
Mechanism of Mutation-Induced Effects on the Catalytic Function of TEV Protease: A Molecular Dynamics Study


Abstract
:
1. Introduction
2. Results
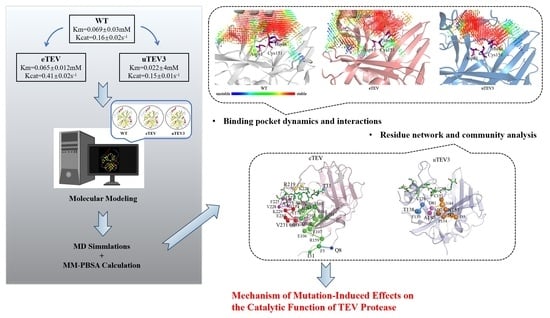
2.1. Overview of TEVp Structures and MD Simulations
2.2. Investigation of Binding Pocket Dynamics and Interactions
2.3. Residue Network and Community Analysis
3. Discussion
4. Materials and Methods
4.1. Structure Preparation
4.2. Molecular Dynamic Simulation
4.3. Binding Energy Calculations
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Beygmoradi, A.; Homaei, A.; Hemmati, R.; Fernandes, P. Recombinant protein expression: Challenges in production and folding related matters. Int. J. Biol. Macromol. 2023, 233, 123407. [Google Scholar] [CrossRef]
- Sun, M.; Gao, A.X.; Liu, X.; Yang, Y.; Ledesma-Amaro, R.; Bai, Z. High-throughput process development from gene cloning to protein production. Microb. Cell Factories 2023, 22, 182. [Google Scholar] [CrossRef]
- Cesaratto, F.; Burrone, O.R.; Petris, G. Tobacco Etch Virus protease: A shortcut across biotechnologies. J. Biotechnol. 2016, 231, 239–249. [Google Scholar] [CrossRef]
- Nunn, C.M.; Jeeves, M.; Cliff, M.J.; Urquhart, G.T.; George, R.R.; Chao, L.H.; Tscuchia, Y.; Djordjevic, S. Crystal structure of tobacco etch virus protease shows the protein C terminus bound within the active site. J. Mol. Biol. 2005, 350, 145–155. [Google Scholar] [CrossRef]
- Raran-Kurussi, S.; Cherry, S.; Zhang, D.; Waugh, D.S. Removal of Affinity Tags with TEV Protease. Methods Mol. Biol. 2017, 1586, 221–230. [Google Scholar] [PubMed]
- Miladi, B.; Bouallagui, H.; Dridi, C.; El Marjou, A.; Boeuf, G.; Di Martino, P.; Dufour, F.; Elm’Selmi, A. A new tagged-TEV protease: Construction, optimisation of production, purification and test activity. Protein Expr. Purif. 2011, 75, 75–82. [Google Scholar] [CrossRef]
- Wehr, M.C.; Rossner, M.J. Split protein biosensor assays in molecular pharmacological studies. Drug Discov. Today 2016, 21, 415–429. [Google Scholar] [CrossRef] [PubMed]
- Renna, P.; Ripoli, C.; Dagliyan, O.; Pastore, F.; Rinaudo, M.; Re, A.; Paciello, F.; Grassi, C. Engineering a switchable single-chain TEV protease to control protein maturation in living neurons. Bioeng. Transl. Med. 2022, 7, e10292. [Google Scholar] [CrossRef]
- Rawlings, N.D.; Waller, M.; Barrett, A.J.; Bateman, A. MEROPS: The database of proteolytic enzymes, their substrates and inhibitors. Nucleic Acids Res. 2014, 42, D503–D509. [Google Scholar] [CrossRef] [PubMed]
- Kostallas, G.; Löfdahl, P.Å.; Samuelson, P. Substrate profiling of tobacco etch virus protease using a novel fluorescence-assisted whole-cell assay. PLoS ONE 2011, 6, e16136. [Google Scholar] [CrossRef] [PubMed]
- Parks, T.D.; Howard, E.D.; Wolpert, T.J.; Arp, D.J.; Dougherty, W.G. Expression and purification of a recombinant tobacco etch virus NIa proteinase: Biochemical analyses of the full-length and a naturally occurring truncated proteinase form. Virology 1995, 210, 194–201. [Google Scholar] [CrossRef] [PubMed]
- van den Berg, S.; Löfdahl, P.A.; Härd, T.; Berglund, H. Improved solubility of TEV protease by directed evolution. J. Biotechnol. 2006, 121, 291–298. [Google Scholar] [CrossRef]
- Kapust, R.B.; Tözsér, J.; Fox, J.D.; Anderson, D.E.; Cherry, S.; Copeland, T.D.; Waugh, D.S. Tobacco etch virus protease: Mechanism of autolysis and rational design of stable mutants with wild-type catalytic proficiency. Protein Eng. 2001, 14, 993–1000. [Google Scholar] [CrossRef]
- Cabrita, L.D.; Gilis, D.; Robertson, A.L.; Dehouck, Y.; Rooman, M.; Bottomley, S.P. Enhancing the stability and solubility of TEV protease using in silico design. Protein Sci. 2007, 16, 2360–2367. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhu, G.F.; Ren, S.Y.; Han, Y.G.; Luo, Y.; Du, L.F. Insight into the structural stability of wild type and mutants of the tobacco etch virus protease with molecular dynamics simulations. J. Mol. Model. 2013, 19, 4865–4875. [Google Scholar] [CrossRef] [PubMed]
- Denard, C.A.; Paresi, C.; Yaghi, R.; McGinnis, N.; Bennett, Z.; Yi, L.; Georgiou, G.; Iverson, B.L. YESS 2.0, a Tunable Platform for Enzyme Evolution, Yields Highly Active TEV Protease Variants. ACS Synth. Biol. 2021, 10, 63–71. [Google Scholar] [CrossRef]
- Sanchez, M.I.; Ting, A.Y. Directed evolution improves the catalytic efficiency of TEV protease. Nat. Methods. 2020, 17, 167–174. [Google Scholar] [CrossRef]
- Chen, Z.; Zhang, X.; Peng, C.; Wang, J.; Xu, Z.; Chen, K.; Shi, J.; Zhu, W. D3Pockets: A Method and Web Server for Systematic Analysis of Protein Pocket Dynamics. J. Chem. Inf. Model. 2019, 59, 3353–3358. [Google Scholar] [CrossRef]
- Schöning-Stierand, K.; Diedrich, K.; Ehrt, C.; Flachsenberg, F.; Graef, J.; Sieg, J.; Penner, P.; Poppinga, M.; Ungethüm, A.; Rarey, M. ProteinsPlus: A comprehensive collection of web-based molecular modeling tools. Nucleic Acids Res. 2022, 50, W611–W615. [Google Scholar] [CrossRef]
- Felline, A.; Seeber, M.; Fanelli, F. webPSN v2.0: A webserver to infer fingerprints of structural communication in biomacromolecules. Nucleic Acids Res. 2020, 48, W94–W103. [Google Scholar] [CrossRef]
- Hu, J.; Chen, Y.; Ren, Y.; Xiao, W.; Hu, Y.; Yu, X.; Fan, J. Combination of the mutations for improving activity of TEV protease in inclusion bodies. Bioprocess. Biosyst. Eng. 2021, 44, 2129–2139. [Google Scholar] [CrossRef] [PubMed]
- Yi, L.; Gebhard, M.C.; Li, Q.; Taft, J.M.; Georgiou, G.; Iverson, B.L. Engineering of TEV protease variants by yeast ER sequestration screening (YESS) of combinatorial libraries. Proc. Natl. Acad. Sci. USA 2013, 110, 7229–7734. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Cai, X.; Qi, Z.; Rong, L.; Cheng, B.; Fan, J. In vivo and in vitro characterization of TEV protease mutants. Protein Expr. Purif. 2012, 83, 157–163. [Google Scholar] [CrossRef] [PubMed]
- Nam, H.; Hwang, B.J.; Choi, D.Y.; Shin, S.; Choi, M. Tobacco etch virus (TEV) protease with multiple mutations to improve solubility and reduce self-cleavage exhibits enhanced enzymatic activity. FEBS Open Bio. 2020, 10, 619–626. [Google Scholar] [CrossRef] [PubMed]
- Zlobin, A.; Golovin, A. Between Protein Fold and Nucleophile Identity: Multiscale Modeling of the TEV Protease Enzyme-Substrate Complex. ACS Omega 2022, 7, 40279–40292. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Vishweshwaraiah, Y.L.; Dokholyan, N.V. Design and engineering of allosteric communications in proteins. Curr. Opin. Struct. Biol. 2022, 73, 102334. [Google Scholar] [CrossRef]
- Wang, J.; Jain, A.; McDonald, L.R.; Gambogi, C.; Lee, A.L.; Dokholyan, N.V. Mapping allosteric communications within individual proteins. Nat. Commun. 2020, 11, 3862. [Google Scholar] [CrossRef]
- Felline, A.; Seeber, M.; Fanelli, F. PSNtools for standalone and web-based structure network analyses of conformational ensembles. Comput. Struct. Biotechnol. J. 2022, 20, 640–649. [Google Scholar] [CrossRef]
- Song, Y.; DiMaio, F.; Wang, R.Y.; Kim, D.; Miles, C.; Brunette, T.; Thompson, J.; Baker, D. High-resolution comparative modeling with RosettaCM. Structure 2013, 21, 1735–1742. [Google Scholar] [CrossRef]
- Raman, S.; Vernon, R.; Thompson, J.; Tyka, M.; Sadreyev, R.; Pei, J.; Kim, D.; Kellogg, E.; DiMaio, F.; Lange, O.; et al. Structure prediction for CASP8 with all-atom refinement using Rosetta. Proteins 2009, 77 (Suppl. S9), 89–99. [Google Scholar] [CrossRef]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef] [PubMed]
- MacKerell, A.D., Jr.; Banavali, N.; Foloppe, N. Development and current status of the CHARMM force field for nucleic acids. Biopolymers 2000, 56, 257–265. [Google Scholar] [CrossRef]
- Nayar, D.; Agarwal, M.; Chakravarty, C. Comparison of Tetrahedral Order, Liquid State Anomalies, and Hydration Behavior of mTIP3P and TIP4P Water Models. J. Chem. Theory Comput. 2011, 7, 3354–3367. [Google Scholar] [CrossRef] [PubMed]
- Kumari, R.; Kumar, R.; Open Source Drug Discovery Consortium; Lynn, A. g_mmpbsa—A GROMACS tool for high-throughput MM-PBSA calculations. J. Chem. Inf. Model. 2014, 54, 1951–1962. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Enzyme | Mutation Sites | Km (mM) | Kcat (s−1) | Kcat/Km (mM−1s−1) |
|---|---|---|---|---|
| WT | — | 0.069 ± 0.03 | 0.16 ± 0.02 | 2.23 ± 1.02 [16] |
| eTEV | S3I, P8Q, S31T, T173A, V219R, A231V | 0.065 ± 0.012 | 0.41 ± 0.02 | 6.31 ± 1.2 [16] |
| uTEV3 | I138T, S153N, T180A | 0.022 ± 4 | 0.15 ± 0.01 | 6.82 [17] |
| WT (Residue)– Peptide (Residue) | eTEV (Residue)– Peptide (Residue) | uTEV3 (Residue)– Peptide (Residue) | |
|---|---|---|---|
| Hydrogen Bonds | Lys141-Glu2 | His214-Glu2 | His214-Glu2 |
| Tyr178-Glu2 | Ser170-Leu4 | Lys215-Glu2 | |
| His214-Glu2 | Lys215-Leu4 | Asn171-Asn3 | |
| Asn171-Asn3 | Phe217-Leu4 | Phe172-Asn3 | |
| Phe172-Asn3 | Ala169-Tyr5 | Lys215-Asn3 | |
| Ser170-Leu4 | Ser170-Tyr5 | Ser170-Leu4 | |
| Lys215-Leu4 | Asn174-Tyr5 | Lys215-Leu4 | |
| Val216-Leu4 | Phe217-Tyr5 | Val216-Leu4 | |
| Phe217-Leu4 | Ser168-Phe6 | Phe217-Leu4 | |
| Ala169-Tyr5 | Phe217-Phe6 | Ala169-Tyr5 | |
| Ser170-Tyr5 | Gly149-Gln7 * | Ser170-Tyr5 | |
| Asn174-Tyr5 | Cys151-Gln7 * | Asn174-Tyr5 | |
| Phe217-Tyr5 | His167-Gln7 * | Phe217-Tyr5 | |
| Ser168-Phe6 | Ser168-Gln7 * | Phe217-Phe6 | |
| Phe217-Phe6 | Ser170-Gln7 * | Ser168-Phe6 | |
| His46-Gln7 * | Thr31-Ser8 * | Thr146-Gln7 * | |
| Ser168-Gln7 * | Thr31-Gly9 | Gly149-Gln7 * | |
| Cys151-Ser8 * | Gly149-Gly9 | Gln150-Gln7 * | |
| Ser31-Gly9 | Cys151-Gln7 * | ||
| Gly149-Gly9 | His167-Gln7 * | ||
| Ser168-Gln7 * | |||
| Ser31-Ser8 * | |||
| Gly149-Ser8 * | |||
| Ser31-Gly9 | |||
| Hydrophobic Interactions | Ala169-Leu4 | His214-Glu2 | Ala169-Leu4 |
| Tyr178-Leu4 | Ala169-Leu4 | Tyr178-Leu4 | |
| His214-Leu4 | Tyr178-Leu4 | Val216-Leu4 | |
| Val216-Leu4 | Val216-Leu4 | Lys220-Tyr5 | |
| Lys220-Tyr5 | Lys220-Tyr5 | Phe225-Tyr5 | |
| His46-Phe6 | His46-Phe6 | His46-Phe6 | |
| Val216-Phe6 | Ala169-Phe6 | Ala169-Phe6 | |
| Val216-Phe6 | Val216-Phe6 |
| System | WT | eTEV | uTEV3 |
|---|---|---|---|
| ΔGgas | −255.39 ± 2.29 | −267.71 ± 0.86 | −240.72 ± 0.59 |
| ΔGsolv | 188.58 ± 1.70 | 202.19 ± 0.79 | 169.44 ± 0.57 |
| Δtotal | −66.81 ± 0.63 | −65.51 ± 0.24 | −71.28 ± 0.20 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, J.; Xu, Y.; Wang, X.; Li, J.; Hua, Z. Mechanism of Mutation-Induced Effects on the Catalytic Function of TEV Protease: A Molecular Dynamics Study. Molecules 2024, 29, 1071. https://doi.org/10.3390/molecules29051071
Wang J, Xu Y, Wang X, Li J, Hua Z. Mechanism of Mutation-Induced Effects on the Catalytic Function of TEV Protease: A Molecular Dynamics Study. Molecules. 2024; 29(5):1071. https://doi.org/10.3390/molecules29051071
Chicago/Turabian StyleWang, Jingyao, Yicong Xu, Xujian Wang, Jiahuang Li, and Zichun Hua. 2024. "Mechanism of Mutation-Induced Effects on the Catalytic Function of TEV Protease: A Molecular Dynamics Study" Molecules 29, no. 5: 1071. https://doi.org/10.3390/molecules29051071
APA StyleWang, J., Xu, Y., Wang, X., Li, J., & Hua, Z. (2024). Mechanism of Mutation-Induced Effects on the Catalytic Function of TEV Protease: A Molecular Dynamics Study. Molecules, 29(5), 1071. https://doi.org/10.3390/molecules29051071