (A) | 15.8 | 159.1 | 163.0 (E) | 12.6 | 163.1 |
| C1 | 155.2 (A) | 24.8 | 150.4 | 157.3 (E) | 21.9 | 154.0 |
| C16 | 151.0 (A) | 23.9 | 151.2 | 157.8 (E) | 12.6 | 163.1 |
| C4 | 148.8 (A) | 28.5 | 146.9 | 149.9 (E) | 32.1 | 144.0 |
| C9 | 147.6 (A) | 27.8 | 147.5 | 145.5 (E) | 33.6 | 142.5 |
| C11 | 146.2 (A) | 27.9 | 147.4 | 147.0 (E) | 28.4 | 147.6 |
| C8 | 143.5 (A) | 27.7 | 147.6 | 144.9 (E) | 30.5 | 145.5 |
| C3 | 139.2 (A) | 37.4 | 138.3 | 140.3 (E) | 34.4 | 141.7 |
| C2 | 133.6 (A) | 40.3 | 135.4 | 136.8 (E) | 40.7 | 135.5 |
| C12 | 120.7 (A) | 46.5 | 129.5 | 134.6 (E) | 34.9 | 141.2 |
| C13 | 127.9 (A) | 43.5 | 132.3 | 127.3 (E) | 43.2 | 133.0 |
| C31 | _ | 41.5 | 134.2 | _ | 41.6 | 134.6 |
| C10 | 101.3 (E) | 79.6 | 97.4 | 112.6 (E) | 63.6 | 113.0 |
| C15 | 108.9 (E) | 65.4 | 111.2 | 102.6 (E) | 72.8 | 104.0 |
| C5 | 97.2 (E) | 81.3 | 95.7 | 96.2 (E) | 73.4 | 103.4 |
| C20 | 92.0 (E) | 83.3 | 93.8 | 93.2 (E) | 86.0 | 91.0 |
| C81 | 114.9 (A) | 57.6 | 118.7 | 64.9 (E) | 113.8 | 63.7 |
| C17 | 52.8 (A) | 128.8 | 49.8 | 53.9 (E) | 129.5 | 48.3 |
| C18 | 48.0 (A) | 130.7 | 47.9 | 49.8 (E) | 125.8 | 51.9 |
| C7 | 44.5 (A) | 134.6 | 44.2 | _ | 42.2 | 134.0 |
| C171 | 29.9 (A) | 152.0 | 27.4 | 31.7 (E) | 148.0 | 30.1 |
| Photosynthetic RCs | Primary Electron Donor | Primary Electron Acceptor | OCSD | ||||
|---|---|---|---|---|---|---|---|
| Cofactor | SACS (ppm) | Em (V) | Cofactor | SACS (ppm) | Em (V) | Δδ (ppm) (Acc.-Don.) | |
| Heliobacteria | BChl g′ | 2167.6 | +0.225 a | 81-OH-Chl aF | 2189.9 | −0.85 d | +22.3 |
| Purple bacteria | BChl a | 2174.1 | +0.5 b | BPhe a | 2156.1 | −0.75 e | −18.0 |
| Photosystem II | Chl a | 2183.9 | +1.2 c | Phe a | 2132.5 | −0.61 f | −51.5 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, Y.; Alia, A.; Kurle-Tucholski, P.; Wiebeler, C.; Matysik, J. Electronic Structures of Radical-Pair-Forming Cofactors in a Heliobacterial Reaction Center. Molecules 2024, 29, 1021. https://doi.org/10.3390/molecules29051021
Kim Y, Alia A, Kurle-Tucholski P, Wiebeler C, Matysik J. Electronic Structures of Radical-Pair-Forming Cofactors in a Heliobacterial Reaction Center. Molecules. 2024; 29(5):1021. https://doi.org/10.3390/molecules29051021
Chicago/Turabian StyleKim, Yunmi, A. Alia, Patrick Kurle-Tucholski, Christian Wiebeler, and Jörg Matysik. 2024. "Electronic Structures of Radical-Pair-Forming Cofactors in a Heliobacterial Reaction Center" Molecules 29, no. 5: 1021. https://doi.org/10.3390/molecules29051021
APA StyleKim, Y., Alia, A., Kurle-Tucholski, P., Wiebeler, C., & Matysik, J. (2024). Electronic Structures of Radical-Pair-Forming Cofactors in a Heliobacterial Reaction Center. Molecules, 29(5), 1021. https://doi.org/10.3390/molecules29051021