Abstract
We disclose a direct approach to the diastereoselective synthesis of phosphorus substituted N-acylaziridines based on a one-pot ZnCl2-catalyzed Joullié–Ugi three-component reaction of phosphorylated 2H-azirines, carboxylic acids and isocyanides. Hence, this robust protocol offers rapid access to an array of N-acylaziridines in moderate-to-good yields and up to 98:2 dr for substrates over a wide scope. The relevance of this synthetic methodology was achieved via a gram-scale reaction and the further derivatization of the nitrogen-containing three-membered heterocycle. The diastereo- and regioselective ring expansion of the obtained N-acylaziridines to oxazole derivatives was accomplished in the presence of BF3·OEt2 as an efficient Lewid acid catalyst.
1. Introduction
Multicomponent reactions (MCRs) are highly convergent processes that can be used to create new potent bioactive molecules. On the basis of green chemistry [1], MCRs benefit from efficiency, energy, time and atom economies, use environmentally favorable conditions and decrease the amount of byproducts and/or waste [2,3,4,5,6,7,8]. In addition, MCRs provide one of the most attractive prospects concerning complexity and diversity for the preparation of chemical libraries compared with other traditional synthetic organic methods. A large number of MCRs have been developed and are currently in a promising place in the chemist’s toolbox of sustainable synthetic methodologies.
Among them, the more versatile isocyanide-based MCRs form the backbone of today’s MCRs, such as the Ugi four-component reaction (U-4CR), which comprises the condensation of primary amines, carbonyl compounds, carboxylic acids and isocyanides to furnish dipeptide-like structures [9,10,11,12,13]. As the U-4CR mechanism occurred via the in situ formation of an imine intermediate, the employment of a cyclic imine instead of the amine and the carbonyl components in the Ugi protocol is a simple concept that supplies the robust Joullié–Ugi three-component reaction (JU-3CR). The Ugi variant was first reported by Joullié et al. [14,15] in 1982, and it is an appealing synthetic methodology not only because nitrogen-containing heterocycles can be directly attained in a single step, but also because of its greater stereochemical control [16]. The ring strain and the impossibility of imine E/Z isomerization contribute to a greater diastereoselectivity. Likewise, Joullié–Ugi adducts are privileged structures of interest in medicinal chemistry due to the synergistic effect of peptidic moieties linked to the nitrogen heterocycles leading to products with unique pharmaceutical activities [17,18,19,20].
Due to a better understanding of the benefits of covalent binding mechanisms and to the FDA support of effective and innocuous covalent drugs, there is great interest in covalent binding therapeutics [21,22,23]. Targeted covalent inhibitors are designed by incorporating an electrophile into a ligand that would bind the target protein. The integrated electrophile, acting as “warheads” [24], including ketone, α,β-unsaturated carbonyl, nitrile, ester, epoxide or aziridine, binds irreversibly to endogenous nucleophilic functionalities, including lysine, tyrosine, serine, cysteine and threonine, among others, on the target protein, introducing a covalent interaction. In this regard, aziridines, well known for their robust alkylating properties, possess the potential to function as potent covalent drugs by virtue of their ability to serve as DNA cross-linking agents. This is achieved through the nucleophilic ring opening of the three-membered nitrogen-containing heterocycle [25]. Aziridines are an important class of synthetic targets because they often exhibit a broad range of biological activities; for example, aziridine-containing mitomycin C (I) [26], azinomycin A (II) [27] and imexon (III) [28] show antitumor activity (Figure 1). Other natural aziridines, also known as aziridine alkaloids, display antibacterial and antimicrobial activity against selected microorganisms. For instance, aziridine-2-phosphonates IV have been claimed to show antibacterial properties [29] (Figure 1). We have recently reported the synthesis of phosphorus-substituted N-acylaziridines V [30] and VI [31], which exhibited a very good cytotoxic effect inhibiting the growth of human tumor cell line A549 (adenocarcinomic human alveolar basal epithelial cells). Recently, oxazoles have been emerged as the critical pharmacophore for various biological and medicinal applications. They serve as the key structural motif in numerous naturally occurring compounds and exhibit a broad spectrum of pharmacological properties, such as anti-cancer, anti-tubercular, anti-bacterial, anti-fungal, anti-parasitic and anti-viral properties, among others [32]. Hence, they can be utilized as primary building blocks in the pharmaceutical sector for the synthesis of several drugs. Likewise, from a biological perspective, organophosphorus compounds are very interesting due to their ability to modify the reactivity of heterocycles and regulate essential biological functions [33]. Thus, the anticancer agents 1,3-oxazol-4-ylphosphonium perchlorates VII [34], or the oxazole-phosphine oxide derivative VIII [31], and the oxazol-4-yl-phosphonate derivative IX displaying interesting anti-human cytomegalovirus (HCMV) properties [35], are only some examples of phosphorus-substituted oxazole derivatives with high potential for medical applications.
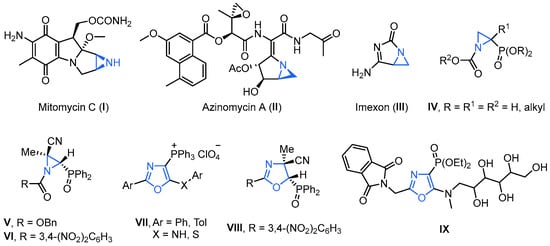
Figure 1.
Aziridine-containing natural products and selected examples of phosphorus-substituted oxazole derivatives with high potential for medical applications.
Many of the reported JU-3CR examples in the literature primarily employ 5, 6 or 7-membered cyclic imines for the preparation of pyrrolidine [36,37], thiazolidine [38], indolines [39,40] (Scheme 1, Equation (1)), piperidine [36,41] or oxazepine [42] peptidomimetics (Scheme 1, Equation (2)). However, only two examples have been reported in relation to the use of 2H-azirines as cyclic imines in the three-component Joullié–Ugi reaction for the synthesis of N-acylaziridines. Kanizsai et al. [43] first described the Lewis acid-promoted version of the Joullié–Ugi reaction using 2H-azirine-2-carboxylates for the diastereoselective synthesis of N-acylaziridines-2-carboxamide derivatives (Scheme 1, Equation (3)). Furthermore, a domino JU-3CR/diastereoselective ring expansion reaction was applied in the preparation of oxazolines from 2-phenyl-2H-azirines [44] (Scheme 1, Equation (4)).
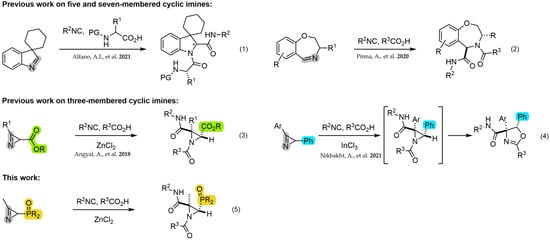
Scheme 1.
JU-3CR on three, five and seven-membered cyclic imines [39,42,43,44].
We have been previously involved in the chemistry of phosphorus-substituted 2H-azirines for the synthesis of phosphorylated cyanoaziridines [30] and their ring expansion [31], hybrid molecules such as azirino[2,1-b]benzo[e][1,3]oxazines [45] or α-aminophosphonic acid derivatives [46]. In continuation of our previous research works, as depicted in (Scheme 1, Equation (5)), here, we describe a diastereoselective approach to phosphorylated N-acylaziridine 2-carboxamide derivatives through the JU-3CR using phosphorus-substituted 2H-azirines such as 3-membered cyclic imines, carboxylic acids and isocyanides (Figure 2).
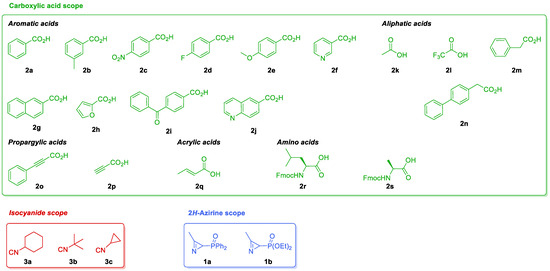
Figure 2.
The scope of phosphorus-substituted 2H-azirines 1, carboxylic acids 2 and isocyanides 3 tested in the JU-3CR.
2. Results
As outlined in Table 1, we started our investigation with the optimization of the three-component reaction conditions of 2H-azirine phosphine oxide 1a, benzoic acid (2a) and cyclohexyl isocyanide (3a) in THF at 60 °C. The Joullié–Ugi reaction without a catalyst led to the obtention of only a 10% yield of N-acylaziridine phosphine oxide 4a (entry 1). It is well known that the activation of 2H-azirines by Lewis acids may significantly enhance their reactivity [47,48]. Thus, we next explored the Lewis or Brønsted acid-mediated JU-3CR. Only a 14% yield of 4a could be achieved when PTSA was used as the Brønsted acid catalyst in this process (entry 2). Trifluoromethanesulfonic acid (TfOH, entry 3) showed moderate catalytic activity since the reaction proceeded smoothly in THF at 60 °C to give the product 4a in a 35% yield. Increasing the amount of TfOH from 10 mol% to 25 mol% did not improve the yield of compound 4a. The JU-3CR was carried out in the presence of different Lewis acids. For instance, Ti(OiPr)4, Sc(OTf)3, InCl3, BF3·OEt2, MgBr2 and ZnCl2·H2O were not suitable for the current reaction since compound 4a could not be detected, and only the starting 2H-azirine 1a or decomposition products were recovered instead (entries 4–9). In general, the most active catalyst for the JU-3CR of 1a with carboxylic acid 2a and isocyanide 3a was found to be ZnCl2 (Table 1, entry 10), which is consistent with literature reports of other JU-3CRs involving 2H-azirine-2-carboxylates [43] Therefore, the use of ZnCl2 (25 mol%, entry 10) resulted in the formation of N-acylaziridine 4a in a 60% chemical yield, together with a small amount of the pyrazine that proceed from the thermal treatment of the corresponding 2H-azirine phosphine oxide 1a [49]. This process yielded product 4a in a diastereomeric ratio of 96:4.

Table 1.
Reaction condition optimization a.
The effect of the solvent on the JU-3CR was also tested. The ZnCl2 catalyst in this process was incompatible with some solvents such as MeOH or MeCN (entries 11 and 12), and in both cases, no reaction product was observed. The degree of consumption of the starting 2H-azirine 1a was found to depend on reaction temperature. In the reaction conducted at –10 °C (entry 13), a significant amount of unreacted 2H-azirine 1a was recovered and the expected product 4a was obtained in a low yield. The JU-3CR was found to work better when the reaction was carried out at room temperature, and the almost complete conversion of the 2H-azirine 1a was then observed (entry 14). Finally, different amounts of ZnCl2 were examined, which showed that decreasing the amount of ZnCl2 (10 mol%, entry 15) led to the desired product 4a in a low yield together with α-ketamide derived from the nucleophilic addition of the carboxylic acid to 2H-azirine 1a [50]. Increasing the amount of ZnCl2 up to 30 mol% (entry 16) did not affect the yield of compound 4a.
Given that the results of this preliminary investigation seemed to define ZnCl2 as the best catalyst in the JU-3CR in THF and room temperature as the best reaction condition, we adopted these conditions for further studies. Then, a range of N-acylaziridine phosphine oxides 4 with diverse substitution patterns (Figure 2) on the aziridine ring were prepared. A considerable selection of aromatic carboxylic acid partners 2 were well tolerated in the JU-3CR with 1a and 3 as coupling partners (Scheme 2). Both electron-donating (OMe) and electron-withdrawing groups (F, NO2) at the para-phenyl position of aromatic carboxylic acid 2 yielded desired products 4c, 4d, 4e, 4j and 4k in 20–74% yields and very good diastereoselectivities. Among them, 4-fluor derivatives 4d and 4j were achieved with the best yields (74%).
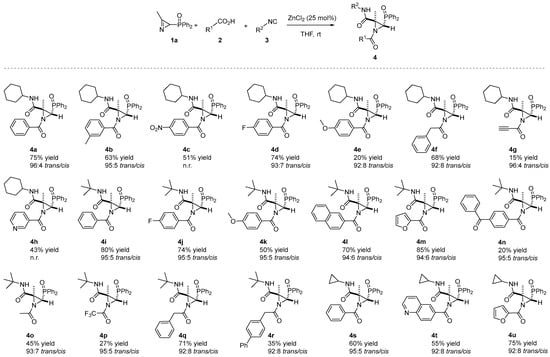
Scheme 2.
Substrate scope of the N-acylaziridine phosphine oxides 4 in the Joullié–Ugi three-component reaction. See the Supporting Information for experimental details. Reactions were carried out on a 0.5 mmol scale; 2H-azirine 1a (0.5 mmol), carboxylic acid 2 (1.3 eq.), isocyanide 3 (1.3 eq.), ZnCl2 (25 mol%) and solvent (0.5 mL). Diastereomeric ratio (trans/cis) determined from the crude reaction mixture via 1H NMR. It was not possible to determine the diastereomeric ratio of compounds 4c and 4h as the crude reaction NMR spectra were not clean enough.
The electron-donating group (Me) at the meta-phenyl position of aromatic carboxylic acid also furnished N-acylaziridine 4b in a 63% yield (Scheme 2). Other aromatic carboxylic acids such as 2-naphthoic acid 2g or 4-benzoylbenzoic acid 2i were selected as suitable candidates for this transformation, providing products 4 in moderate-to-good yields. Even heteroaromatic carboxylic acids were well tolerated in the Joullié–Ugi three-component reaction. For instance, nicotinic acid 2f, 2-furoic acid 2h and or quinoline-6-carboxylic acid 2j gave desired products 4h, 4m, 4t and 4u in 43–85% yields. Phenylacetic acid 2m yielded N-acylaziridines 4f and 4q in a 68 and 71% yield, respectively. However, worse yields of N-acylaziridines 4o, 4p and 4r, derived, respectively, from acetic acid 2k, trifluoroacetic acid 2l and 4-biphenylacetic acid 2n were attained (Scheme 2). Moreover, propargylic acids, such as propiolic acid 2p, can also be subjected to the JU-3CR to give compound 4g in a low yield but with high diastereoselectivity (96:4 trans:cis dr). This confirms the strength of the carboxylic acid scope in the JU-3CR.
In order to assess the applicability of the JU-3CR, we next investigated the scope of this process with regard to the isocyanide partner. Besides the cyclohexyl isocyanide (3a), other aliphatic isocyanides such as tert-butyl isocyanide (3b) or cyclopropyl isocyanide (3c) have been tested in the JU-3CR. All the isocyanides studied are well tolerated, giving the N-azylaziridines 4 in a moderate-to-good yield (see Scheme 2). The isocyanide partner does not affect the outcome of this protocol. For instance, compare the chemical yields of 4a, 4i and 4s (60–80% yields), 4d and 4j (74% yield) or 4m and 4u, which proceeded smoothly in 85 and 75% yields (Scheme 2).
A careful examination of the spectroscopic data of the crude reaction mixture of N-acylaziridine 4a, showed two well-resolved doublets in the 1H NMR spectrum corresponding to the H3 methine proton of the aziridine ring, which is consistent with the presence of two diastereoisomers. The major diastereoisomer appears at δH ~ 3.81 ppm with the coupling constant 2JPH = 24.0 Hz, and the minor one at δH ~ 3.35 ppm with a lower coupling constant of 2JPH = 21.7 Hz in a ratio of 96:4. Substrates 4 were extensively characterized on the basis of their 1H, 13C, 31P, 19F NMR and 2D NMR spectra and HRMS (see the Supporting Information). The most characteristic signals for N-acylaziridine 4a (major diastereoisomer) in the 1H NMR spectrum are the two well-resolved doublets at δH ~ 5.76 ppm (3JHH = 8.1 Hz) and δH ~ 3.81 ppm with a coupling constant of 2JPH = 24.0 Hz, corresponding to the NH of amide group and the H3 methine proton of the aziridine ring, respectively. A singlet at δH ~ 1.96 ppm was attributed to the methyl group. In the 13C NMR spectrum, the formation of compound 4a is evident from the presence of two carbonyl groups at δC ~ 176.2 (3JPC = 3.3 Hz) and 165.6 ppm, while the quaternary carbon C2 appears as a doublet at δC ~ 49.6 ppm (2JPC = 3.0 Hz) and the methine carbon C3 shows a chemical shift at δC ~ 42.0 ppm with a large coupling constant (1JPC = 100.4 Hz).
Since it was not possible to assign the stereochemistry of N-acylaziridines 4 via 1H and 13C NMR, their structure has been unambiguously determined via X-ray diffraction analysis [51,52,53], establishing the trans-relationship between the amide group at the C2 position and the phosphorus moiety at the C3 position of the major diastereoisomer 4i. The CIF data are presented in the Supporting Information, and the ORTEP drawing of 4i is shown in Figure 3.
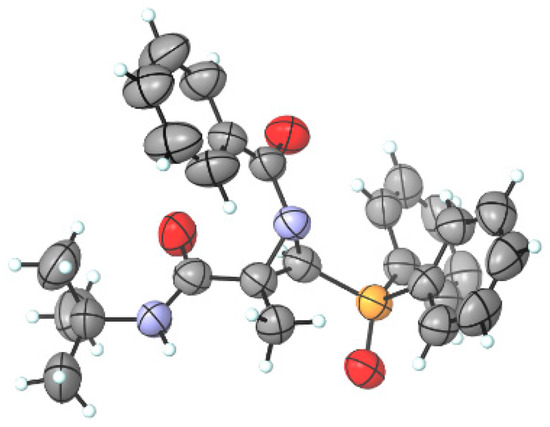
Figure 3.
ORTEP diagram of functionalized N-acylaziridine phosphine oxide 4i (H, white; C, grey; O, red; N, blue; P, orange) (2S,3S enantiomer shown).
The ZnCl2-catalyzed Joullié–Ugi three-component reaction was extended to the use of 2H-azirine phosphonate 1b. Then, 2H-azirine 1b reacted with a series of carboxylic acids 2 and isocyanides 3 in THF at room temperature in the presence of ZnCl2 (25 mol%). As outlined in Scheme 3, aromatic (2a and 2d), heteroaromatic (2h and 2j), aliphatic (2m and 2n), propargylic (2o) and acrylic (2q) acids are allowable in the JU-3CR, yielding the desired N-acylaziridine phosphonates 5 in chemical yields ranging from 35 to 76%.
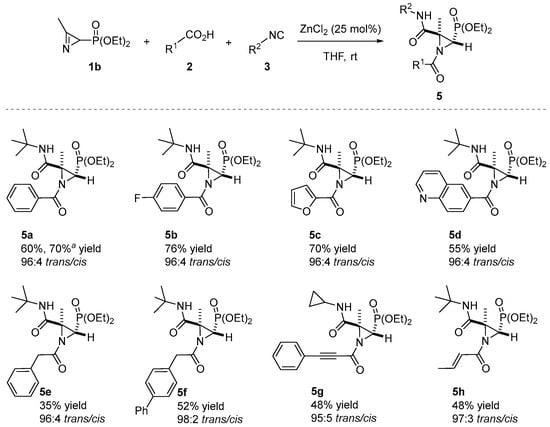
Scheme 3.
Substrate scope of the N-acylaziridine phosphonates 5 in the Joullié–Ugi three-component reaction. See the Supporting Information for experimental details. Reactions were carried out on a 0.5 mmol scale; 2H-azirine 1b (0.5 mmol), carboxylic acid 2 (1.3 eq.), isocyanide 3 (1.3 eq.), ZnCl2 (25 mol%) and solvent (0.5 mL). a Yield of isolated compound 5a after crystallization, at a 4 mmol scale. The diastereomeric ratio (trans/cis) was determined from the crude reaction mixture via 1H NMR.
Encouraged by the abovementioned obtained results of the Joullié–Ugi three-component reaction between phosphorylated 2H-azirines 1, carboxylic acids 2 and isocyanides 3, we further investigated the substrate scope using N-Fmoc-protected amino acids as carboxylic acid partners for the preparation of phosphorylated aziridine peptidomimetics. To our delight, it was found that the reaction proceeded smoothly when 2H-azirine phosphonate 1b reacted with tert-butyl isocyanide (3b) and Fmoc-Leu (2r, R1 = iBu) in the standard conditions to yield derivative 6a in a 65% isolated yield (Scheme 4). The 1H NMR spectrum of the crude reaction mixture confirmed the presence of two well-resolved doublets at δH = 3.03 and 2.90 ppm with a coupling constant of 2JPH = 18.6 and 18.4 Hz, respectively, for the H3 methine proton of the aziridine ring of both trans-diastereoisomers, in a ratio of 1:1: Conversely, another doublet appeared at δH = 2.71 ppm with a lower coupling constant of 2JPH = 14.7 Hz corresponding to H3 of the cis-diastereoisomer, while the fourth doublet corresponding to the other cis-diastereoisomer appeared overlapped in the range of δH = 2.83–2.75 ppm. The diastereomeric ratio between trans- and cis-diastereoisomers is approximately 92:8. After purification via flash-chromatography, it was possible to identity both trans-diastereoisomers of 6a. The Joullié–Ugi reaction between 2H-azirine 1b, tert-butyl isocyanide (3b) and Fmoc-Ala (2s, R1 = Me) using 25 mol% of ZnCl2 in THF and at room temperature led to the formation of derivative 6b in a lower yield (Scheme 4). Via the 1H NMR of the crude compound, it was possible to determine the 1:1 ratio between both trans-diastereoisomers. Nevertheless, in this case, determining the diastereomeric ratio (trans/cis) was infeasible.
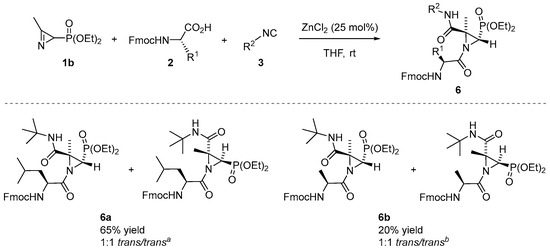
Scheme 4.
Substrate scope of phosphorylated aziridine peptidomimetics 6 through the Joullié–Ugi three-component reaction using N-Fmoc amino acids. See the Supporting Information for experimental details. Reactions were carried out on a 1 mmol scale; 2H-azirine 1b (1 mmol), Fmoc-protected amino acid 2r or 2s (1.3 eq.), isocyanide 3b (1.3 eq.), ZnCl2 (25 mol%) and solvent (1 mL). a Diastereomeric ratio (trans/trans) determined from the crude reaction mixture via 1H NMR. The diastereomeric ratio between trans- and cis-diastereoisomers is approximately 92:8. b The diastereomeric ratio (trans/trans) determined from the crude reaction mixture via 1H NMR. The diastereomeric ratio between trans and cis-diastereoisomers could not be determined.
The gram-scale synthesis of phosphorylated N-acylaziridines was accomplished, as shown in Scheme 3. The use of 4.0 mmol of 2H-azirine phosphonate 1b, under the JU standard conditions, gave N-acylaziridine phosphonate 5a in a 70% yield (1.11 g) after recrystallization.
Scheme 5 outlines a plausible mechanism for the JU-3CR. This process carried out in a polar solvent, suggesting the formation of polar intermediates, is compatible with a stepwise mechanism. The addition of Lewis acids (in our case ZnCl2) increases the electrophilicity of the iminic C–N double bond in 2H-azirine 1. Thus, the electrophilic imine and nucleophilic carboxylic acid 2 add to the carbon atom of isocyanide 3. The amino group of the adduct thus formed promotes the irreversible Mumm rearrangement in the presence of a zinc catalyst and the intramolecular acylation of the amine nitrogen atom, which after subsequent hydroxylimine → amide tautomerization leads to phosphorylated N-acylaziridines 4, 5 or 6.
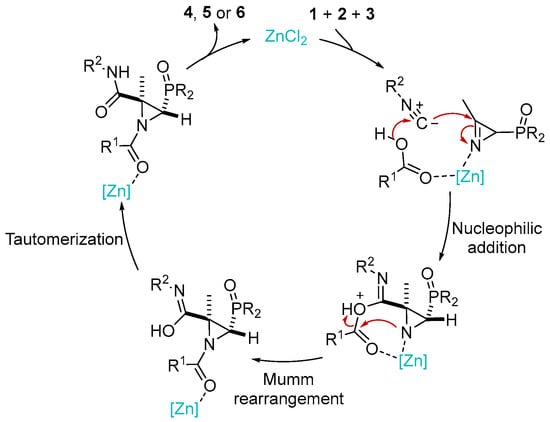
Scheme 5.
Plausible mechanism for the synthesis of phosphorus substituted N-Acylaziridines 4, 5 and 6 through the Joullié–Ugi three-component reaction.
We performed further derivatization in order to illustrate the utility of the Joullié–Ugi adducts. Thus, we explored the isomerization reaction of N-acylaziridines 4 and 5 to oxazole derivatives. For this purpose, and taking into account that the regio- and stereochemical outcomes of these rearrangements strongly depend on the reaction conditions, as well as the substitution pattern of the N-acylaziridine, we started exploring thermal conditions for the ring opening of compounds 4 and 5. Thus, phosphorus-substituted N-acylaziridine 4a was heated in refluxing CHCl3. Under these conditions, the corresponding oxazole derivative was not observed, and the unreacted starting substrate was recovered instead. Next, the rearrangement of 4a was also tested under nucleophilic conditions [31,54,55,56,57]. When 4a reacted with 0.2 equivalents of NaI in THF at 60 °C, as in the previous case, no satisfactory results were attained, observing only decomposition products. Likewise, the isomerization of the N-acylaziridine to oxazole derivative under mild acidic conditions was examined. Compound 4a was treated with both Brønsted acids, including p-toluenesulfonic acid (PTSA), and Lewis acids, including BF3·OEt2. Only the use of BF3·OEt2 gave satisfactory results. Hence, when N-acylaziridine 4a reacted in the presence of 1.2 equivalents of BF3·OEt2 in MeCN at 90 °C and under microwave irradiation for 10 min, the formation of 4-diphenylphosphoryl-4,5-dihydrooxazole-5-carboxamide 7a was achieved in a very good yield and in a regio- and diastereoselective fashion (Scheme 6).
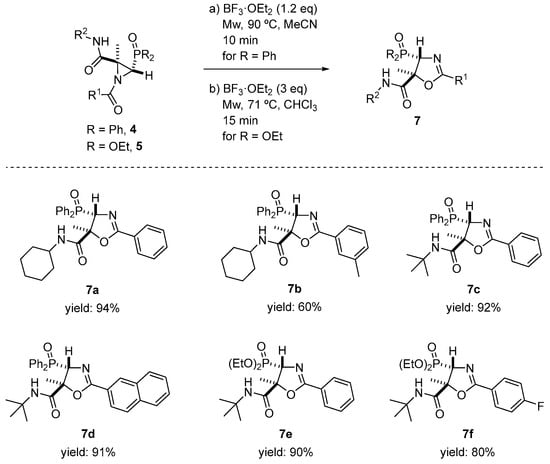
Scheme 6.
Phosphorus-substituted oxazole derivatives 7 through the ring expansion of N-acylaziridines 4 or 5. See the Supporting Information for experimental details. Reactions were carried out on a 0.12–0.80 mmol scale; N-acylaziridine 4 or 5 (0.12–0.80 mmol), BF3·OEt2 (1.2 eq. for N-acylaziridines 4 derived from phosphine oxide and 3 eq. for N-acylaziridines 5 derived from phosphonate) and solvent (20 mL/mmol).
Spectroscopic data confirmed the isomerization of N-acylaziridine 4a into oxazole derivative 7a. While the 1H NMR spectrum of 4a shows a signal for the methyl group at δH = 1.96 ppm and the methine hydrogen resonates at δH = 3.81 ppm as a well-resolved doublet (2JPH = 24 Hz, see above), in dihydrooxazole-5-carboxamide 7a, these signals appear at δH = 1.63 and 5.32 ppm as a singlet and a well-resolved doublet with a much lower coupling constant (2JPH = 6.0 Hz), respectively. Similarly, other N-acylaziridines derived from phosphine oxide 4b, 4i and 4l reacted with BF3·OEt2 under the same reaction conditions, providing 60, 92 and 91% yields of oxazole derivatives 7b–d (Scheme 6). This synthetic methodology was extended to the use of N-acylaziridines 5 derived from phosphonate. Thus, the ring expansion of 5a and 5b easily occurred via the slight excess of BF3·OEt2 (3 equivalents) in CHCl3 at 71 °C and under microwave irradiation for 15 min, to obtain regio- and diasteroselective 7e and 7f in high yields (Scheme 6).
Since it was not possible to assign the stereochemistry of oxazole derivatives 7 via 1H and 13C NMR, their structure has been unambiguously determined via X-ray diffraction analysis [51,52,53], establishing not only the regioselectivity of the isomerization process, but also the anti-relationship between the amide group at the C5 position and the phosphorus moiety at the C4 position of 7a (Figure 4).
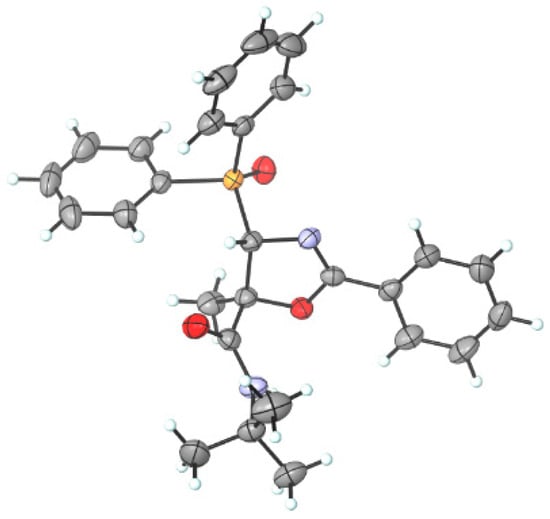
Figure 4.
ORTEP diagram of functionalized diphenylphosphoryl oxazole derivative 7a (H, white; C, grey; O, red; N, blue; P, orange) (4S,5S enantiomer shown).
A reasonable mechanism that would explain the formation of 7 is exemplified in Scheme 7. First, BF3·OEt2 would activate the carbonyl group of N-acylaziridine 4 or 5, thus assisting the ring-opening reaction, through the N–C2 bond of N-acylaziridine, with the concomitant generation of the most stable carbocation. This intermediate enables the ring expansion of N-acylaziridine 4 or 5 to oxazole derivative 7, as the only regio- and diastereoisomer.
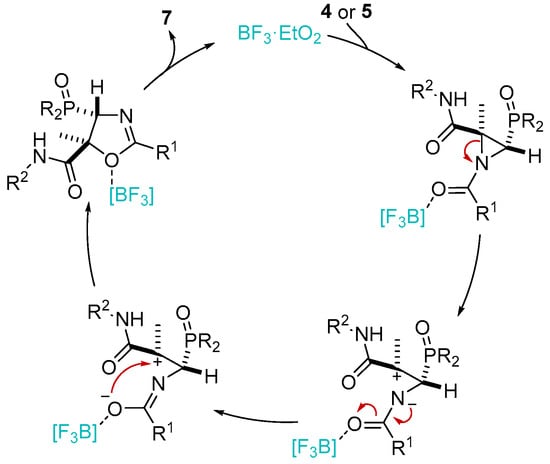
Scheme 7.
Rational mechanism for the stereospecific and regioselective ring expansion of N-acylaziridines 4 or 5 to oxazole derivatives 7.
3. Materials and Methods
3.1. General Experimental Information
Solvents for extraction and chromatography were reagent-grade. All solvents used in reactions were freshly distilled and dried over 4 Å molecular sieves before use. Unless otherwise mentioned, all other solvents and chemicals were purchased from commercial vendors and recrystallized or distilled as necessary, or used without further purification. All reactions were performed under an atmosphere of dry nitrogen. The reaction progress was monitored via 31P NMR or analytical thin-layer chromatography (TLC) performed on precoated Merck silica gel 60 F254 TLC aluminum plates, and spot-visualized with UV light or permanganate stain. Melting points were uncorrected. 1H (400 MHz), 13C (100 MHz), 19F (376 MHz) and 31P NMR (160 MHz) spectra were recorded using a Bruker Avance 400 (400 MHz) spectrometer in CDCl3 at room temperature. Chemical shifts (δ) are reported in parts per million (ppm) with the internal chloroform signal at 7.26 ppm as a standard for 1H, the internal chloroform signal at 77.2 ppm as a standard for 13C, the external fluorotrichloromethane (CFCl3) signal at 0.0 ppm for 19F or the external H3PO4 (50%) signal at 0.0 ppm as a standard for 31P NMR spectra. All coupling constants (J) values are reported in Hz. 19F and 13C NMR spectra were recorded in a broadband decoupled mode from hydrogen nuclei. Distortionless enhanced polarization transfer (DEPT) supported peak assignments for 13C NMR. Data for 1H NMR spectra are reported for the following: chemical shift, multiplicity, coupling constant and integration. Multiplicity abbreviations are reported as s = singlet; d = doublet; t = triplet; q = quartet; m = multiplet; dd = double doublet; bs = broad singlet. IR spectra were measured using a Nicolet iS10 Termo Scientific spectrometer using an attenuated total reflectance technique (ATR). Absorbance frequencies are given at maximum of intensity in cm–1. High-resolution mass spectra (HRMS) were obtained via the positive-ion electrospray ionization (ESI) method with a time of flight Q-TOF method. Data are reported in the form m/z (intensity relative to base = 100). 2H-Azirine phosphine oxide 1a [58] and phosphonate 1b [59] were prepared according to procedures in the literature and characterized using NMR spectra.
3.2. Experimental Procedure and Characterization Data for N-Acylaziridine Phosphine Oxide 4
In a flame-dried flask, the corresponding carboxylic acid 2 (0.65 mmol, 1.3 eq.), isocyanide 3 (0.65 mmol, 1.3 eq.) and 1M diethyl ether solution of ZnCl2 (0.12 mL; 0.12 mmol; 0.25 eq.) were added to 0.5 mL of dry THF. Then, 2H-azirine phosphine oxide 1a (0.50 mmol, 1 eq.) was added at room temperature. The reaction mixture was stirred until TLC showed the disappearance of 2H-azirine 1a (1–24 h). The solvent was removed under vacuum, and the residue was dissolved in dichloromethane (5 mL) and washed with water (2 × 5 mL). The organic layer was dried over anhydrous MgSO4 and filtered, and the solvent was evaporated under reduced pressure. The resulting residue was purified via flash column chromatography (SiO2, hexanes/AcOEt) to yield compounds 4.
(2S*,3S*)-1-Benzoyl-N-cyclohexyl-3-(diphenylphosphoryl)-2-methylaziridine-2-carboxamide (4a) (182 mg, 75%) was obtained as a white solid from carboxylic acid 2a (79 mg, 0.65 mmol), isocyanide 3a (81 µL mg, 0.65 mmol) and 2H-azirine 1a (127 mg, 0.50 mmol), as described in the general procedure. The crude product was purified via flash column chromatography (SiO2, AcOEt/hexanes 50:50) to give the title compound 4a. mp 249–251 °C; IR (neat) vmax 3281, 3070, 2923, 2848, 1685, 1651, 1549, 1285, 1193 cm–1; 1H NMR (400 MHz, CDCl3) δ 8.01–7.95 (m, 4H, ArH), 7.84–7.82 (m, 2H, ArH), 7.62–7.47 (m, 7H, ArH), 7.40–7.37 (m, 2H, ArH), 5.76 (d, 2JHH = 8.2 Hz, 1H, HC-NH), 3.81 (d, 2JPH = 24.0 Hz, 1H, CH-P), 3.50–3.40 (m, 1H, HC-NH), 1.96 (s, 3H, CH3), 1.76–1.73 (m, 1H, cHex), 1.63–1.59 (m, 1H, cHex), 1.53–1.45 (m, 2H, cHex), 1.27–0.98 (m, 5H, cHex), 0.73–0.63 (m, 1H, cHex) ppm; 13C {1H} NMR (100 MHz, CDCl3) δ 176.9 (d, 3JPC = 3.1 Hz), 165.6), 134.2, 132.6 (d, 1JPC = 103.4 Hz, Cquat), 132.5, 132.4, 132.4, 132.3, 132.3, 131.8, 131.7, 131.2, 131.1, 129.1, 129.0, 128.8, 128.6, 128.5, 49.7 (d, 2JPC = 2.7 Hz, Cquat), 49.1, 42.2 (d, 1JPC = 101.0 Hz), 32.5, 32.4, 25.4, 24.7, 24.6, 15.5 ppm; 31P NMR (160 MHz, CDCl3) δ 24.1 ppm; ESI-HRMS (CI) m/z calculated for C29H32N2O3P ([M + H]+), 487.2151; found 487.21567.
(2S*,3S*)-N-Cyclohexyl-3-(diphenylphosphoryl)-2-methyl-1-(3-methylbenzoyl)aziridine-2-carboxamide (4b) (158 mg, 63%) was obtained as a white solid from carboxylic acid 2b (88 mg, 0.65 mmol), isocyanide 3a (81 µL, 0.65 mmol) and 2H-azirine 1a (127 mg, 0.50 mmol), as described in the general procedure. The crude product was purified via flash column chromatography (SiO2, AcOEt/hexane 50:50) to give the title compound 4b. mp 201–203 °C; IR (neat) vmax 3303, 3056, 2939, 1679, 1643, 1538, 1293, 1191 cm–1; 1H NMR (400 MHz, CDCl3) δ 7.90–7.85 (m, 4H, ArH), 7.59 (s, 1H, ArH), 7.59–7.39 (m, 7H, ArH), 7.21–7.13 (m, 2H, ArH), 5.81 (d, 3JHH = 8.2 Hz, 1H, HC-NH), 3.78 (d, 2JPC = 24.2 Hz, 1H, CH-P), 3.42–3.33 (m, 1H, HC-NH), 2.26 (s, 3H, CH3), 1.86 (s, 3H, CH3), 1.65–1.63 (m, 1H, cHex), 1.52–1.49 (m, 1H, cHex), 1.43–1.36 (m, 2H, cHex), 1.18–0.90 (m, 5H, cHex), 0.70–0.61 (m, 1H, cHex) ppm; 13C {1H} NMR (100 MHz, CDCl3) δ 176.2 (d, 3JPC = 3.3 Hz), 165.6, 138.2, 133.9, 133.2, 132.3, 132.2, 132.2, 132.1, 131.7, 131.6 131.1, 131.0, 129.1, 129.0, 128.9, 128.7, 128.6, 128.2, 125.5, 49.6 (d,2 JPC = 3.0 Hz), 49.0, 42.0 (d, 1JPC = 100.4 Hz), 32.5, 32.4, 25.3, 24.7, 24.6, 21.4, 15.4 ppm; 31P NMR (160 MHz, CDCl3) δ 23.9 ppm; ESI-HRMS (CI) m/z calculated for C30H34N2O3P ([M + H]+), 501.2307; found 501.2308.
(2S*,3S*)-N-Cyclohexyl-3-(diphenylphosphoryl)-2-methyl-1-(4-nitrobenzoyl)aziridine-2-carboxamide (4c) (135 mg, 51%) was obtained as a white solid from carboxylic acid 2c (108 mg, 0.65 mmol), isocyanide 3a (81 µL, 0.65 mmol) and 2H-azirine 1a (127 mg, 0.50 mmol), as described in the general procedure. The crude product was purified via flash column chromatography (SiO2, AcOEt/hexane 50:50) to give the title compound 4c. mp 254–256 °C; IR (neat) vmax 3281, 3078, 2923, 1737, 1687, 1649, 1596, 1554, 1501, 1390, 1285, 1193 cm–1; 1H NMR (400 MHz, CDCl3) δ 8.20 (d, 3JHH = 8.7 Hz, 2H, ArH), 7.96–7.78 (m, 6H, ArH), 7.59–7.48 (m, 6H, ArH), 5.88 (d, 3JHH = 7.8 Hz, 1H, HC-NH), 3.68 (d, 2JPH = 23.5 Hz, 1H, CH-P), 3.49–3.38 (m, 1H, HC-NH), 1.95 (s, 3H, CH3), 1.73 (d, 3JHH = 12.2 Hz, 1H, cHex), 1.60 (d, 3JHH = 14.4 Hz, 1H, cHex), 1.52–1.45 (m, 2H, cHex), 1.34 (d, 3JHH = 12.4, 1H, cHex), 1.23–1.03 (m, 4H, cHex), 0.83–0.75, 1H, cHex) ppm; 13C {1H} NMR (100 MHz, CDCl3) δ 175.2 (d, 3JPC = 3.2 Hz), 165.7, 150.0, 139.8, 132.6, 136.6, 132.6, 132.5, 132.2 (d, 1JPC = 103.9 Hz), 131.6, 131.5, 131.3 (d, 1JPC = 105.2 Hz), 131.1, 131.0, 129.3, 129.2, 129.1, 128.9, 128.8. 123.7, 50.0 (d, 2JPC = 2.73 Hz), 49.4, 42.7 (d, 1JPC = 99.2 Hz), 32.6, 32.5, 25.3, 24.7, 24.6, 15.3 ppm; 31P NMR (160 MHz, CDCl3) δ 23.5 ppm; ESI-HRMS (CI) m/z calculated for C29H31N3O5P ([M + H]+), 532.2001; found 532.1980.
(2S*,3S*)-N-Cyclohexyl-3-(diphenylphosphoryl)-1-(4-fluorobenzoyl)-2-methylaziridine-2-carboxamide (4d) (186 mg, 74%) was obtained as a white solid from carboxylic acid 2d (91 mg, 0.65 mmol), isocyanide 3a (81 µL, 0.65 mmol) and 2H-azirine 1a (127 mg, 0.50 mmol), as described in the general procedure. The crude product was purified via flash column chromatography (SiO2, AcOEt/hexane 50:50) to give the title compound 4d. mp 255–257 °C; IR (neat) vmax 3284, 3018, 2939, 1688, 1650, 1590, 1286, 1102 cm–1; 1H NMR (400 MHz, CDCl3) δ 7.96–7.91 (m, 4H, ArH), 7.82 (dd, 3JHH = 8.8, 4JHH = 5.4 Hz, 2H), 7.60–7.46 (m, 6H, ArH), 7.04 (t, 3JHH = 8.6 Hz, 3JHF = 8.6 Hz, 2H, ArH), 5.77 (d, 3JHH = 8.2 Hz, 1H, HC-NH), 3.75 (d, 2JPH = 24.0 Hz, 1H, CH-P), 3.48 (m, 1H, HC-NH), 1.93 (s, 3H, CH3), 1.75–1.72 (m, 1H, cHex), 1.62–1.59 (m, 1H, cHex), 1.53–1.48 (m, 2H, cHex), 1.22–1.17 (m, 2H, cHex), 1.12–1.00 (m, 3H, cHex), 0.79–0.70 (m, 1H, cHex) ppm; 13C {1H}NMR (100 MHz, CDCl3) δ 175.7 (d, 3JPC = 3.2 Hz), 165.6, 165.4 (d, 1JCF = 253.3 Hz), 132.5 (d, 1JPC = 103.6 Hz, Cquat), 132.4, 132.4, 132.4, 132.3, 132.2, 131.7, 131.1, 131.0, 130.9, 130.8, 130.6 (d, 4JCF = 3.0 Hz), 129.1, 129.0, 128.8, 115.7, 115.5, 49.7 (d, 2JPC = 3.0 Hz), 49.0, 42.2 (d, 1JPC = 100.4 Hz), 32.5, 32.5, 25.4, 24.7, 24.6, 15.4 ppm; 31P NMR (160 MHz, CDCl3) δ 24.4 ppm; 19F NMR (376 MHz, CDCl3) δ –106.5 ppm; ESI-HRMS (CI) m/z calculated for C29H31FN2O3P ([M + H]+), 505.2056; found 505.2043.
(2S*,3S*)-N-Cyclohexyl-3-(diphenylphosphoryl)-1-(4-methoxybenzoyl)-2-methylaziridine-2-carboxamide (4e) was (51 mg, 20%) obtained as a white solid from carboxylic acid 2e (100 mg, 0.65 mmol), isocyanide 3a (81 µL, 0.65 mmol) and 2H-azirine 1a (127 mg, 0.50 mmol), as described in the general procedure. The crude product was purified via flash column chromatography (SiO2, AcOEt/hexane 50:50) to give the title compound 4e. mp 251–252 °C; IR (neat) vmax 3278, 3075, 2923, 1685, 1651, 1596, 1549, 1282, 1196, 1121 cm–1; 1H NMR (400 MHz, CDCl3) δ 7.96–7.92 (m, 4H, ArH), 7.77 (d, 3JHH = 8.9 Hz, 2H, ArH), 7.57–7.44 (m, 6H, ArH), 6.84 (d, 3JHH = 8.9 Hz, 2H, ArH), 5.82 (d, 3JHH = 7.9 Hz, 1H, HC-NH), 3.80 (s, 3H, OCH3), 3.76 (d, 1JPC = 24.2 Hz, 1H, CH-P), 3.51–3.40 (m, 1H, HC-NH), 1.92 (s, 3H, CH3), 1.73–1.69 (m, 1H, cHex), 1.59–1.56 (m, 1H, cHex), 1.59–1.47 (m, 2H, cHex), 1.31–0.96 (m, 5H, cHex), 0.77–0.68 (m, 1H, cHex) ppm; 13C {1H} NMR (100 MHz, CDCl3) δ 176.0 (d, 3JPC = 3.3 Hz), 165.6, 163.1, 133.2, 132.3, 132.3, 132.2, 132.2, 131.8, 131.7, 131.3, 131.1, 131.0, 130.5, 129.0, 128.9, 128.7, 128.6, 126.7, 113.7, 55.5, 49.7 (d, 2JPC = 2.9 Hz, Cquat), 49.0, 41.9 (d, 1JPC = 101.0 Hz), 32.5, 32.4, 25.4, 24.7, 24.6, 15.5 ppm; 31P NMR (160 MHz, CDCl3) δ 24.0 ppm; ESI-HRMS (CI) m/z calculated for C30H34N2O4P ([M + H]+), 517.2256; found 517.2257.
(2S*,3S*)-N-Cyclohexyl-3-(diphenylphosphoryl)-2-methyl-1-(2-phenylacetyl)aziridine-2-carboxamide (4f) (170 mg, 68%) was obtained as a white solid from carboxylic acid 2m (88 mg, 0.65 mmol), isocyanide 3a (81 µL, 0.65 mmol) and 2H-azirine 1a (127 mg, 0.50 mmol), as described in the general procedure. The crude product was purified via flash column chromatography (SiO2, AcOEt/hexane 60:40) to give the title compound 4f. mp 245–247 °C; IR (neat) vmax 3311, 3056, 2978, 1691, 1653, 1524, 1254, 1182 cm–1; 1H NMR (400 MHz, CDCl3) δ 7.97–7.91 (m, 2H, ArH), 7.85–7.80 (m, 2H, ArH), 7.54–7.41 (m, 6H, ArH), 7.22–7.18 (m, 3H, ArH), 7.12 (dd, 3JHH = 7.6, 4JHH = 1.8, 2H, ArH), 5.99 (bs, 1H, HC-NH), 3.74–3.63 (m, 1H, HC-NH), 3.68 (d, 2JHH = 16.4 Hz, 1H, CH2), 3.57 (d, 2JHH = 16.5 Hz, 1H, CH2), 3.40 (d, 2JPH = 23.7 Hz, 1H, CH-P), 1.91–1.87 (m, 1H, cHex), 1.83–1.79 (m, 1H, cHex), 1.70–1.57 (m, 3H, cHex), 1.39–1.25 (m, 2H, cHex), 1.31 (s, 3H, CH3), 1.19–1.09 (m, 3H, cHex); 13C {1H} NMR (100 MHz, CDCl3) δ 180.8 (d, 3JPC = 2.1 Hz), 166.8, 134.2, 133.2, 132.3, 132.3, 132.2, 132.1, 131.7, 131.6, 131.2 (d, 1JPC = 103.5 Hz) 131.0, 130.7, 130.1, 129.0, 128.9, 128.6, 128.5, 128.4, 126.9, 49.5, 49.1 (d, 2JPC = 2.9 Hz), 44.4, 42.2 (d, 1JPC = 101.3 Hz), 32.9, 32.7, 25.3, 24.9, 24.8, 24.7, 14.8 ppm; 31P NMR (160 MHz, CDCl3) δ 24.4 ppm; ESI-HRMS (CI) m/z calculated for C30H34N2O3P ([M + H]+), 501.2307; found 501.2290.
(2S*,3S*)-N-Cyclohexyl-3-(diphenylphosphoryl)-2-methyl-1-propioloylaziridine-2-carboxamide (4g) (33 mg, 15%) was obtained as a pale orange solid from carboxylic acid 2p (40 µL, 0.65 mmol), isocyanide 3a (81 µL, 0.65 mmol) and 2H-azirine 1a (127 mg, 0.50 mmol), as described in the general procedure. The crude product was purified via flash column chromatography (SiO2, AcOEt/hexane 50:50) to give the title compound 4g. mp 152–153 °C; IR (neat) vmax 3270, 3059, 2923, 2098, 1674, 1658, 1587, 1371, 1188 cm–1; 1H NMR (400 MHz, CDCl3) δ 7.91–7.86 (m, 4H, ArH), 7.57–7.51 (m, 6H, ArH), 6.06 (d, 3JHH = 8.1 Hz, 1H, HC-NH), 3.81–3.75 (m, 1H, HC-NH), 3.67 (d, 2JPH = 23.9 Hz, 1H, CH-P), 2.80 (s, 1H, C≡CH), 1.93–1.92 (m, 2H, cHex), 1.85 (s, 3H, CH3), 1.77–1.73 (m, 2H, cHex), 1.68–1.58 (m, 1H, cHex), 1.41–1.26 (m, 2H, cHex), 1.25–1.11 (m, 3H, cHex) ppm; 13C {1H} NMR (100 MHz, CDCl3) δ 165.5, 160.3, 132.7, 132.6, 132.5, 132.5, 131.9 (d, 1JPC = 103.4 Hz), 131.8, 131.7, 131.2 (d, 1JPC = 105.3 Hz), 131.1, 131.0, 129.2, 129.0, 128.8, 128.7, 76.7, 49.8, 49.3 (d, 2JPC = 2.2 Hz), 43.9 (d, 1JPC = 98.0 Hz), 33.2, 32.8, 25.5, 24.9, 24.9, 14.7 ppm; 31P NMR (160 MHz, CDCl3) δ 22.8 ppm; ESI-HRMS (CI) m/z calculated for C25H28N2O3P ([M + H]+), 435.1838; found 435.1838.
(2S*,3S*)-N-Cyclohexyl-3-(diphenylphosphoryl)-2-methyl-1-nicotinoylaziridine-2-carboxamide (4h) (104 mg, 43%) was obtained as a white solid from carboxylic acid 2f (80 mg, 0.65 mmol), isocyanide 3a (81 µL, 0.65 mmol) and 2H-azirine 1a (127 mg, 0.50 mmol), as described in the general procedure. The crude product was purified via flash column chromatography (SiO2, AcOEt/hexane 50:50) to give the title compound 4h. mp 234–236 °C; IR (neat) vmax 3275, 3078, 3053, 2925, 1690, 1651, 1543, 1318, 1199, 1127 cm–1; 1H NMR (400 MHz, CDCl3) δ 8.95 (s, 1H, ArH), 8.68 (d, 3JHH = 4.60 Hz, 1H, ArH), 8.14 (d, 3JHH = 8.0 Hz, 1H, ArH), 7.96–7.90 (m, 4H, ArH), 7.57–7.48 (m, 6H, ArH), 7.34 (dd, 3JHH = 7.7 Hz, 3JHH = 4.60 Hz, 1H, ArH), 5.86 (bs, 1H, HC-NH), 3.76 (d, 2JPH = 23.5 Hz, 1H, CH-P), 3.45–3.37 (m, 1H, HC-NH), 1.95 (s, 3H, CH3), 1.72 (d, 3JHH = 11.7 Hz, 1H, cHex), 1.61–1.57 (m, 1H, cHex), 1.51–1.45 (m, 2H, cHex), 1.27–0.99 (m, 5H, cHex), 0.78–0.69 (m, 1H, cHex) ppm; 13C {1H} NMR (100 MHz, CDCl3) δ 175.4 (d, 3JPC = 3.4 Hz), 165.5, 152.9, 149.4, 132.5, 132.5, 132.5, 132.4, 132.4 (d, 1JPC = 103.8 Hz), 131.7, 131.4 (d, 1JPC = 105.4 Hz), 131.6, 131.1, 131.0, 130.0), 129.1, 129.0, 128.8, 128.7, 123.6, 50.0, 49.4, 42.5 (d, 1JPC = 100.1 Hz), 32.5, 32.5, 25.3, 24.7, 24.6, 15.3 ppm; 31P NMR (160 MHz, CDCl3) δ 23.7 ppm; ESI-HRMS (CI) m/z calculated for C28H31N3O3P ([M + H]+), 488.2103; found 488.2102.
(2S*,3S*)-1-Benzoyl-N-(tert-butyl)-3-(diphenylphosphoryl)-2-methylaziridine-2-carboxamide (4i) (184 mg, 80%) was obtained as a white solid from carboxylic acid 2a (79 mg, 0.65 mmol), isocyanide 3b (79 µL, 0.65 mmol) and 2H-azirine 1a (127 mg, 0.50 mmol), as described in the general procedure. The crude product was purified via flash column chromatography (SiO2, AcOEt/hexane 50:50) to give the title compound 4i. mp 177–179 °C; IR (neat) vmax 3319, 3050, 2967, 2923, 1699, 1682, 1699, 1601, 1535, 1232, 1190 cm–1; 1H NMR (400 MHz, CDCl3) δ 8.03–7.93 (m, 4H, ArH), 7.80–7.78 (m, 2H, ArH), 7.59–7.45 (m, 7H, ArH), 7.36 (t, 3JHH = 7.6 Hz, 2H, ArH), 5.66 (s, 1H, NH), 3.80 (d, 2JPH = 23.6 Hz, 1H, CH-P), 1.89 (s, 3H, CH3), 0.98 (s, 9H, tBu) ppm; 13C {1H} NMR (100 MHz, CDCl3) δ 177.1 (d, 3JPC = 3.1 Hz), 165.3, 134.3, 132.9 (d, 1JPC = 103.5 Hz), 132.5, 132.3, 132.3, 132.2, 132.2, 131.8, 131.7, 131.6 (d, 1JPC = 105.3 Hz) 131.2, 131.1, 129.0, 128.9, 128.7, 128.6, 128.5, 128.4, 52.1, 50.0 (d, 2JPC = 3.0 Hz, Cquart), 42.3 (d, 1JPC = 101.3 Hz), 28.1, 15.8 ppm; 31P NMR (160 MHz, CDCl3) 23.9 ppm; δ ESI-HRMS (CI) m/z calculated for C27H30N2O3P ([M + H]+), 461.1994; found 461.1995.
2S*,3S*-N-(tert-Butyl)-3-(diphenylphosphoryl)-1-(4-fluorobenzoyl)-2-methylaziridine-2-carboxamide (4j) (177mg, 74%) was obtained as a white solid from carboxylic acid 2d (91 mg, 0.65 mmol), isocyanide 3b (76 µL, 0.65 mmol) and 2H-azirine 1a (127 mg, 0.50 mmol), as described in the general procedure. The crude product was purified via flash column chromatography (SiO2, AcOEt/hexane 50:50) to give the title compound 4j. mp 221–223 °C; IR (neat) vmax 3297, 3053, 2964, 1685, 1665, 1524, 1282, 1196 cm–1; 1H NMR (400 MHz, CDCl3) δ 8.01–7.91 (m, 4H, ArH), 7.83–7.79 (m, 2H, ArH), 7.59–7.44 (m, 6H, ArH), 7.04 (t, 3JHH = 8.6 Hz, 3JHF = 8.6 Hz, 2H, ArH), 5.69 (s, 1H, NH), 3.77 (d, 2JPH = 23.5 Hz, 1H, CH-P), 1.89 (s, 3H, CH3), 1.03 (s, 9H, tBu) ppm; 13C {1H} NMR (100 MHz, CDCl3) δ 176.0 (d, 3JPC = 2.8 Hz), 165.4 (d, 1JCF = 253.3 Hz), 165.3, 133.3, 132.4, 132.3, 132.3, 132.1, 131.8, 131.7, 131.2, 131.1, 131.0, 130.9, 130.8 (d, 4JCF = 3.0 Hz), 129.1, 129.0, 128.8, 128.7, 115.6 (d, 2JCF = 21.9 Hz, ArC), 52.3, 50.1, 42.2 (d, 1JPC = 101.0 Hz, 28.2, 15.7 ppm; 31P NMR (160 MHz, CDCl3) 24.0 ppm; 19F NMR (376 MHz, CDCl3) δ –106.5 ppm; ESI-HRMS (CI) m/z calculated for C27H29FN2O3P ([M + H]+), 479.1900; found, 479.1904.
(2S*,3S*)-N-(tert-Butyl)-3-(diphenylphosphoryl)-1-(4-methoxybenzoyl)-2-methylaziridine-2-carboxamide(4k) (123 mg, 50%) was obtained as a white solid from carboxylic acid 2e (100 mg, 0.65 mmol), isocyanide 3b (76 µL, 0.65 mmol) and 2H-azirine 1a (127 mg, 0.50 mmol), as described in the general procedure. The crude product was purified via flash column chromatography (SiO2, AcOEt/hexane 45:55) to give the title compound 4k. mp > 275 °C; IR (neat) vmax 3250, 2961, 1671, 1660, 1601, 1066 cm–1; 1H NMR (400 MHz, CDCl3) δ 8.01–7.92 (m, 4H, ArH), 7.77 (d, 3JHH = 8.9 Hz, 2H, ArH), 7.54–7.45 (m, 6H, ArH), 6.85 (d, 3JHH = 8.9 Hz, 2H, ArH), 5.67 (s, 1H, NH), 3.82 (s, 3H, CH3), 3.79 (d, 2JPH = 23.8 Hz, 1H, CH-P), 1.89 (s, 3H, CH3), 1.01 (s, 9H, tBu) ppm; 13C {1H} NMR (100 MHz, CDCl3) δ 176.2 (d, 3JPC = 3.3 Hz), 165.3, 163.1 (Cquat), 133.0 (d, 1JPC = 103.3 Hz), 132.3, 132.2, 132.2, 131.8, 131.7 (ArC), 131.2 (Cquat), 131.2, 131.1, 130.6, 129.0, 128.9, 128.7, 128.6, 126.9, 113.7, 55.6, 52.1, 50.1 (d, 2JPC = 3.1 Hz, Cquat), 42.0 (d, 1JPC = 101.5 Hz), 28.2, 15.8 ppm; 31P NMR (160 MHz, CDCl3) δ 24.1 ppm ESI-HRMS (CI) m/z calculated for C28H32N2O4P ([M + H]+), 491.2100; found 491.2101.
(2S*,3S*)-1-(2-Naphthoyl)-N-(tert-butyl)-3-(diphenylphosphoryl)-2-methylaziridine-2-carboxamide (4l) (178 mg, 70%) was obtained as a white solid from carboxylic acid 2g (112 mg, 0.65 mmol), isocyanide 3b (76µL, 0.65 mmol) and 2H-azirine 1a (127 mg, 0.50 mmol), as described in the general procedure. The crude product was purified via flash column chromatography (SiO2, AcOEt/hexane 50:50) to give the title compound 4l. mp 265–267 °C; IR (neat) vmax 3381, 3061, 2970, 1682, 1662, 1529, 1293, 1196 cm–1; 1H NMR (400 MHz, CDCl3) δ 8.33 (s, 1H, ArH), 8.04–7.93 (m, 4H, ArH), 7.88–7.81 (m, 4H, ArH), 7.58–7.43 (m, 8H, ArH) 5.66 (s, 1H, NH), 3.84 (d, 2JPH = 23.6 Hz, 1H, CH-P), 1.98 (s, 3H, CH3), 0.88 (s, 9H, tBu) ppm; 13C {1H} NMR (100 MHz, CDCl3) δ 177.2 (d, 3JPC = 3.3 Hz), 165.4, 135.4, 133.0 (d, 1JPC = 103.6 Hz), 132.5, 132.4, 132.3, 132.3, 132.2, 131.8, 131.7, 131.2, 131.1, 129.6, 129.3, 129.1, 129.0, 128.7, 128.6, 128.3, 128.2, 127.9, 126.8, 124.7, 51.1, 50.2 (d, 2JPC = 2.9 Hz), 42.4 (d, 1JPC = 100.5 Hz), 28.1, 15.7 ppm; 31P NMR (160 MHz, CDCl3) δ 23.9 ppm; ESI-HRMS (CI) m/z calculated for C31H32N2O3P [M + H]+), 511.2151; found 511.2152.
(2S*,3S*)-N-(tert-Butyl)-3-(diphenylphosphoryl)-1-(furan-2-carbonyl)-2-methylaziridine-2-carboxamide (4m) (191 mg, 85%) was obtained as a white solid from carboxylic acid 2h (73 mg, 0.65 mmol), isocyanide 3b (76 µL, 0.65 mmol) and 2H-azirine 1a (127 mg, 0.50 mmol), as described in the general procedure. The crude product was purified via flash column chromatography (SiO2, AcOEt/hexane 45:55) and recrystallized from diethyl ether to give the title compound 4m. mp 222–224 °C; IR (neat) vmax 3317, 3056, 2959, 1674, 1576, 1296, 1118 cm–1; 1H NMR (400 MHz, CDCl3) δ 8.02–7.91 (m, 4H, ArH), 7.59–7.45 (m, 6H), 7.31 (dd, 3JHH = 1.7 Hz, 4JHH = 0.8 Hz, 1H), 7.14 (dd, 3JHH = 3.5, 4JHH = 0.9 Hz, 1H), 6.44 (dd, 3JHH = 3.5 Hz, 3JHH = 1.7 Hz, 1H), 5.85 (s, 1H, NH), 3.69 (d, 2JPH = 23.3 Hz, 1H, CH-P), 1.86 (s, 3H, CH3), 1.18 (s, 9H, tBu) ppm; 13C {1H} NMR (100 MHz, CDCl3) δ 166.4 (d, 3JPC = 3.5 Hz), 166.0, 148.7, 145.0, 133.0 (d, 1JPC = 103.5 Hz), 132.3, 132.2, 132.1, 131.8, 131.7, 131.1, 131.0, 129.0, 128.9, 128.7, 128.6, 116.5, 112.3, 52.0, 49.8 (d, 2JPC = 3.2 Hz, Cquat), 42.3 (d, 1JPC = 101.0 Hz), 28.3, 15.6 ppm; 31P NMR (160 MHz, CDCl3) δ 23.6 ppm; ESI-HRMS (CI) m/z calculated for C25H28N2O4P ([M + H]+), 451.1787; found 451.1789.
(2S*,3S*)-1-(4-Benzoylbenzoyl)-N-(tert-butyl)-3-(diphenylphosphoryl)-2-methylaziridine-2-carboxamide (4n) (57 mg, 20%) was obtained as a white solid from carboxylic acid 2i (147 mg, 0.65 mmol), isocyanide 3b (76 µL, 0.65 mmol) and 2H-azirine 1b (127 mg, 0.50 mmol), as described in the general procedure. The crude product was purified via flash column chromatography (SiO2, AcOEt/hexane 50:50) to give the title compound 4n. mp 103–105 °C; IR (neat) vmax 3385, 3062, 2974, 1694, 1656, 1524, 1273, 1188 cm–1; 1H NMR (400 MHz, CDCl3) δ 8.02–7.88 (m, 6H, ArH), 7.78–7.74 (m, 4H, ArH), 7.63–7.46 (m, 9H, ArH), 5.71 (s, 1H, NH), 3.78 (d, 2JPH = 23.3 Hz, 1H, CH-P), 1.91 (s, 3H, CH3), 1.04 (s, 9H, tBu) ppm; 13C {1H} NMR (100 MHz, CDCl3) δ 196.0, 176.5 (d, 3JPC = 3.3 Hz), 165.5, 140.8, 137.5, 137.1, 133.0, 132.7 (d, 1JPC = 103.7 Hz), 132.5, 132.4, 132.4, 132.3, 131.7, 131.6, 131.5 (d, 1JPC = 105.4 Hz), 131.1, 131.0, 130.2, 129.9, 129.1, 129.0, 128.8, 128.7, 128.6, 128.3, 52.3, 50.1 (d, 3JPC = 2.8 Hz), 42.7 (d, 1JPC = 100.2 Hz), 28.2, 15.6 ppm; 31P NMR (160 MHz, CDCl3) δ 25.7 ppm; ESI-HRMS (CI) m/z calculated for C34H34N2O4P ([M + H]+), 565.2256; found 565.2252.
(2S*,3S*)-1-Acetyl-N-(tert-butyl)-3-(diphenylphosphoryl)-2-methylaziridine-2-carboxamide (4o) (89 mg, 45%) was obtained as a white solid from carboxylic acid 2k (38 µL, 0.65 mmol), isocyanide 3b (76 µL, 0.65 mmol) and 2H-azirine 1a (127 mg, 0.50 mmol), as described in the general procedure. The crude product was purified via flash column chromatography (SiO2, AcOEt/hexane 50:50) and recrystallized from diethyl ether to give the title compound 4o. mp 195–197 °C; IR (neat) vmax 3358, 3056, 2984, 1699, 1665, 1540, 1274, 1116 cm–1; 1H NMR (400 MHz, CDCl3) δ 8.06–7.76 (m, 4H, ArH), 7.69–7.35 (m, 6H, ArH), 5.90 (s, 1H, NH), 3.46 (d, 2JPH = 24.8 Hz, 1H, CH-P), 1.94 (s, 3H, CH3), 1.76 (s, 3H, CH3), 1.33 (s, 9H, tBu) ppm; 13C {1H} NMR (100 MHz, CDCl3) δ 179.8 (d, 3JPC = 2.9 Hz), 166.5, 132.5 (d, 1JPC = 103.5 Hz), 132.4, 132.3 131.7, 131.7 (d, 1JPC = 104.3 Hz), 131.6, 131.1, 130.0, 129.1, 129.0, 128.8, 128.7, 52.4, 48.4 (d, 2JPC = 3.3 Hz), 42.9 (d, 1JPC = 100.4 Hz), 28.6, 24.3, 15.4 ppm; 31P NMR (160 MHz, CDCl3) δ 23.8 ppm; ESI-HRMS (CI) m/z calculated for C22H28N2O3P ([M + H]+), 399.1838; found 399.1837.
(2S*,3S*)-N-(tert-Butyl)-3-(diphenylphosphoryl)-2-methyl-1-(2,2,2-trifluoroacetyl)aziridine-2-carboxamide (4p) (61 mg, 27%,) was obtained as a white solid from carboxylic acid 2l (50 µL, 0.65 mmol), isocyanide 3b (76 µL, 0.65 mmol) and 2H-azirine 1a (127 mg, 0.50 mmol), as described in the general procedure. The crude product was purified via flash column chromatography (SiO2, AcOEt/hexane 75:25) to give the title compound 4p. mp 172–174 °C; IR (neat) vmax 3346, 3059, 2921, 1732, 1664, 1538, 1399, 1204, 1138 cm–1; 1H NMR (400 MHz, CDCl3) δ 7.95–7.90 (m, 2H, ArH), 7.85–7.79 (m, 2H, ArH), 7.58–7.45 (m, 6H, ArH), 5.91 (s, 1H, NH), 3.42 (d, 2JPH = 21.0 Hz, 1H, CH-P), 1.75 (s, 3H, CH3), 1.29 (s, 9H, tBu) ppm; 13C {1H} NMR (100 MHz, CDCl3) δ 167.3 (qd, 2JCF = 38.2 Hz, 3JPC = 3.4 Hz), 166.1, 132.7, 132.7, 132.7, 132.6, 132.1 (d, 1JPC = 104.7 Hz), 131.4, 130.9, 130.8, 129.9, 129.2, 129.1, 128.9, 128.8, 115.37 (q, 1JCF = 287.7 Hz), 55.9, 52.6 (d, 2JPC = 3.5 Hz, Cquat), 41.8 (d, 1JPC = 98.3 Hz), 28.3, 14.8 ppm; 19F NMR (376 MHz, CDCl3) δ –75.5 ppm; 31P NMR (160 MHz, CDCl3) δ 23.4 ppm; ESI-HRMS (CI) m/z calculated for C22H25F3N2O3P ([M + H]+), 453.1555; found 453.1543.
(2S*,3S*)-N-(tert-Butyl)-3-(diphenylphosphoryl)-2-methyl-1-(2-phenylacetyl)aziridine-2-carboxamide (4q) (158 mg, 71%) was obtained as a white solid from carboxylic acid 2m (88 mg, 0.65 mmol), isocyanide 3b (76 µL mg, 0.65 mmol) and 2H-azirine 1a (127 mg, 0.50 mmol), as described in the general procedure. The crude product was purified via flash column chromatography (SiO2, AcOEt/hexane 50:50) to give the title compound 4q. mp: 236–238 °C; IR (neat) vmax 3286, 3056, 2956, 1693, 1657, 1549, 1293, 1188 cm–1; 1H NMR (400 MHz, CDCl3) δ 8.01 (m, 2H, ArH), 7.91–7.77 (m, 2H, ArH), 7.59–7.41 (m, 6H, ArH), 7.27–7.21 (m, 3H, ArH), 7.16 (d, 3JHH = 7.5, 2H, ArH), 5.68 (s, 1H, NH), 3.71 (d, 2JHH = 16.5 Hz, 1H, CH2), 3.61 (d, 2JHH = 16.5 Hz, 1H, CH2), 3.41 (d, 2JPH = 23.5 Hz, 1H, CH-P), 1.35 (s, 9H, tBu), 1.32 (s, 3H, CH3) ppm; 13C {1H} NMR (100 MHz, CDCl3) δ 180.8 (d, 3JPC = 2.7 Hz), 166.6, 134.2, 133.3, 132.8 (d, 1JPC = 103.5 Hz), 132.2, 132.2, 132.0, 132.0, 131.6, 131.5, 131.3 (d, 1JPC = 104.8 Hz), 130.9, 130.9, 130.1, 128.9, 128.8, 128.5, 128.4, 128.4, 126.9, 52.3, 49.3 (d, 2JPC = 3.5 Hz, Cquat), 44.4, 42.2 (d, 1JPC = 101.4 Hz), 28.5, 15.0 ppm; 31P NMR (160 MHz, CDCl3) δ 23.9 ppm; ESI-HRMS (CI) m/z calculated for C28H32N2O3P ([M + H]+), 475.2151; found 475.21341.
(2S*,3S*)-1-(2-([1,1’-Biphenyl]-4-yl)acetyl)-N-(tert-butyl)-3-(diphenylphosphoryl)-2-methylaziridine-2-carboxamide (4r) (96 mg, 35%) was obtained as a white solid from carboxylic acid 2n (137 mg, 0.65 mmol), isocyanide 3b (76 µL, 0.65 mmol) and 2H-azirine 1a (127 mg, 0.50 mmol), as described in the general procedure. The crude product was purified via flash column chromatography (SiO2, AcOEt/hexane 50:50) to give the title compound 4r. mp 256–258 °C; IR (neat) vmax 3363, 3056, 2977, 1698, 1675, 1533, 1197 cm–1; 1H NMR (400 MHz, CDCl3) δ 8.02–7.97 (m, 2H, ArH), 7.88–7.83 (m, 2H, ArH), 7.56–7.41 (m, 12H, ArH), 7.34 (t, 3JHH = 7.3 Hz, 1H, ArH), 7.22 (d, 3JHH = 7.9 Hz, 2H, ArH), 5.74 (s, 1H, NH), 3.71 (d, 2JHH = 16.6 Hz, 1H, CH2) 3.64 (d, 2JHH = 16.6 Hz, 1H, CH2), 3.41 (d, 2JPC = 21.9 Hz, 1H, CH-P), 1.40 (s, 3H, CH3), 1.38 (s, 9H, tBu) ppm; 13C {1H} NMR (100 MHz, CDCl3) δ 181.0 (d, 3JPC = 2.9 Hz), 166.8, 141.0, 140.0, 133.4, 132.4, 132.4, 132.2, 132.2, 131.8, 131.7, 131.3 (d, 1JPC = 104.9 Hz), 131.1, 131.0, 130.6, 129.0, 128.9, 128.9, 128.7, 128.6, 127.4, 127.2, 127.2, 52.5, 49.6 (d, 2JPC = 3.6 Hz), 44.2, 42.3 (d, 1JPC = 101.3 Hz), 28.7, 15.2 ppm; 31P NMR (160 MHz, CDCl3) δ 24.2 ppm; ESI-HRMS (CI) m/z calculated for C34H36N2O3P ([M + H]+), 551.2464; found 551.2459.
(2S*,3S*)-1-Benzoyl-N-cyclopropyl-3-(diphenylphosphoryl)-2-methylaziridine-2-carboxamide (4s) (133 mg, 60%) was obtained as a white solid from carboxylic acid 2a (79 mg, 0.65 mmol), isocyanide 3c (43 µL, 0.65 mmol) and 2H-azirine 1a (127 mg, 0.50 mmol), as described in the general procedure. The crude product was purified via flash column chromatography (SiO2, AcOEt/hexane 20:80) to give the title compound 4s. mp 203–205 °C; IR (neat) vmax 3256, 3059, 2923, 1674, 1579, 1299, 1182 cm–1; 1H NMR (400 MHz, CDCl3) δ 8.04–7.88 (m, 4H, ArH), 7.81 (d, 3JHH = 7.5 Hz, 2H, ArH), 7.64–7.44 (m, 7H, ArH), 7.38 (t, 3JHH = 7.6 Hz, 2H, ArH), 6.30 (s, 1H, HC-NH) 3.80 (d, 2JPC = 23.8 Hz, 1H, CH-P), 2.46–2.29 (m, 1H, HC-NH), 1.93 (s, 3H, CH3), 0.68–0.43 (m, 2H, CH2), 0.40–0.21 (m, 1H, CH2), 0.00 to –0.17 (m, 1H, CH2) ppm; 13C {1H} NMR (100 MHz, CDCl3) δ 176.7 (d, 3JPC = 3.2 Hz), 168.0, 134.1, 132.6 (d, 1JPC = 103.5 Hz), 132.6, 132.4, 132.4, 132.3, 132.3, 132.1, 131.7, 131.6, 131.1, 131.1, 129.1, 129.0, 128.8, 128.6, 128.5, 128.5, 49.5 (d, 2JPC = 3.0 Hz, Cquat), 42.4 (d, 1JPC = 100.5 Hz), 23.1, 15.4, 6.5, 6.4 ppm; 31P NMR (160 MHz, CDCl3) δ 26.4 ppm; ESI-HRMS (CI) m/z calculated for C26H26N2O3P ([M + H]+), 445.1681; found 445.1682.
(2S*,3S*)-N-Cyclopropyl-3-(diphenylphosphoryl)-2-methyl-1-(quinoline-6-carbonyl)aziridine-2-carboxamide (4t) (136 mg, 55%) obtained as a white solid from carboxylic acid 2j (112 mg, 0.65 mmol), isocyanide 3c (43 µL, 0.65 mmol) and 2H-azirine 1a (127 mg, 0.50 mmol), as described in the general procedure. The crude product was purified via flash column chromatography (SiO2, AcOEt/hexane 20:80) to give the title compound 4t. mp 212–214 °C; IR (neat) vmax 3195, 3045, 2953, 1674, 1660, 1551, 1290, 1177 cm–1; 1H NMR (400 MHz, CDCl3) δ 8.98 (d, 4JHH = 3.0 Hz, 1H, ArH), 8.37 (s, 1H, ArH), 8.16 (d, 3JHH = 9.5 Hz, 1H, ArH), 8.08–8.03 (m, 2H, ArH), 7.98–7.86 (m, 4H, ArH), 7.60–7.48 (m, 4H, ArH), 7.46–7.41 (m, 3H, ArH), 6.20 (d, 2JHH = 22.1 Hz, 1H, HC-NH), 3.78 (d, 2JPC = 23.7 Hz, 1H, CH-P), 2.41–2.34 (m, 1H, HC-NH), 0.62–0.44 (m, 2H, CH2), 0.29–0.23 (m, 1H, CH2), 0.04 to –0.10 (m, 1H, CH2); 13C {1H} NMR (100 MHz, CDCl3) δ 176.1 (d, 3JPC = 3.1 Hz), 168.3, 152.5, 149.9, 137.5, 132.5 (d, 1JPC = 103.5 Hz), 132.5, 132.5, 132.4, 132.4, 132.2, 131.5 (d, 1JPC = 105.3 Hz), 129.9, 129.7, 129.2, 129.0, 128.8, 128.7, 128.1, 127.6, 122.0, 49.7 (d, 3JPC = 2.8 Hz, Cquat), 42.5 (d, 1JPC = 99.9 Hz), 23.2, 15.3, 6.6, 6.6 ppm; 31P NMR (160 MHz, CDCl3) δ 23.8 ppm; ESI-HRMS (CI) m/z calculated for C29H27N3O3P ([M + H]+), 496.1790; found 496.1788.
(2S*,3S*)-N-Cyclopropyl-3-(diphenylphosphoryl)-1-(furan-2-carbonyl)-2-methylaziridine-2-carboxamide (4u) (162 mg, 75%) was obtained as a white solid from carboxylic acid 2h (73 mg, 0.65 mmol), isocyanide 3c (43 µL, 0.65 mmol) and 2H-azirine 1a (127 mg, 0.50 mmol), as described in the general procedure. The crude product was purified via flash column chromatography (SiO2, AcOEt/hexane 80:20) to give the title compound 4u. mp 127–129 °C; IR (neat) vmax 3236, 3114, 2961, 1674, 1579, 1290, 1171 cm–1; 1H NMR (400 MHz, CDCl3) δ 7.98–7.91 (m, 4H, ArH), 7.59–7.46 (m, 6H, ArH), 7.27 (d, 4JHH = 1.0 Hz, 1H, ArH), 7.15 (dd, 3JHH = 3.5 Hz, 4JHH = 0.8 Hz, 1H, ArH), 6.46 (dd, 3JHH = 3.6 Hz, 3JHH = 1.7 Hz, 1H, ArH), 6.24 (s, 1H, HC-NH), 3.70 (d, 2JPH = 23.6 Hz, 1H, CH-P), 2.60–2.54 (m, 1H, HC-NH), 1.90 (s, 3H, CH3), 0.76–0.64 (m, 2H, CH2), 0.49–0.42 (m, 1H, CH2), 0.32–0.26 (m, 1H, CH2) ppm; 13C {1H} NMR (100 MHz, CDCl3) δ 168.6, 165.8 (d, 3JPC = 3.4 Hz), 148.3, 145.0, 132.5 (d, 1JPC = 103.5 Hz), 132.3, 132.3, 132.2, 132.1, 131.7, 131.6, 131.0, 130.9, 129.0, 128.8, 128.6, 128.5, 116.7, 112.3, 48.9, 42.5 (d, 1JPC = 99.9 Hz), 29.7, 23.2, 15.1, 6.6, 6.6 ppm; 31P NMR (160 MHz, CDCl3) δ 24.6 ppm; ESI-HRMS (CI) m/z calculated for C24H24N2O4P ([M + H]+), 435.1474; found 434.1463.
3.3. Experimental Procedure and Characterization Data for N-Acylaziridine Phosphonates 5
In a flame-dried flask, the corresponding carboxylic acid 2 (0.65 mmol, 1.3 eq.), isocyanide 3 (0.65 mmol, 1.3 eq.) and a 1M diethyl ether solution of ZnCl2 (0.12 mL, 0.12 mmol, 0.25 eq.) were added to 0.5 mL of dry THF. Then, 2H-azirine phosphonate 1b (0.50 mmol, 1 eq.) was added at room temperature. The reaction mixture was stirred until TLC showed the disappearance of 2H-azirine 1b (1–24 h). The solvent was removed under vacuum, and the residue was dissolved in dichloromethane (5 mL) and washed with water (2 × 5 mL). The organic layer was dried over anhydrous MgSO4 and filtered, and the solvent was evaporated under reduced pressure. The resulting residue was purified via flash column chromatography (SiO2, hexanes/AcOEt) to yield compounds 5.
Diethyl ((2S*,3S*)-1-benzoyl-3-(tert-butylcarbamoyl)-3-methylaziridin-2-yl)phosphonate (5a) (118 mg, 60%) was obtained as a white solid from carboxylic acid 2a (79 mg, 0.65 mmol), isocyanide 3b (76 µL, 0.65 mmol) and 2H-azirine 1b (96 mg, 0.50 mmol), as described in the general procedure. The crude product was purified via flash column chromatography (SiO2, AcOEt/hexane 45:55) to give the title compound 5a. mp 140–141 °C; IR (neat) vmax 3306, 3071, 2966, 1688, 1665, 1633, 1236 cm–1; 1H NMR (400 MHz, CDCl3) δ 7.83 (d, 3JHH = 7.3 Hz, 2H, ArH), 7.48 (d, 3JHH = 7.3 Hz, 1H, ArH), 7.39 (t, 3JHH = 7.5 Hz, 2H, ArH), 5.75 (s, 1H, NH), 4.33–4.10 (m, 4H, OCH2CH3), 3.25 (d, 2JPC = 17.2 Hz, 1H, CH-P), 1.94 (s, 3H, CH3), 1.39–1.35 (m, 6H, OCH2CH3), 1.01 (s, 9H, tBu) ppm; 13C {1H} NMR (100 MHz, CDCl3) δ 176.3 (d, 3JPC = 4.7 Hz), 165.2, 134.1, 132.5, 128.5, 128.4, 63.6 (d, 2JPC = 6.9 Hz), 62.6 (d, 2JPC = 6.9 Hz), 52.1, 48.4 (d, 2JPC = 2.8 Hz), 38.8 (d, 1JPC = 202.0 Hz), 28.1, 16.5 (d, 3JPC = 6.5 Hz), 16.4 (t, 3JPC = 6.5 Hz), 15.4 ppm; 31P NMR (160 MHz, CDCl3) δ 17.3 ppm; ESI-HRMS (CI m/z calcd. For C19H30N2O5P ([M + H]+), 397.1892; found 397.1891.
Diethyl ((2S*,3S*)-3-(tert-butylcarbamoyl)-1-(4-fluorobenzoyl)-3-methylaziridin-2-yl)phosphonate (5b) (158 mg, 76%) was obtained as a needle-shaped crystal from carboxylic acid 2d (91 mg, 0.65 mmol), isocyanide 3b (76 µL, 0.65 mmol) and 2H-azirine 1b (96 mg, 0.50 mmol), as described in the general procedure. The crude product was purified via flash column chromatography (SiO2, AcOEt/hexane 50:50) to give the title compound 5b. mp 99–101 °C; IR (neat) vmax 3391, 2982, 1686, 1602, 1525, 1288, 1240 cm–1; 1H NMR (400 MHz, CDCl3) δ 7.85 (dd, 2JHH = 8.8 Hz, 3JHF = 5.4 Hz, 2H, ArH), 7.08 (t, 2JHF = 8.6 Hz, 2JHH = 8.6 Hz, 2H, ArH), 5.76 (s, 1H, NH), 4.42–4.05 (m, 4H, OCH2CH3), 3.24 (d, 2JPC = 17.1 Hz, 1H, CH-P), 1.94 (s, 3H, CH3), 1.55–1.20 (m, 6H, OCH2CH3), 1.06 (s, 9H, tBu) ppm; 13C {1H} NMR (100 MHz, CDCl3) δ 175.2 (d, 3JPC = 4.7 Hz), 165.4 (d, 1JCF = 253.4 Hz), 165.2, 130.9 (d, 3JCF = 9.1 Hz, 130.5 (d, 4JCF = 3.0 Hz), 115.5 (d, 2JCF = 21.8 Hz), 63.6 (d, 2JPC = 6.1 Hz), 62.7 (d, 2JPC = 6.6 Hz), 52.2, 48.4 (d, 2JPC = 2.8 Hz), 38.7 (d, 1JPC = 202.4 Hz), 28.2, 16.5 (d, 2JPC = 6.6 Hz), 16.5 (d, 2JPC = 6.6 Hz) 15.4 ppm; 31P NMR (160 MHz, CDCl3) δ 17.1 ppm; 19F NMR (376 CDCl3) δ –106.4 ppm; ESI-HRMS (CI) m/z calcd. For C19H29FN2O5P ([M + H]+), 415.1798; found 415.1806.
Diethyl ((2S*,3S*)-3-(tert-butylcarbamoyl)-1-(furan-2-carbonyl)-3-methylaziridin-2-yl)phosphonate (5c) (135 mg, 70%) was obtained as a white solid from carboxylic acid 2h (73 mg, 0.65 mmol), isocyanide 3b (76 µL, 0.65 mmol) and 2H-azirine 1b (96 mg, 0.50 mmol), as described in the general procedure. The crude product was purified via flash column chromatography (SiO2, AcOEt/hexane 50:50) to give the title compound 5c. mp 94–96 °C; IR (neat) vmax 3246, 3062, 2986, 1701, 1679, 1652, 1448, 1188, 1121 cm–1; 1H NMR (400 MHz, CDCl3) δ 7.44–7.42 (m, 1H, ArH), 7.13–7.11 (m, 1H, ArH), 6.59–6.11 (m, 1H, ArH), 5.97 (s, 1H, NH), 4.40–3.69 (m, 4H, OCH2CH3), 3.12 (d, 2JPC = 18.2 Hz, 1H, CH-P), 1.89 (s, 3H, CH3), 1.35 (t, 3JHH = 7.1 Hz, 6H, OCH2CH3), 1.15 (s, 9H, tBu) ppm; 13C {1H} NMR (100 MHz, CDCl3) δ 165.7 (d, 3JPC = 5.1 Hz), 165.7 (d, 3JPC = 1.1 Hz), 148.4, 145.2, 116.5, 112.2, 63.8 (d, 2JPC = 6.4 Hz), 63.0 (d, 2JPC = 6.5 Hz), 52.1, 48.5 (d, 2JPC = 3.0 Hz), 39.2 (d, 1JPC = 203.7 Hz), 28.2, 16.3 (d, 3JPC = 5.0 Hz), 16.2 (d, 3JPC = 5.1 Hz), 15.3 ppm; 31P NMR (160 MHz, CDCl3) δ 16.9 ppm; ESI-HRMS (CI) m/z calcd. for C17H28N2O6P ([M + H]+), 387.1685; found 387.1668.
Diethyl ((2S*,3S*)-3-(tert-butylcarbamoyl)-3-methyl-1-(quinoline-6-carbonyl)aziridin-2-yl)phosphonate (5d) (123 mg, 55%) was obtained as a white solid from carboxylic acid 2j (112 mg, 0.65 mmol), isocyanide 3b (76 µL, 0.65 mmol) and 2H-azirine 1b (96 mg, 0.50 mmol), as described in the general procedure. The crude product was purified via flash column chromatography (SiO2, CH2Cl2/MeOH 99:1) to give the title compound 5d. mp 152–153 °C; IR (neat) vmax 3412, 2922, 1676, 1665, 1528, 1287, 1157 cm–1; 1H NMR (400 MHz, CDCl3) δ 8.99 (dd, 3JHH = 4.2 Hz, 4JHH = 1.7 Hz, 1H, ArH), 8.42 (dd, 4JHH = 1.8 Hz, 4JHH = 0.9 Hz, 1H), 8.26–8.23 (m, 1H, ArH), 8.13–8.08 (m, 2H, ArH), 7.46 (ddd, 3JHH = 8.3 Hz, 3JHH = 4.2 Hz, 4JHH = 1.0 Hz, 1H, ArH), 5.76 (s, 1H, NH), 4.30–4.23 (m, 4H, OCH2CH3), 3.32 (d, 2JPC = 17.0 Hz, 1H, CH-P), 2.02 (s, 3H, CH3), 1.42–1.38 (m, 6H, OCH2CH3), 0.95 (s, 9H, tBu) ppm; 13C {1H} NMR (100 MHz, CDCl3) δ 175.9 (d, 3JPC = 4.7 Hz), 165.3, 152.5, 149.9, 137.4, 132.1, 129.9, 129.7, 128.1, 127.6, 122.0, 63.7 (d, 2JPC = 6.2 Hz), 62.7 (d, 2JPC = 6.6 Hz), 52.2, 48.6 (d, 2JPC = 2.8 Hz), 39.2 (d, 1JPC = 201.9 Hz), 28.2, 16.6, (d, 3JPC = 6.6 Hz), 16.5, (d, 3JPC = 6.9 Hz), 15.5 ppm; 31P NMR (160 MHz, CDCl3) δ 17.0 ppm; ESI-HRMS (CI) m/z calcd. for C22H31N3O5P ([M + H]+), 448.2001; found 448.1994.
Diethyl ((2S*,3S*)-3-(tert-butylcarbamoyl)-3-methyl-1-(2-phenylacetyl)aziridin-2-yl)phosphonate (5e) (71 mg, 35%) was obtained as a white crystalline solid from carboxylic acid 2m (88 mg, 0.65 mmol), isocyanide 3b (76 µL, 0.65 mmol) and 2H-azirine 1b (96 mg, 0.50 mmol), as described in the general procedure. The crude product was purified via flash column chromatography (SiO2, AcOEt/hexane 50:50) to give the title compound 5e. mp 124–126 °C; IR (neat) vmax 3312, 3061, 2985, 1688, 1669, 1565, 1236, 1028 cm–1; 1H NMR (400 MHz, CDCl3) δ 7.43–6.85 (m, 5H, ArH), 5.76 (s, 1H, NH), 4.37–4.09 (m, 4H, OCH2CH3), 3.73 (d, 2JHH = 16.3, 1H, CH2), 3.66 (d, 2JHH = 16.4 Hz, 1H, CH2), 2.89 (d, 2JPC = 18.3 Hz, 1H, CH-P), 1.49 (s, 3H, CH3), 1.42–1.22 (m, 15H), 1.32 (s, 9H, tBu) ppm; 13C {1H} NMR (100 MHz, CDCl3) δ 180.1 (d, 3JPC = 4.2 Hz), 166.6, 134.3, 130.2, 128.6, 127.1, 63.5 (d, 2 JPC = 6.3 Hz), 62.7 (d, 2JPC = 6.5 Hz), 52.3, 48.0 (d, 2JPC = 3.3 Hz), 44.4, 38.8 (d, 1JPC = 201.5 Hz), 28.6, 16.3 (d, 3JPC = 6.3 Hz), 16.3 (d, 3JPC = 7.8, Hz), 14.8 (CH3) ppm; 31P NMR (160 MHz, CDCl3) δ 16.7 ppm; ESI-HRMS (CI) m/z calcd. for C20H32N2O5P+ ([M + H]+), 411.2049; found 411.2046.
Diethyl ((2S*,3S*)-1-(2-([1,1’-biphenyl]-4-yl)acetyl)-3-(tert-butylcarbamoyl)-3-methylaziridin-2-yl)phosphonate (5f) (126 mg, 52%) was obtained as a white solid from carboxylic acid 2n (137 mg, 0.65 mmol), isocyanide 3b (76 µL, 0.65 mmol) and 2H-azirine 1b (96 mg, 0.50 mmol), as described in the general procedure. The crude product was purified via flash column chromatography (SiO2, AcOEt/hexane 50:50) to give the title compound 5f. mp 150–152 °C; IR (neat) vmax 3361, 3056, 2975, 1699, 1654, 1526, 1252 cm–1; 1H NMR (400 MHz, CDCl3) δ 7.56 (dd, 3JHH = 10.7 Hz, 3JHH = 7.8 Hz, 4H, ArH), 7.43 (t, 3JHH = 7.6 Hz, 2H, ArH), 7.34 (d, 3JHH = 8.1 Hz, 3H, ArH), 5.77 (s, 1H, NH), 4.25–4.15 (m, 4H, OCH2CH3), 3.75 (d, 2JHH = 16.3 Hz, 1H, CH2), 3.69 (d, 2JHH = 16.3 Hz, 1H, CH2), 2.91 (d, 2JPC = 18.2 Hz, 1H, CH-P), 1.55 (s, 3H, CH3), 1.37–1.33 (m, 6H, OCH2CH3) 1.36 (s, 9H, tBu) ppm; 13C {1H} NMR (100 MHz, CDCl3) δ 180.1 (d, 3JPC = 4.3 Hz), 166.6, 140.9, 140.1, 133.4, 130.6, 128.9, 127.4, 127.2, 127.0, 63.5 (d, 2JPC = 6.3 Hz), 62.7 (d, 2JPC = 6.5 Hz), 52.4, 47.9 (d, 2JPC = 3.3 Hz), 44.0, 38.9 (d, 1JPC = 201.3 Hz), 28.7, 16.5 (d, 3JPC = 6.2 Hz), 16.5 (d, 3JPC = 6.3 Hz), 14.9 (CH3) ppm; 31P NMR (160 MHz, CDCl3) δ 16.6 ppm; ESI-HRMS (CI) m/z calcd. for C26H36N2O5P ([M + H]+), 487.2362; found 487.2340.
Diethyl ((2S*,3S*)-3-(cyclopropylcarbamoyl)-3-methyl-1-(3-phenylpropioloyl)aziridin-2-yl)phosphonate (5g) (96 mg, 48%) was obtained as a white wax from carboxylic acid 2o (95 mg, 0.65 mmol), isocyanide 3b (76 µL, 0.65 mmol) and 2H-azirine 1b (96 mg, 0.50 mmol), as described in the general procedure. The crude product was purified via flash column chromatography (SiO2, AcOEt/hexane 60:40) to give the title compound 5g. mp 133–135 °C; IR (neat) vmax 3332, 2987, 2204, 1681, 1672, 1538, 1286, 1187, 1016 cm–1; 1H NMR (400 MHz, CDCl3) δ 7.55–7.53 (m, 2H, ArH), 7.45–7.41 (m, 1H, ArH), 7.38–7.33 (m, 2H, ArH), 6.56 (d, 3JHH = 2.9 Hz, 1H, HC-NH), 4.29–4.17 (m, 4H, OCH2CH3), 3.26 (d, 2JPC = 17.1 Hz, 1H, CH-P), 2.75–2.69 (m, 1H, HC-NH), 1.86 (s, 3H, CH3), 1.36–1.32 (m, 6H, OCH2CH3), 0.79–0.74 (m, 2H, CH2), 0.58–0.54 (m, 2H, CH2) ppm; 13C {1H} NMR (100 MHz, CDCl3) δ 167.9, 161.0 (d, 3JPC = 5.6 Hz), 133.0, 130.8, 128.7, 119.9, 89.3, 83.1, 63.6 (d, 2 JPC = 6.3 Hz), 62.8 (d, 2JPC = 6.5 Hz), 48.1 (2JPC = 2.04 Hz) 40.8 (d, 1JPC = 200.6 Hz), 23.6, 16.5 (d, 3JPC = 6.1 Hz), 14.6, 6.7, 6.6 ppm; 31P NMR (160 MHz, CDCl3) δ 15.9 ppm; ESI-HRMS (CI) m/z calcd. for C20H26N2O5P ([M + H]+), 405.1579; found 405.1570.
Diethyl ((2S*,3S*)-1-((E)-but-2-enoyl)-3-(tert-butylcarbamoyl)-3-methylaziridin-2-yl)phosphonate (5h) (96 mg, 48%) was obtained as an oil from carboxylic acid 2q (56 mg, 0.65 mmol), isocyanide 3b (76 µL, 0.65 mmol) and 2H-azirine 1b (96 mg, 0.50 mmol), as described in the general procedure. The crude product was purified via flash column chromatography (SiO2, AcOEt/hexane 50:50) to give the title compound 5h. Rf: 0.50 (AcOEt); IR (neat) vmax 3362, 2977, 1689, 1645, 1523, 1288, 1244, 1192, 1027 cm–1; 1H NMR (400 MHz, CDCl3) δ 6.90 (dq, 3JHH = 15.5 Hz, 3JHH = 6.9 Hz, 1H, CH3CH=CH) 6.00 (s, 1H, NH), 5.95 (dd, 3JHH = 15.5 Hz, 4JHH = 1.7 Hz, 1H, CH3CH=CH) 4.38–4.14 (m, 4H, OCH2CH3), 3.03 (d, 2JPC = 17.1 Hz, 1H, CH-P), 1.88 (dd, 3JHH = 6.9 Hz, 4JHH = 1.7 Hz, 3H, CH3-CH=), 1.89 (s, 3H, CH3), 1.43–1.33 (m, 6H, OCH2CH3), 1.34 (s, 9H, tBu) ppm; 13C {1H} NMR (100 MHz, CDCl3) δ 173.6 (d, 3JPC = 4.8 Hz), 165.9, 143.4, 125.4, 63.3 (d, 2JPC = 6.3 Hz), 62.6 (d, 2JPC = 6.5 Hz), 52.1, 47.3 (d, 2JPC = 2.8 Hz), 38.7 (d, 1JPC = 200.8 Hz), 28.5, 18.1, 16.4 (d, 3JPC = 6.0 Hz), 16.4 (d, 3JPC = 6.2, Hz), 14.9 ppm; 31P NMR (160 MHz, CDCl3) δ 17.1 ppm; ESI-HRMS (CI) m/z calcd. for C16H30N2O5P ([M + H]+), 361.1892; found 361.1878.
3.4. Gram Scale Procedure of N-Acylaziridine 5a
In a flame-dried flask, benzoic acid 2a (635 mg, 5.2 mmol, 1.3 eq.), tert-butyl isocyanide 3b (588 µL, 5.2 mmol, 1.3 eq.) and a 1M diethyl ether solution of ZnCl2 (1.0 mL, 1 mmol, 0.25 eq.) were added to 4 mL of dry THF. Then, 2H-azirine phosphonate 1b (764 mg, 4 mmol, 1 eq.) was added at room temperature. The reaction mixture was stirred until TLC showed the disappearance of 2H-azirine 1b (4 h). The solvent was removed under vacuum, and the residue was dissolved in dichloromethane (15 mL) and washed with saturated NaHCO3 solution (15 mL) and water (2 × 15 mL). The organic layer was dried over anhydrous MgSO4 and filtered, and the solvent was evaporated under reduced pressure. The resulting residue was recrystallized from pentane to yield the desired N-acylaziridine 5a (1.11 g, 70%).
3.5. Experimental Procedure and Characterization Data for Phosphorylated Aziridine Peptidomimetics 6
In a flame-dried flask, corresponding amino acid 2r or 2s (1.3 mmol, 1.3 eq.), tert-butyl isocyanide 3b (1.3 mmol, 1.3 eq.) and a 1M diethyl ether solution of ZnCl2 (0.25 mL, 0.25 mmol, 0.25 eq.) were added to 1 mL of dry THF. Then, 2H-azirine phosphonate 1b (1 mmol, 1 eq.) was added at room temperature. The reaction mixture was stirred until TLC showed the disappearance of 2H-azirine 1b (6 h). The solvent was removed under vacuum, and the residue was dissolved in dichloromethane (5 mL) and washed with water (2 × 5 mL). The organic layer was dried over anhydrous MgSO4 and filtered, and the solvent was evaporated under reduced pressure. The resulting residue was purified via flash column chromatography (SiO2, hexanes/AcOEt) to yield the title compounds.
(9H-Fluoren-9-yl)methyl ((S)-1-((2S,3S)-2-(tert-butylcarbamoyl)-3-(diethoxyphosphoryl)-2-methylaziridin-1-yl)-4-methyl-1-oxopentan-2-yl)carbamate and (9H-fluoren-9-yl)methyl ((S)-1-((2R,3R)-2-(tert-butylcarbamoyl)-3-(diethoxyphosphoryl)-2-methylaziridin-1-yl)-4-methyl-1-oxopentan-2-yl)carbamate (6a) (407 mg, 65%) yielded (50/50) of a mixture of two diastereoisomers, 6aA and 6aB, obtained as oils from amino acid 2r (459 mg, 1.3 mmol), tert-butyl isocyanide 3b (152 µL, 1.3 mmol) and 2H-azirine 1b (191 mg, 1 mmol), as described in the general procedure. The crude product was purified via flash column chromatography (SiO2, AcOEt/hexane 40:60) to give the title compound as a mixture of two diastereoisomers (6aA and 6aB). Rf: 0.60 (AcOEt); IR (neat) vmax 3444, 3328, 3067, 2961, 1701, 1668, 1521, 1257, 1218, 1016 cm–1; 1H NMR (400 MHz, CDCl3) δ 7.75 (d, 3JHH = 7.5 Hz, 4H, ArH)A+B, 7.60–7.58 (m, 1H, 4H, ArH)A+B, 7.39 (t, 3JHH = 7.4 Hz, 4H, ArH)A+B, 7.32–7.28 (m, 3JHH = 7.4 Hz, 4H, ArH)A+B, 5.99 (s, 1H, NH)A, 5.89 (s, 1H, NH)B, 5.28 (d, 3JHH = 8.7 Hz, NHFmoc)A, 5.11 (d, 3JHH = 9.2 Hz, NHFmoc)B, 4.49–4.18 (m, 16H, CHα + OCH2CH3 + CH2Fmoc + CHFmoc)A+B, 2.99 (d, 2JPH = 18.1 Hz, 1H, CH-P)B, 2.85 (d, 2JPH = 17.7 Hz, 1H, CH-P)A, 1.80–1.57 (m, 12H, CH3 + CHCH2)A+B, 1.38–1.29 (m, 30H, OCH2CH3 + tBu)A+B, 0.97–0.93 (m, 12H, CH3)A+B ppm; 13C {1H} NMR (100 MHz, CDCl3) δ 181.8 (d, 3JPC = 4.3 Hz)A, 181.7 (d, 3JPC = 4.3 Hz,)B, 166.7A, 166.0B, 155.8A, 155.7B, 143.9B, 143.8B, 143.8A, 141.4B, 141.3A, 141.3B, 141.3A, 127.8B, 127.7A, 127.7B, 127.6A, 127.1A, 127.1A, 127.1B, 127.0B, 125.1A, 125.0B, 124.9B, 120.1B, 120.0B, 119.9A, 66.8B, 66.6A, 63.5 (d, 2JPC = 6.6 Hz)A, 63.3 (d, 2JPC = 6.3 Hz)B, 62.7 (d, 2JPC = 6.4 Hz)A, 62.7 (d, 2JPC = 6.0 Hz)B, 54.9A, 54.2B, 52.4A, 52.3B, 48.8 (d, 2JPC = 3.3 Hz)A, 48.4 (d, 2JPC = 3.2 Hz)B, 47.2, 47.2, 42.4A, 42.3B, 38.62 (d, 1JPC = 201.8 Hz)A, 37.83 (d, 1JPC = 201.1, Hz)B, 28.5B, 28.4A, 24.6A, 24.5B, 23.4B, 23.2A, 21.8A, 21.6B, 16.4A+B, 15.2A, 15.1B ppm; 31P NMR (160 MHz, CDCl3) δ 6aB 16.9, 6aA 16.2 ppm; ESI-HRMS (CI) m/z calcd. for C33H47N3O7P ([M + H]+), 628.3152; found 628.3143.
(9H-Fluoren-9-yl)methyl ((S)-1-((2S,3S)-2-(tert-butylcarbamoyl)-3-(diethoxyphosphoryl)-2-methylaziridin-1-yl)-1-oxopropan-2-yl)carbamate and (9H-fluoren-9-yl)methyl ((S)-1-((2R,3R)-2-(tert-butylcarbamoyl)-3-(diethoxyphosphoryl)-2-methylaziridin-1-yl)-1-oxopropan-2-yl)carbamate (6b) (114 mg, 20%) yielded (50/50) of a mixture of two diastereoisomers, 6bA and 6bB, obtained as oils from amino acid 2s (405 mg, 1.3 mmol), tert-butyl isocyanide 3b (152 µL, 1.3 mmol) and 2H-azirine 1b (191 mg, 1 mmol), as described in the general procedure. The crude product was purified via flash column chromatography (SiO2, AcOEt/hexane 40:60) to give the title compound as a mixture of two diastereoisomers (6bA and 6bB). Rf: 0.60 (AcOEt); IR (neat) vmax 3450, 3278, 3075, 2925, 1682, 1651, 1288, 1196 cm–1;1H NMR (400 MHz, CDCl3) δ 7.76–7.74 (m, 4H, ArH), 7.59–7.57 (m, 4H, ArH), 7.41–7.37 (m, 4H, ArH), 7.33–7.27 (m, 4H, ArH), 6.01 (s, 1H, NH), 5.90 (s, 1H, NH), 5.47 (d, 3JHH = 7.7 Hz, 1H, NHFmoc), 5.33 (d, 3JHH = 7.8 Hz, 1H, NHFmoc), 4.48–4.32 (m, 16H, CHα + OCH2CH3 + CH2Fmoc + CHFmoc), 2.99 (d, 2JPC = 18.1 Hz, 1H, CH-P), 2.87 (d, 2JPC = 17.8 Hz, 1H, CH-P), 1.72 (s, 3H, CH3), 1.68 (s, 3H, CH3), 1.46 (d, 3JHH = 6.9 Hz, 3H, CH3), 1.41 (d, 3JHH = 7.0 Hz, 3H, CH3), 1.37–1.35 (m, 12H, OCH2CH3) 1.33 (s, 9H, tBu), 1.30 (s, 9H, tBu) ppm; 13C {1H} NMR (100 MHz, CDCl3) δ 182.1 (d, 3JPC = 4.4 Hz), 181.6 (d, 3JPC = 4.1 Hz), 166.7, 166.0, 155.7, 155.5, 155.5, 144.0, 143.9, 143.9, 141.5, 141.4, 141.3, 127.9, 127.8, 127.8, 127.7, 127.2, 127.1, 127.1, 127.1, 125.1, 125.1, 125.1, 124.9, 120.2, 120.1, 120.0, 120.0, 66.9, 66.8, 63.7 (d, 2JPC = 6.4 Hz), 63.5 (d, 2JPC = 6.3 Hz), 52.6, 52.5, 52.3, 51.7, 49.0, 48.3 (d, 2JPC = 3.1 Hz), 47.2, 38.8 (d, 1JPC = 202.7 Hz), 37.9 (d, 1JPC = 201.8 Hz), 28.6, 28.4, 19.5, 18.3, 16.5, 16.4, 16.4, 16.4, 16.3, 16.3, 16.3, 15.3, 15.0 ppm; 31P NMR (160 MHz, CDCl3) δ 16.8, 16.1 ppm; ESI-HRMS (CI) m/z calcd. for C30H41N3O7P ([M + H]+), 586.2682; found 586.2659.
3.6. Experimental Procedure and Characterization Data for Phosphorus Substituted Oxazole Derivatives 7
Method A: Corresponding Joullié–Ugi adduct 4 derived from phosphine oxide (1 eq.) was dissolved in 20 mL/mmol of acetonitrile, and BF3·OEt2 (1.2 eq.) was added. The solution was heated under microwave irradiation at 90 °C for 10 min. The solvent was evaporated, and the crude product was diluted with AcOEt, washed with saturated NaHCO3 (5 mL) and extracted with AcOEt (3 × 5 mL). The organic layer was dried over anhydrous MgSO4 and filtered, and the solvent was evaporated under reduced pressure. The resulting residue was purified via flash column chromatography (SiO2, hexanes/AcOEt) or via crystallization to yield the corresponding oxazole derivative. Method B: Corresponding Joullié–Ugi adduct 5 derived from phosphonate (1 eq.) was dissolved in 20 mL/mmol of chloroform, and BF3·OEt2 (3 eq.) was added. The solution was heated under microwave irradiation at 71 °C for 15 min. The solvent was evaporated, and the crude product was diluted with AcOEt, washed with saturated NaHCO3 (5 mL) and extracted with AcOEt (3 × 5 mL). The organic layer was dried over anhydrous MgSO4 and filtered, and the solvent was evaporated under reduced pressure. The resulting residue was purified via flash column chromatography (SiO2, hexanes/AcOEt) to yield the corresponding oxazole derivative.
(4S*,5S*)-N-Cyclohexyl-4-(diphenylphosphoryl)-5-methyl-2-phenyl-4,5-dihydrooxazole-5-carboxamide (7a) (105 mg, 94%) was obtained as a white solid from Joullié–Ugi adduct 4a (111 mg, 0.23 mmol) and BF3·OEt2 (35 µL, 0.28 mmol), as described in the general procedure in method A. The crude product was purified via flash column chromatography (SiO2, AcOEt/hexane 50:50) to give title compound 7a. mp 209–211 °C; IR (neat) vmax 3325, 2936, 1659, 1589, 1251, 1218, 1151 cm–1; 1H NMR (400 MHz, CDCl3) δ 8.15–8.10 (m, 2H, ArH), 7.97–7.90 (m, 4H, ArH), 7.56–7.40 (m, 9H, ArH), 6.91 (d, 3JHH = 7.9 Hz, 1H, HC-NH), 5.32 (d, 2JPC = 6.0 Hz, 1H, CH-P), 3.77–3.70 (m, 1H, HC-HN), 1.89–1.87 (m, 1H, cHex), 1.82–1.79 (m, 1H, cHex), 1.69–1.54 (m, 3H, cHex), 1.63 (s, 3H, CH3), 1.36–1.11 (m, 5H, cHex) ppm; 13C {1H} NMR (100 MHz, CDCl3) δ 171.8 (d, 3JPC = 9.4 Hz), 164.1 (d, 3JPC = 10.7 Hz), 132.9, 132.4, 132.2, 132.2, 132.1, 131.9, 131.9, 131.9, 131.8, 131.4, 131.3, 128.9, 128.8, 128.5, 128.5, 128.4, 128.3, 127.0 (d, 4JPC = 1.6 Hz), 88.7, 71.6 (d, 1JPC = 81.0 Hz), 48.3, 32.8, 32.7, 25.5, 24.7, 24.6, 21.4 (d, 3JPC = 6.9 Hz) ppm; 31P NMR (160 MHz, CDCl3) δ 25.6 ppm; ESI-HRMS (CI) m/z calculated for C29H32N2O3P ([M + H]+), 487.2151; found 487.2154.
(4S*,5S*)-N-Cyclohexyl-4-(diphenylphosphoryl)-5-methyl-2-(3-methylphenyl)-4,5-dihydrooxazole-5-carboxamide (7b) (36 mg, 60%) was obtained as a white solid from Joullié–Ugi adduct 4b (60 mg, 0.12 mmol) and BF3·OEt2 (18 µL, 0.144 mmol), as described in the general procedure in method A. The crude product was purified via flash column chromatography (SiO2, AcOEt/hexane 70:30) to give title compound 7b. mp 203–205 °C; IR (neat) vmax 3300, 3059, 1660, 1637, 1527, 1194 cm–1; 1H NMR (400 MHz, CDCl3) δ 8.17–8.12 (m, 2H, ArH), 7.96–7.94 (m, 2H, ArH), 7.79 (s, 2H, ArH), 7.58–7.35 (m, 8H, ArH), 6.94 (d, 3JHH = 8.1 Hz, 1H, HC-NH), 5.33 (d, 2JPC = 6.2 Hz, 1H CH-P), 3.77–3.75 (m, 1H, HC-NH), 2.39 (s, 3H, CH3), 1.92–1.81 (m, 2H, cHex), 1.73–1.54 (m, 6H, cHex + CH3), 1.37–1.36 (m, 2H cHex), 1.27–1.15 (m, 3H, cHex) ppm; 13C {1H} NMR (100 MHz, CDCl3) δ 171.7 (d, 3JPC = 9.5 Hz), 164.2 (d, 3JPC = 10.5 Hz), 138.3, 132.9, 132.8, 132.3, 132.1, 131.8, 131.8, 131.7, 131.3, 131.2, 128.9, 128.8, 128.7, 128.4, 128.4, 128.3, 126.8 (d, 4JPC = 1.6 Hz), 125.5, 88.5, 71.7 (d, 1JPC = 81 Hz), 48.2, 32.7, 32.7, 25.4, 24.6, 24.5, 21.3, 21.3 ppm; 31P NMR (160 MHz, CDCl3) δ 25.5 ppm; ESI-HRMS (CI) m/z calculated for C30H34N2O3P ([M + H]+), 501.2307; found 501.2303.
(4S*,5S*)-N-(tert-Butyl)-4-(diphenylphosphoryl)-5-methyl-2-phenyl-4,5-dihydrooxazole-5-carboxamide (7c) (63 mg, 92%) was obtained as a white solid from Joullié–Ugi adduct 4i (69 mg 0.15 mmol) and BF3·OEt2 (22 µL, 0.18 mmol), as described in the general procedure described in method A. The crude product was recrystallized from the diethyl ether–pentane mixture to give 7c. mp 206–207 °C; IR (neat) vmax 3306, 3075, 2920, 1662, 1629, 1549, 1196, 1118 cm–1; 1H NMR (400 MHz, CDCl3) δ 8.15–8.10 (m, 2H, ArH), 7.97–7.90 (m, 4H, ArH), 7.57–7.39 (m, 9H, ArH), 6.87 (s, 1H, NH), 5.32 (d, 2JPC = 6.2 Hz, 1H, CH-P), 1.61 (s, 3H, CH3), 1.33 (s, 9H, tBu) ppm; 13C {1H} NMR (100 MHz, CDCl3) δ 171.8 (d, 3JPC = 9.5 Hz), 164.1 (d, 3JPC = 10.7 Hz), 132.6 (d, 1JPC = 66.6 Hz), 132.9, 132.3, 132.1, 132.1, 132.1, 132.0, 131.8, 131.8, 131.8, 131.7, 131.3, 131.2, 128.9, 128.4, 128.5, 128.4, 128.3, 128.3, 127.0, 88.8, 71.4 (d, 1JPC = 80.7 Hz), 51.4, 28.7, 21.3 (d, 3JPC = 6.9 Hz) ppm; 31P NMR (160 MHz, CDCl3) δ 25.3 ppm; ESI-HRMS (CI) m/z calculated for C27H30N2O3P ([M + H]+), 461.1994; found 461.1995.
(4S*,5S*)-N-(tert-Butyl)-4-(diphenylphosphoryl)-5-methyl-2-(naphthalen-2-yl)-4,5-dihydrooxazole-5-carboxamide (7d) (74 mg, 91%) was obtained as a white solid from Joullié–Ugi adduct 4l (81 mg, 0.16 mmol) and BF3·OEt2 (25 µL, 0.20 mmol), as described in the general procedure in method A. The crude product was recrystallized from diethyl ether-pentane mixture to give title compound 7d. mp 199–202 °C; IR (neat) vmax 3297, 2929, 1667, 1521, 1193, 1117 cm–1; 1H NMR (400 MHz, CDCl3) δ 8.45 (s, 1H, ArH), 8.18–8.13 (m, 2H, ArH), 8.06–8.04 (m, 1H ArH), 8.01–7.96 (m, 2H, ArH), 7.92–7.86 (m, 3H, ArH), 7.57–7.42 (m, 8H, ArH), 7.02 (s, 1H, NH), 5.38 (d, 2JPH = 6.7 Hz, 1H, CH-P), 1.66 (s, 3H, CH3), 1.35 (s, 9H, tBu) ppm; 13C {1H} NMR (100 MHz, CDCl3) δ 171.7 (d, 3JPC = 9.1 Hz), 164.3 (d, 3JPC = 10.9 Hz), 135.1, 133.0, 132.7, 132.3, 132.3, 132.2, 132.0, 132.0, 131.9, 131.8, 131.4, 131.3, 131.2, 129.1, 129.0, 128.9, 128.5, 128.4, 128.1, 127.9, 126.9, 124.7, 124.3 (d, 4JPC = 1.6 Hz), 89.0, 71.7 (d, 1JPC = 80.6 Hz), 51.5, 28.7, 21.4 (d, 3JPC = 6.8 Hz) ppm; 31P NMR (160 MHz, CDCl3) δ 25.4 ppm; ESI-HRMS (CI) m/z calculated for C31H32N2O3P ([M + H]+), 511.2151; found 511.2134.
Diethyl ((4S*,5S*)-5-(tert-butylcarbamoyl)-5-methyl-2-phenyl-4,5-dihydrooxazol-5-yl)phosphonate (7e) (285 mg, 90%) was obtained as a white solid from Joullié–Ugi adduct 5a (316 mg, 0.80 mmol) and BF3·OEt2 (296 µL, 2.40 mmol), as described in the general procedure, method B. The crude product was purified via flash column chromatography (SiO2, AcOEt/hexane 65:35) to give title compound 7e. mp 99–102 °C; IR (neat) vmax 3294, 3067, 2975, 1665, 1637, 1538, 1252, 1213, 1074, 1052 cm–1; 1H NMR (400 MHz, CDCl3) δ 7.97–7.95 (m, 2H, ArH), 7.52 (d, 3JHH = 7.4 Hz, 1H, ArH), 7.44–7.41 (m, 2H, ArH), 6.31 (s, 1H, NH), 4.81 (d, 2JPC = 17.3 Hz, 1H, CH-P), 4.36–4.18 (m, 4H, OCH2CH3), 1.89 (d, 4JPH = 0.6, 3H, CH3), 1.39–1.34 (m, 6H, OCH2CH3), 1.31 (s, 9H, tBu) ppm; 13C {1H} NMR (100 MHz, CDCl3) δ 172.2 (d, 3JPC = 14.9 Hz), 163.7 (d, 3JPC = 12.0 Hz), 132.1, 128.6, 128.3, 127.1 (d, 4JPC = 2.6 Hz), 87.4, 69.9 (d, 1JPC = 160.4 Hz), 63.8 (d, 2JPC = 6.7 Hz), 62.8 (d, 2JPC = 7.4 Hz), 51.3, 28.7, 20.9 (d, 3JPC = 6.2 Hz), 16.5 (t, 3JPC = 5.6 Hz) ppm; 31P NMR (160 MHz, CDCl3) δ 18.5 ppm; ESI-HRMS (CI) m/z calculated for C19H30N2O5P ([M + H]+), 397.1892; found 397.1878.
Diethyl ((4S*,5S*)-5-(tert-butylcarbamoyl)-2-(4-fluorophenyl)-5-methyl-4,5-dihydrooxazol-4-yl)phosphonate (7f) (53 mg, 80%) was obtained as a yellowish oil obtained from Joullié–Ugi adduct 5b (66 mg, 0.16 mmol) and BF3·OEt2 (57 µL, 0.48 mmol), as described in the general procedure method B. The crude product was purified via flash column chromatography (SiO2, AcOEt/hexane 65:35) to give title compound 7f. Rf: 0.70 (AcOEt); IR (neat) vmax 3311, 3067, 2978, 1649, 1587, 1252, 1227, 1040 cm–1; 1H NMR (400 MHz, CDCl3) δ 8.00 (dd, 3JHH = 8.8 Hz, 4JHF = 5.4 Hz, 2H, ArH), 7.14 (t, 3JHH = 8.7 Hz, 3JHF = 8.7 Hz, 2H, ArH), 6.31 (s, 1H, NH), 4.82 (d, 2JPC = 17.3 Hz, 1H, CH-P), 4.40–4.19 (m, 4H, OCH2CH3), 1.91 (s, 3H, CH3), 1.45–1.35 (m, 6H, OCH2CH3), 1.34 (s, 9H, tBu) ppm; 13C {1H} NMR (100 MHz, CDCl3) δ 171.9 (d, 3JPC = 14.9 Hz), 165.1 (d, 1JCF = 253.2 Hz), 163.5 (d, 3JPC = 12.0 Hz), 130.6, 130.5, 123.2 (t, 4JCF = 2.9 Hz), 115.9, 115.7, 87.6 (d, 2JPC = 1.9 Hz), 69.7 (d, 1JPC = 160.7 Hz), 63.6 (d, 3JPC = 6.7 Hz), 62.8 (d, 2JPC = 7.3 Hz), 51.3, 28.6, 20.8 (d, 3JPC = 6.1 Hz), 16.5 (t, 3JPC = 5.6 Hz) ppm; 19F NMR (376 MHz, CDCl3) δ –106.8 ppm; 31P NMR (160 MHz, CDCl3) δ 18.4 ppm; ESI-HRMS (CI) m/z calculated for C19H29FN2O5P ([M + H]+), 415.1798; found 415.1771.
4. Conclusions
In conclusion, we developed a novel strategy to efficiently access to phosphorylated N-acylaziridines through the zinc chloride-catalyzed Joullié–Ugi three-component reaction of phosphorus-substituted 2H-azirines, carboxylic acids and isocyanides. Most of the JU-3CR proceeded smoothly in THF for a few hours, giving exclusive diastereoselectivity and satisfactory yields. This protocol was applicable to a wide range of substrates, including 2H-azirines derived from phosphine oxide and phosphonate, various aromatic and heteroaromatic, aliphatic, acrylic and propargylic acids, and isocyanide with different alkyl substitutions. Even N-Fmoc protected amino acids as carboxylic acid partners are well tolerated and phosphorylated aziridine peptidomimetics are achieved in a simple procedure. This strategy for the preparation of phosphorylated N-acylaziridines represents a valued method owing to the high degree of diastereoselectivity observed, the high atom economy and the reaction stages. The synthetic potential of this JU-3CR was established with the preparative-scale reaction and useful transformations of the JU-3CR adducts. The regio- and stereospecific ring expansion of N-acylaziridines to oxazole derivatives was accomplished in the presence of BF3·OEt2 as an efficient Lewis acid catalyst.
Supplementary Materials
The following supporting information can be downloaded at https://www.mdpi.com/article/10.3390/molecules29051023/s1: 1H, 13C{1H}, 19F, 31P NMR spectra of synthesized compounds 4, 5, 6 and 7; Figure S1: ORTEP diagram of compound 4i with thermal displacement parameters drawn at a 50% probability; Table S1: Crystal data and structure refinement for 4i; Figure S2: ORTEP diagram of compound 7a with thermal displacement parameters drawn at a 50% probability; Table S2: Crystal data and structure refinement for 7a.
Author Contributions
Conceptualization, F.P. and J.M.d.l.S.; methodology, J.A. and I.O.; validation, J.A. and I.O.; formal analysis J.A. and I.O.; investigation, J.A. and I.O.; resources, F.P. and J.M.d.l.S.; data curation, J.A. and A.M.O.d.R.; writing—original draft preparation, J.M.d.l.S.; writing—review and editing, I.O., A.M.O.d.R., F.P. and J.M.d.l.S.; visualization, J.A., I.O., A.M.O.d.R. and J.M.d.l.S.; supervision, A.M.O.d.R. and J.M.d.l.S.; project administration, A.M.O.d.R. and J.M.d.l.S.; funding acquisition, F.P. and J.M.d.l.S. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Ministerio de Ciencia, Innovación y Universidades (MCIU–Madrid) (PID2021-122558OB-I00, UE) and Gobierno Vasco (GV, IT1701-22; UPV-EHU).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this study are available in the Supplementary Materials file or on request from the corresponding author (1H, 13C, 19F, 31P, 2D-NMR and HRMS spectra).
Acknowledgments
The authors wish to thank the technical and human support provided by SGIker (UPV/EHU/ERDF, EU).
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Cioc, R.C.; Ruijter, E.; Orru, R.V.A. Multicomponent reactions: Advanced tools for sustainable organic synthesis. Green Chem. 2014, 16, 2958–2975. [Google Scholar] [CrossRef]
- Parvin, T. Multicomponent reactions using C,N-binucleophilic nature of aminopyrazoles: Construction of pyrazole-fused heterocycles. Top. Curr. Chem. 2023, 381, 19. [Google Scholar] [CrossRef] [PubMed]
- Borah, P.; Borah, G.; Nath, A.C.; Latif, W.; Banik, B.K. Facile multicomponent Mannich reaction towards biologically active compounds. ChemistrySelect 2023, 8, e202203758. [Google Scholar] [CrossRef]
- Ghashghaei, O.; Pedrola, M.; Escolano, C.; Lavilla, R. Heterocycles as inputs in MCRs: An update. In Multicomponent Reactions towards Heterocycles; van der Eycken, E., Sharma, U.K., Eds.; Wiley-VCH: Weinheim, Germany, 2022; pp. 1–43. [Google Scholar]
- John, S.E.; Gulati, S.; Shankaraiah, N. Recent advances in multi-component reactions and their mechanistic insights: A triennium review. Org. Chem. Front. 2021, 8, 4237–4287. [Google Scholar] [CrossRef]
- Younus, H.A.; Al-Rashida, M.; Hammed, A.; Uroos, M.; Salar, U.; Rana, S.; Khan, K.M. Multicomponent reactions (MCR) in medicinal chemistry: A patent review (2010–2020). Expert Opin. Ther. Pat. 2021, 31, 267–289. [Google Scholar] [CrossRef]
- Neochoritis, C.G.; Zhao, T.; Dömling, A. Tetrazoles via multicomponent reactions. Chem. Rev. 2019, 119, 1970–2042. [Google Scholar] [CrossRef]
- Zhu, J.; Wang, Q.; Wang, M.-X. (Eds.) Multicomponent Reactions in Organic Synthesis; Wiley-VCH: Weinheim, Germany, 2015. [Google Scholar]
- Dömling, A. Recent developments in isocyanide based multicomponent reactions in applied chemistry. Chem. Rev. 2006, 106, 17–89. [Google Scholar] [CrossRef]
- Graziano, G.; Stefanachi, A.; Contino, M.; Prieto-Díaz, R.; Ligresti, A.; Kumar, P.; Scilimati, A.; Sotelo, E.; Leonetti, F. Multicomponent reaction-assisted drug discovery: A time- and cost-effective green approach speeding up identification and optimization of anticancer drugs. Int. J. Mol. Sci. 2023, 24, 6581. [Google Scholar] [CrossRef] [PubMed]
- Cores, A.; Clerigué, J.; Orocio-Rodríguez, E.; Menéndez, J.C. Multicomponent reactions for the synthesis of active pharmaceutical ingredients. Pharmaceuticals 2022, 15, 1009. [Google Scholar] [CrossRef]
- Fouad, M.A.; Abdel-Hamid, H.; Ayoup, M.S. Two decades of recent advances of Ugi reactions: Synthetic and pharmaceutical applications. RSC Adv. 2020, 10, 42644–42681. [Google Scholar] [CrossRef] [PubMed]
- Ruijter, E.; Orru, R.V.A. Multicomponent reactions–opportunities for the pharmaceutical industry. Drug Discov. Today Technol. 2013, 10, e15–e20. [Google Scholar] [CrossRef]
- Bowers, M.M.; Caroll, P.; Joullié, M.M. Model studies directed toward the total synthesis of 14-membered cyclopeptide alkaloids: Synthesis of prolyl peptides via a four-component condensation. J. Chem. Soc. Perkin Trans. 1 1989, 857–865. [Google Scholar] [CrossRef]
- Nutt, R.F.; Joullié, M.M. Four-component condensation: A new versatile method for the synthesis of substituted prolyl peptides. J. Am. Chem. Soc. 1982, 104, 5852–5853. [Google Scholar] [CrossRef]
- Katsuyama, A.; Matsuda, A.; Ichikawa, A. Revisited mechanistic implications of the Joullié–Ugi three-component reaction. Org. Lett. 2016, 18, 2552–2555. [Google Scholar] [CrossRef]
- Nazeri, M.T.; Farhid, H.; Mohammadian, R.; Shaabani, A. Cyclic imines in Ugi and Ugi-type reactions. ACS Comb. Sci. 2020, 22, 361–400. [Google Scholar] [CrossRef]
- Gazzotti, S.; Rainoldi, G.; Silvani, A. Exploitation of the Ugi–Joullié reaction in drug discovery and development. Expert Opin. Drug Discov. 2019, 17, 639–652. [Google Scholar] [CrossRef]
- Katsuyama, A.; Yakushiji, F.; Ichikawa, S. Total synthesis of plusbacin A3 and its dideoxy derivative using a solvent-dependent diastereodivergent Joullié–Ugi three-component reaction. J. Org. Chem. 2018, 83, 7085–7101. [Google Scholar] [CrossRef]
- Chapman, T.M.; Davies, I.G.; Gu, B.; Block, T.M.; Scopes, D.I.C.; Hay, P.A.; Courtney, S.M.; McNeill, L.A.; Schofield, C.J.; Davies, B.G. Glyco- and peptidomimetics from three-component Joullié–Ugi coupling show selective antiviral activity. J. Am. Chem. Soc. 2005, 127, 506–507. [Google Scholar] [CrossRef][Green Version]
- Boike, L.; Henning, N.J.; Nomura, D.K. Advances in covalent drug discovery. Nat. Rev. Drug Discov. 2022, 21, 881–898. [Google Scholar] [CrossRef]
- Sutanto, F.; Konstantinidou, M.; Dömling, A. Covalent inhibitors: A rational approach to drug discovery. RSC Med. Chem. 2020, 11, 876–884. [Google Scholar] [CrossRef]
- Bauer, R.A. Covalent inhibitors in drug discovery: From accidental discoveries to avoided liabilities and designed therapies. Drug Discov. Today 2015, 20, 1061–1073. [Google Scholar] [CrossRef] [PubMed]
- Gehringer, M.; Laufer, S.A. Emerging and re-emerging warheads for targeted covalent inhibitors: Applications in medicinal chemistry and chemical biology. J. Med. Chem. 2019, 62, 5673–5724. [Google Scholar] [CrossRef] [PubMed]
- Vaidergorn, M.M.; Carneiro, Z.A.; Lopes, C.D.; de Albuquerque, S.; Reis, F.C.C.; Nikolaou, S.; e Mello, J.F.R.; Genesi, G.L.; Trossini, G.H.G.; Ganesan, A.; et al. β-Amino alcohols and their respective 2-phenyl-N-alkyl aziridines as potential DNA minor groove binders. Eur. J. Med. Chem. 2018, 157, 657–664. [Google Scholar] [CrossRef]
- Wakaki, S.; Marumo, H.; Tomioka, K.; Shimizu, G.; Kato, E.; Kamada, H.; Kudo, S.; Fujimoto, Y. Isolation of new fractions of antitumor mitomycins. J. Antibiot. 1958, 8, 228–235. [Google Scholar]
- Nagaoka, K.; Matsumoto, M.; Ono, J.; Yokoi, K.; Ishizeki, S.; Nakashima, T. Azinomycins A and B, new antitumor antibiotics. I. Producing organism, fermentation, isolation, and characterization. J. Antibiot. 1986, 39, 1527–1532. [Google Scholar] [CrossRef] [PubMed]
- Ismail, F.M.D.; Levitsky, D.O.; Dembitsky, V.M. Aziridine alkaloids as potential therapeutic agents. Eur. J. Med. Chem. 2009, 44, 3373–3387. [Google Scholar] [CrossRef] [PubMed]
- Sakurai, T.; Okamoto, Y.; Fukuda, M. 1-Alkoxycarbonyl-2-aziridinephosphonates. Jp54012364, 1979. Chem. Abstr. 1979, 91, 20707. [Google Scholar]
- Carramiñana, V.; Ochoa de Retana, A.M.; Vélez del Burgo, A.; de los Santos, J.M.; Palacios, F. Synthesis and biological evaluation of cyanoaziridine phosphine oxides and phosphonates with antiproliferative activity. Eur. J. Med. Chem. 2019, 163, 736–746. [Google Scholar] [CrossRef]
- Carramiñana, V.; Ochoa de Retana, A.M.; Palacios, F.; de los Santos, J.M. Synthesis and antiproliferative activity of phosphorus substituted 4-cyanooxazolines, 2-aminocyanooxazolines, 2-iminocyanooxazolidines and 2-aminocyanothiazolines by rearrangement of cyanoaziridines. Molecules 2021, 26, 4265. [Google Scholar] [CrossRef]
- Thakur, A.; Verma, M.; Bharti, R.; Sharma, R. Oxazole and isoxazole: From one-pot synthesis to medical applications. Tetrahedron 2022, 119, 132813. [Google Scholar] [CrossRef]
- Mucha, A.; Kafarski, P.; Berlicki, L. Remarkable potential of the α-aminophosphonate/phosphinate structural motif in medicinal chemistry. J. Med. Chem. 2011, 54, 5955–5980. [Google Scholar] [CrossRef]
- Brusnakov, M.; Golovchenko, O.; Velihina, Y.; Liavynets, O.; Zhirnov, V.; Brovarets, V. Evaluation of anticancer activity of 1,3-oxazol-4-ylphosphonium salts in vitro. ChemMedChem 2022, 17, e20220031. [Google Scholar] [CrossRef] [PubMed]
- Abdurakhmanova, E.R.; Brusnakov, M.Y.; Golovchenko, O.V.; Pilyo, S.G.; Velychko, N.V.; Harden, E.A.; Prichard, M.N.; James, S.H.; Zhirnov, V.V.; Brovarets, V.S. Synthesis and in vitro anticytomegalovirus activity of 5-hydroxyalkylamino-1,3-oxazoles derivatives. Med. Chem. Res. 2020, 29, 1669–1675. [Google Scholar] [CrossRef]
- Szczesniak, P.; Maziarz, E.; Stecko, S.; Furman, B. Synthesis of polyhydroxylated piperidine and pyrrolidine peptidomimetics via one-pot sequential lactam reduction/Joullié–Ugi reaction. J. Org. Chem. 2015, 80, 3621–3633. [Google Scholar] [CrossRef]
- Gulevich, A.V.; Shevchenko, N.E.; Balenkova, E.S.; Röschenthaler, G.-V.; Nenajdenko, V.G. Efficient multicomponent synthesis of α-trifluoromethyl proline, homoproline, and azepan carboxylic acid dipeptides. Synlett 2009, 403–406. [Google Scholar] [CrossRef]
- Rainoldi, G.; Begnini, F.; de Munnik, M.; Lo Presti, L.; Vande Velde, C.M.L.; Orru, R.; Lesma, G.; Ruijter, E.; Silvani, A. Sequential multicomponent strategy for the diastereoselective synthesis of densely functionalized spirooxindole-fused thiazolidines. ACS Comb. Sci. 2018, 20, 98–105. [Google Scholar] [CrossRef] [PubMed]
- Alfano, A.I.; Buommino, E.; Ferraro, M.G.; Irace, C.; Zampella, A.; Lange, H.; Brindisi, M. Coupling interrupted Fischer and multicomponent Joullié–Ugi to chase chemical diversity: From batch to sustainable flow synthesis of peptidomimetics. ChemMedChem 2021, 16, 3795–3809. [Google Scholar] [CrossRef] [PubMed]
- Golubev, P.; Krasavin, M. Sterically constrained and encumbered: An approach to the naturally occurring peptidomimetic tetrahydropyrazino [1,2-a]-indole-1,4-dione core. Eur. J. Org. Chem. 2017, 2017, 1740–1744. [Google Scholar] [CrossRef]
- Maison, W.; Lützen, A.; Kosten, M.; Schlemminger, I.; Westerhoff, O.; Saak, W.; Martens, J. Multicomponent synthesis of tripeptides containing pipecolic acid derivatives: Selective induction of cis- and trans-imide bonds into peptide backbones. J. Chem. Soc. Perkin Trans. 1 2000, 1867–1871. [Google Scholar] [CrossRef]
- Pinna, A.; Basso, A.; Lambruschini, C.; Moni, L.; Riva, R.; Rocca, V.; Banfi, L. Stereodivergent access to all four stereoisomers of chiral tetrahydrobenzo[f][1,4]oxazepines, through highly diastereoselective multicomponent Ugi–Joullié reaction. RSC Adv. 2020, 10, 965–972. [Google Scholar] [CrossRef]
- Angyal, A.; Demjén, A.; Wéber, E.; Kovács, A.K.; Wölfling, J.; Puskás, L.G.; Kanizsai, I. Lewis acid-catalyzed diastereoselective synthesis of multisubstituted N-acylaziridine-2-carboxamides from 2H-azirines via Joullié–Ugi three-component reaction. J. Org. Chem. 2018, 83, 3570–3581. [Google Scholar] [CrossRef]
- Nikbakht, A.; Mohammadi, F.; Mousavi, M.S.; Amiri, K.; Balalai, S.; Rominger, F.; Bijanzadeh, H.R. A domino approach for the synthesis of 4-carboxamide oxazolines from azirines. Synthesis 2021, 53, 4654–4661. [Google Scholar]
- Carramiñana, V.; Ochoa de Retana, A.M.; de los Santos, J.M.; Palacios, F. First synthesis of merged hybrids phosphorylated azirino[2,1-b]benzo[e][1,3]oxazine derivatives as anticancer agents. Eur. J. Med. Chem. 2020, 185, 111771. [Google Scholar] [CrossRef] [PubMed]
- Carramiñana, V.; Ochoa de Retana, A.M.; Palacios, F.; de los Santos, J.M. Synthesis of α-aminophosphonic acid derivatives through the addition of O- and S-nucleophiles to 2H-azirines and their antiproliferative effect on A549 human lung adenocarcinoma cells. Molecules 2020, 25, 3332. [Google Scholar] [CrossRef]
- Suzuki, I.; Takenaka, Y.; Morishita, Y.; Shibata, I. One-step preparation of N-unprotected aziridines from 2H-azirines by addition of ketene silyl acetals catalyzed by Lewis acids. Chem. Lett. 2022, 51, 9–12. [Google Scholar] [CrossRef]
- Timén, A.S.; Somfai, P. Investigation of Lewis acid-catalyzed asymmetric aza-Diels–Alder reactions of 2H-azirines. J. Org. Chem. 2003, 68, 9958–9963. [Google Scholar] [CrossRef] [PubMed]
- Palacios, F.; Ochoa de Retana, A.M.; Gil, J.I.; López de Munain, R. Synthesis of pyrazine-phosphonates and -phosphine oxides from 2H-azirines or oximes. Org. Lett. 2002, 4, 2405–2408. [Google Scholar] [CrossRef] [PubMed]
- Palacios, F.; Aparicio, D.; Ochoa de Retana, A.M.; de los Santos, J.M.; Gil, J.I.; Alonso, J.M. Asymmetric synthesis of 2H-azirines derived from phosphine oxides using solid-supported amines. Ring opening of azirines with carboxylic acids. J. Org. Chem. 2002, 67, 7283–7288. [Google Scholar] [CrossRef] [PubMed]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Cryst. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Sheldrick, G.M. A Short History of SHELX. Acta Cryst. 2008, A64, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. Crystal Structure Refinement with SHELXL. Acta Cryst. 2015, C71, 3–8. [Google Scholar]
- Foglia, T.A.; Gregory, L.M.; Maerker, G. Stereochemistry of the isomerization of N-acyl-2,3-disubstituted aziridines to Δ2-oxazolines. J. Org. Chem. 1970, 35, 3779–3785. [Google Scholar] [CrossRef]
- Heine, H.W.; Kaplan, M.S. Aziridines. XVI. Isomerization of some 1-aroyl-aziridines. J. Org. Chem. 1967, 32, 3069–3074. [Google Scholar] [CrossRef]
- Heine, H.W.; King, D.C.; Portland, L.A. Aziridines. XII. The isomerization of some cis- and trans-1-p-nitrobenzoyl-2,3-substituted aziridines. J. Org. Chem. 1966, 31, 2662–2665. [Google Scholar] [CrossRef]
- Heine, H.W.; Kenyon, W.G.; Johnson, E.M. The isomerization and dimerization of aziridine derivatives. IV. J. Am. Chem. Soc. 1961, 83, 2570–2574. [Google Scholar] [CrossRef]
- Palacios, F.; Ochoa de Retana, A.M.; Gil, J.I.; Ezpeleta, J.M. Simple asymmetric synthesis of 2H-azirines derived from phosphine oxides. J. Org. Chem. 2000, 65, 3213–3217. [Google Scholar] [CrossRef]
- Palacios, F.; Ochoa de Retana, A.M.; Gil, J.I. Easy and efficient synthesis of enantiomerically enriched 2H-azirines derived from phosphonates. Tetrahedron Lett. 2000, 41, 5363–5366. [Google Scholar] [CrossRef]












Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).