
Metal Complexes of Redox Non-Innocent Ligand N,N′-Bis(3,5-di-tertbutyl-2-hydroxy-phenyl)-1,2-phenylenediamine


Abstract
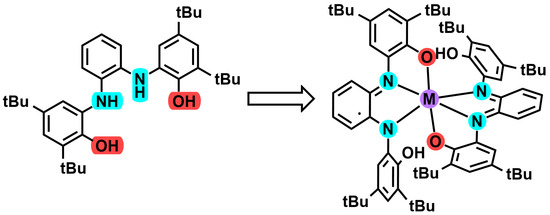
:
1. Introduction
2. Metal Complexes of N,N′-Bis(3,5-di-tert-butyl-2-hydroxyphenyl)-1,2-phenylenediamine
2.1. Group 4 Complexes: Ti, Zr, Hf
2.2. Group 5 and 6 Complexes: V, Mo, W
2.3. Group 7–10 Complexes: Mn, Fe, Co, Ni
2.4. Group 11,12,14 Complexes: Cu, Zn, Sn
2.5. Actinoids: U
2.6. Metalloids: P, Te
3. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Jørgensen, C.K. Differences between the Four Halide Ligands, and Discussion Remarks on Trigonal-Bipyramidal Complexes, on Oxidation States, and on Diagonal Elements of One-Electron Energy. Coord. Chem. Rev. 1966, 1, 164–178. [Google Scholar] [CrossRef]
- Berben, L.A.; De Bruin, B.; Heyduk, A.F. Non-Innocent Ligands. Chem. Commun. 2015, 51, 1553–1554. [Google Scholar] [CrossRef] [PubMed]
- Kaim, W. Chelate Rings of Different Sizes with Non-Innocent Ligands. Dalton Trans. 2019, 48, 8521–8529. [Google Scholar] [CrossRef] [PubMed]
- Kaim, W. “Guilty” Verdict-Evidence for the Noninnocence of Cyanide. Angew. Chem. Int. Ed. 2011, 50, 10498–10500. [Google Scholar] [CrossRef] [PubMed]
- Kaim, W.; Schwederski, B. Non-Innocent Ligands in Bioinorganic Chemistry—An Overview. Coord. Chem. Rev. 2010, 254, 1580–1588. [Google Scholar] [CrossRef]
- Broere, D.L.J.; Plessius, R.; Van Der Vlugt, J.I. New Avenues for Ligand-Mediated Processes-Expanding Metal Reactivity by the Use of Redox-Active Catechol, o-Aminophenol and o-Phenylenediamine Ligands. Chem. Soc. Rev. 2015, 44, 6886–6915. [Google Scholar] [CrossRef] [PubMed]
- Luca, O.R.; Crabtree, R.H. Redox-Active Ligands in Catalysis. Chem. Soc. Rev. 2013, 42, 1440–1459. [Google Scholar] [CrossRef]
- Demir, S.; Jeon, I.R.; Long, J.R.; Harris, T.D. Radical Ligand-Containing Single-Molecule Magnets. Coord. Chem. Rev. 2015, 289–290, 149–176. [Google Scholar] [CrossRef]
- D’Alessandro, D.M. Exploiting Redox Activity in Metal-Organic Frameworks: Concepts, Trends and Perspectives. Chem. Commun. 2016, 52, 8957–8971. [Google Scholar] [CrossRef]
- Praneeth, V.K.K.; Ringenberg, M.R.; Ward, T.R. Redox-Active Ligands in Catalysis. Angew. Chem. Int. Ed. 2012, 51, 10228–10234. [Google Scholar] [CrossRef]
- Pashanova, K.I.; Poddel’sky, A.I.; Piskunov, A.V. Complexes of “Late” Transition Metals of the 3d Row Based on Functionalized o-Iminobenzoquinone Type Ligands: Interrelation of Molecular and Electronic Structure, Magnetic Behaviour. Coord. Chem. Rev. 2022, 459, 214399. [Google Scholar] [CrossRef]
- Poddel’sky, A.I.; Cherkasov, V.K.; Abakumov, G.A. Transition Metal Complexes with Bulky 4,6-Di-Tert-Butyl-N-Aryl(Alkyl)-o-Iminobenzoquinonato Ligands: Structure, EPR and Magnetism. Coord. Chem. Rev. 2009, 253, 291–324. [Google Scholar] [CrossRef]
- Verani, C.N.; Gallert, S.; Bill, E.; Weyhermüller, T.; Wieghardt, K.; Chaudhuri, P. [Tris(o-Iminosemiquinone)Cobalt(III)]—A Radical Complex with an St = 3/2 Ground State. Chem. Commun. 1999, 1747–1748. [Google Scholar] [CrossRef]
- Brown, S.N. Metrical Oxidation States of 2-Amidophenoxide and Catecholate Ligands: Structural Signatures of Metal-Ligand π Bonding in Potentially Noninnocent Ligands. Inorg. Chem. 2012, 51, 1251–1260. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, R. Assigning Ligand Redox Levels in Complexes of 2-Aminophenolates: Structural Signatures. Inorg. Chem. 2020, 59, 12961–12977. [Google Scholar] [CrossRef] [PubMed]
- Chaudhuri, P.; Hess, M.; Müller, J.; Hildenbrand, K.; Bill, E.; Weyhermüller, T.; Wieghardt, K. Aerobic Oxidation of Primary Alcohols (Including Methanol) by Copper(II)− and Zinc(II)−Phenoxyl Radical Catalysts. J. Am. Chem. Soc. 1999, 121, 9599–9610. [Google Scholar] [CrossRef]
- Zelikoff, A.L.; Kopilov, J.; Goldberg, I.; Coates, G.W.; Kol, M. New Facets of an Old Ligand: Titanium and Zirconium Complexes of Phenylenediamine Bis(Phenolate) in Lactide Polymerisation Catalysisf. Chem. Commun. 2009, 6804–6806. [Google Scholar] [CrossRef]
- Liu, X.H.; Yu, H.Y.; Huang, J.Y.; Su, J.H.; Xue, C.; Zhou, X.T.; He, Y.R.; He, Q.; Xu, D.J.; Xiong, C.; et al. Biomimetic Catalytic Aerobic Oxidation of C-Sp(3)-H Bonds under Mild Conditions Using Galactose Oxidase Model Compound CuIIL. Chem. Sci. 2022, 13, 9560–9568. [Google Scholar] [CrossRef]
- Zhang, F.; Zhang, Z.; Hu, J.; Zhu, Y.; Gao, H. Rapid Catalysis for Aerobic Oxidation of Alcohols Based on Nitroxyl-Radical-Free Copper(II) under Ambient Conditions. Ind. Eng. Chem. Res. 2022, 61, 13408–13415. [Google Scholar]
- Lesh, F.D.; Lord, R.L.; Heeg, M.J.; Schlegel, H.B.; Verani, C.N. Unexpected Formation of a Cobalt(III) Phenoxazinylate Electron Reservoir. Eur. J. Inorg. Chem. 2012, 2012, 463–466. [Google Scholar] [CrossRef]
- Salojärvi, E.; Peuronen, A.; Moilanen, J.; Huhtinen, H.; Lindén, J.; Mansikkamäki, A.; Lastusaari, M.; Lehtonen, A. A Diamagnetic Iron Complex and Its Twisted Sister—Structural Evidence on Partial Spin State Change in a Crystalline Iron Complex. Dalt. Trans. 2021, 50, 15831–15840. [Google Scholar] [CrossRef] [PubMed]
- Karnbrock, S.B.H.; Golz, C.; Mata, R.A.; Alcarazo, M. Ligand-Enabled Disproportionation of 1,2-Diphenylhydrazine at a PV-Center**. Angew. Chem. Int. Ed. 2022, 61, e202207450. [Google Scholar] [CrossRef] [PubMed]
- Despagnet-Ayoub, E.; Henling, L.M.; Labinger, J.A.; Bercaw, J.E. Addition of a Phosphine Ligand Switches an N-Heterocyclic Carbene-Zirconium Catalyst from Oligomerization to Polymerization of 1-Hexene. Dalton Trans. 2013, 42, 15544–15547. [Google Scholar] [CrossRef]
- Lalrempuia, R.; Breivik, F.; Törnroos, K.W.; Le Roux, E. Coordination Behavior of Bis-Phenolate Saturated and Unsaturated N-Heterocyclic Carbene Ligands to Zirconium: Reactivity and Activity in the Copolymerization of Cyclohexene Oxide with CO2. Dalton Trans. 2017, 46, 8065–8076. [Google Scholar] [CrossRef]
- Ali, A.; Dhar, D.; Barman, S.K.; Lloret, F.; Mukherjee, R. Nickel(II) Complex of a Hexadentate Ligand with Two o-Iminosemiquinonato(1-) π-Radical Units and Its Monocation and Dication. Inorg. Chem. 2016, 55, 5759–5771. [Google Scholar] [CrossRef] [PubMed]
- Blackmore, K.J.; Lal, N.; Ziller, J.W.; Heyduk, A.F. Group IV Coordination Chemistry of a Tetradentate Redox-Active Ligand in Two Oxidation States. Eur. J. Inorg. Chem. 2009, 2009, 735–743. [Google Scholar] [CrossRef]
- Salojärvi, E.; Peuronen, A.; Lahtinen, M.; Huhtinen, H.; Vlasenko, L.S.; Lastusaari, M.; Lehtonen, A. Series of Near-IR-Absorbing Transition Metal Complexes with Redox Active Ligands. Molecules 2020, 25, 2531. [Google Scholar] [CrossRef] [PubMed]
- Blackmore, K.J.; Lal, N.; Ziller, J.W.; Heyduk, A.F. Catalytic Reactivity of a Zirconium(IV) Redox-Active Ligand Complex with 1,2-Diphenylhydrazine. J. Am. Chem. Soc. 2008, 130, 2728–2729. [Google Scholar] [CrossRef]
- Hänninen, M.M.; Paturi, P.; Tuononen, H.M.; Sillanpää, R.; Lehtonen, A. Heptacoordinated Molybdenum(VI) Complexes of Phenylenediamine Bis(Phenolate): A Stable Molybdenum Amidophenoxide Radical. Inorg. Chem. 2013, 52, 5714–5721. [Google Scholar] [CrossRef]
- Hossain, M.K.; Haukka, M.; Hänninen, M.M.; Lisensky, G.C.; Paturi, P.; Nordlander, E.; Lehtonen, A. An Experimental and Theoretical Study of a Heptacoordinated Tungsten(VI) Complex of a Noninnocent Phenylenediamine Bis(Phenolate) Ligand. Inorg. Chem. Commun. 2018, 93, 149–152. [Google Scholar] [CrossRef]
- Lehtonen, A.; Sapkota, N.; Peuronen, A. Cobalt(III) Complex with a Redox Non-Innocent Bis (o-Aminophenol). Inorg. Chem. Commun. 2024, 159, 111704. [Google Scholar] [CrossRef]
- Cherkasov, V.K.; Piskunov, A.V.; Trofimova, O.Y.; Smolyaninov, I.V.; Fukin, G.K. A New Octacoordinated Tin Complex with Tetradentate Redox-Active Ligands. Dokl. Chem. 2013, 448, 61–65. [Google Scholar] [CrossRef]
- Takeyama, T.; Tsushima, S.; Takao, K. Utility of Redox-Active Ligands for Reversible Multi-Electron Transfer in Uranyl(vi) Complexes. Inorg. Chem. Front. 2023, 10, 4028–4044. [Google Scholar] [CrossRef]
- Volodarsky, S.; Malahov, I.; Bawari, D.; Diab, M.; Malik, N.; Tumanskii, B.; Dobrovetsky, R. Geometrically Constrained Square Pyramidal Phosphoranide. Chem. Sci. 2022, 13, 5957–5963. [Google Scholar] [CrossRef] [PubMed]
- Petrov, P.A.; Kadilenko, E.M.; Sukhikh, T.S.; Eltsov, I.V.; Gushchin, A.L.; Nadolinny, V.A.; Sokolov, M.N.; Gritsan, N.P. A Sterically Hindered Derivative of 2,1,3-Benzotelluradiazole: A Way to the First Structurally Characterised Monomeric Tellurium–Nitrogen Radical Anion. Chem. A Eur. J. 2020, 26, 14688–14699. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.X.; Li, Y.; Huang, F. Persistent and Stable Organic Radicals: Design, Synthesis, and Applications. Chem 2021, 7, 288–332. [Google Scholar] [CrossRef]
- Bawari, D.; Volodarsky, S.; Ginzburg, Y.; Jaiswal, K.; Joshi, P.; Dobrovetsky, R. Intramolecular C-N Bond Activation by a Geometrically Constrained PIII-Centre. Chem. Commun. 2022, 58, 12176–12179. [Google Scholar] [CrossRef]



















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lehtonen, A. Metal Complexes of Redox Non-Innocent Ligand N,N′-Bis(3,5-di-tertbutyl-2-hydroxy-phenyl)-1,2-phenylenediamine. Molecules 2024, 29, 1088. https://doi.org/10.3390/molecules29051088
Lehtonen A. Metal Complexes of Redox Non-Innocent Ligand N,N′-Bis(3,5-di-tertbutyl-2-hydroxy-phenyl)-1,2-phenylenediamine. Molecules. 2024; 29(5):1088. https://doi.org/10.3390/molecules29051088
Chicago/Turabian StyleLehtonen, Ari. 2024. "Metal Complexes of Redox Non-Innocent Ligand N,N′-Bis(3,5-di-tertbutyl-2-hydroxy-phenyl)-1,2-phenylenediamine" Molecules 29, no. 5: 1088. https://doi.org/10.3390/molecules29051088
APA StyleLehtonen, A. (2024). Metal Complexes of Redox Non-Innocent Ligand N,N′-Bis(3,5-di-tertbutyl-2-hydroxy-phenyl)-1,2-phenylenediamine. Molecules, 29(5), 1088. https://doi.org/10.3390/molecules29051088