
Welcoming Neighbour or Inhospitable Host? Selective Second Metal Binding in 5- and 6-Phospha-Substituted Bpy Ligands




Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
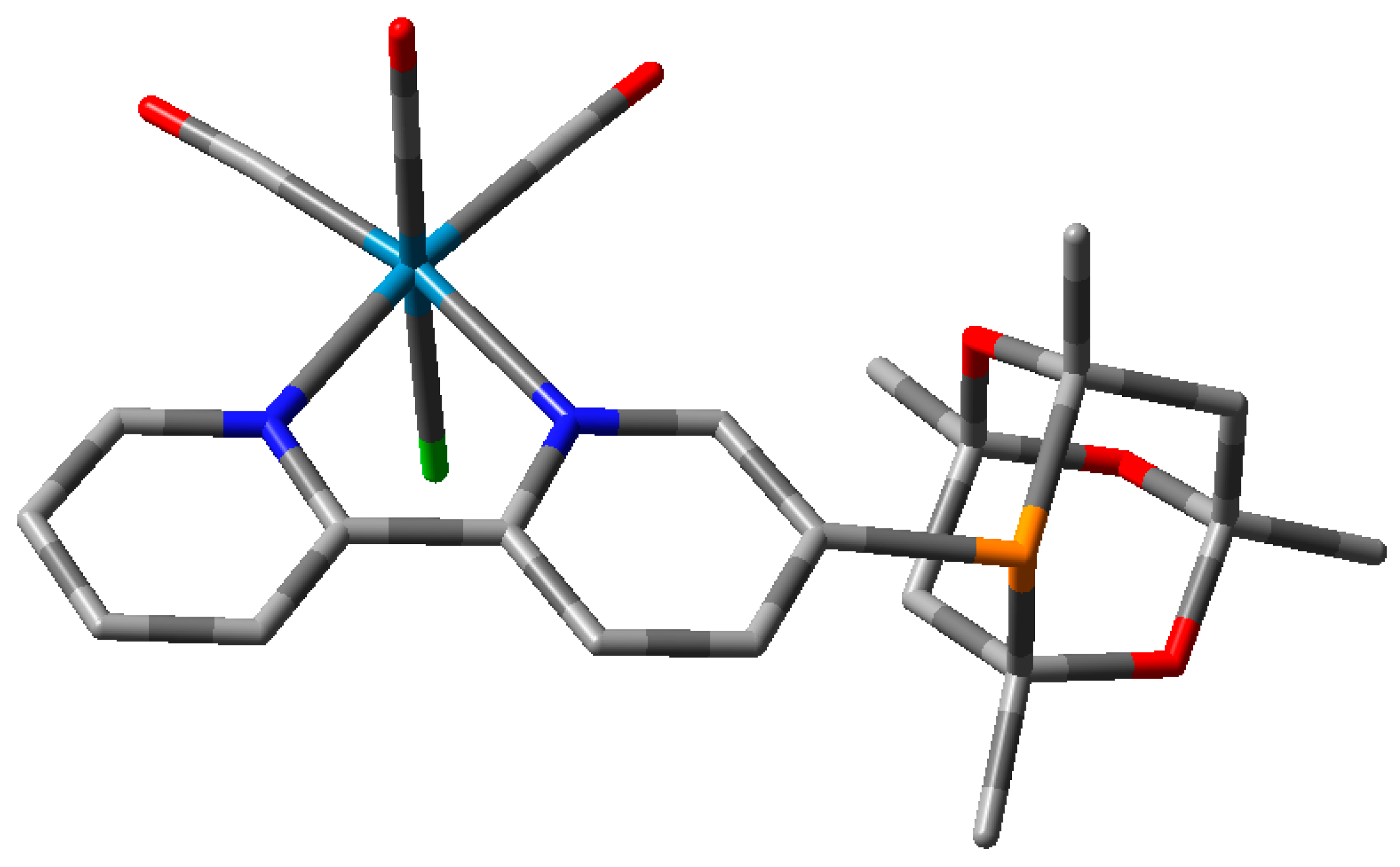
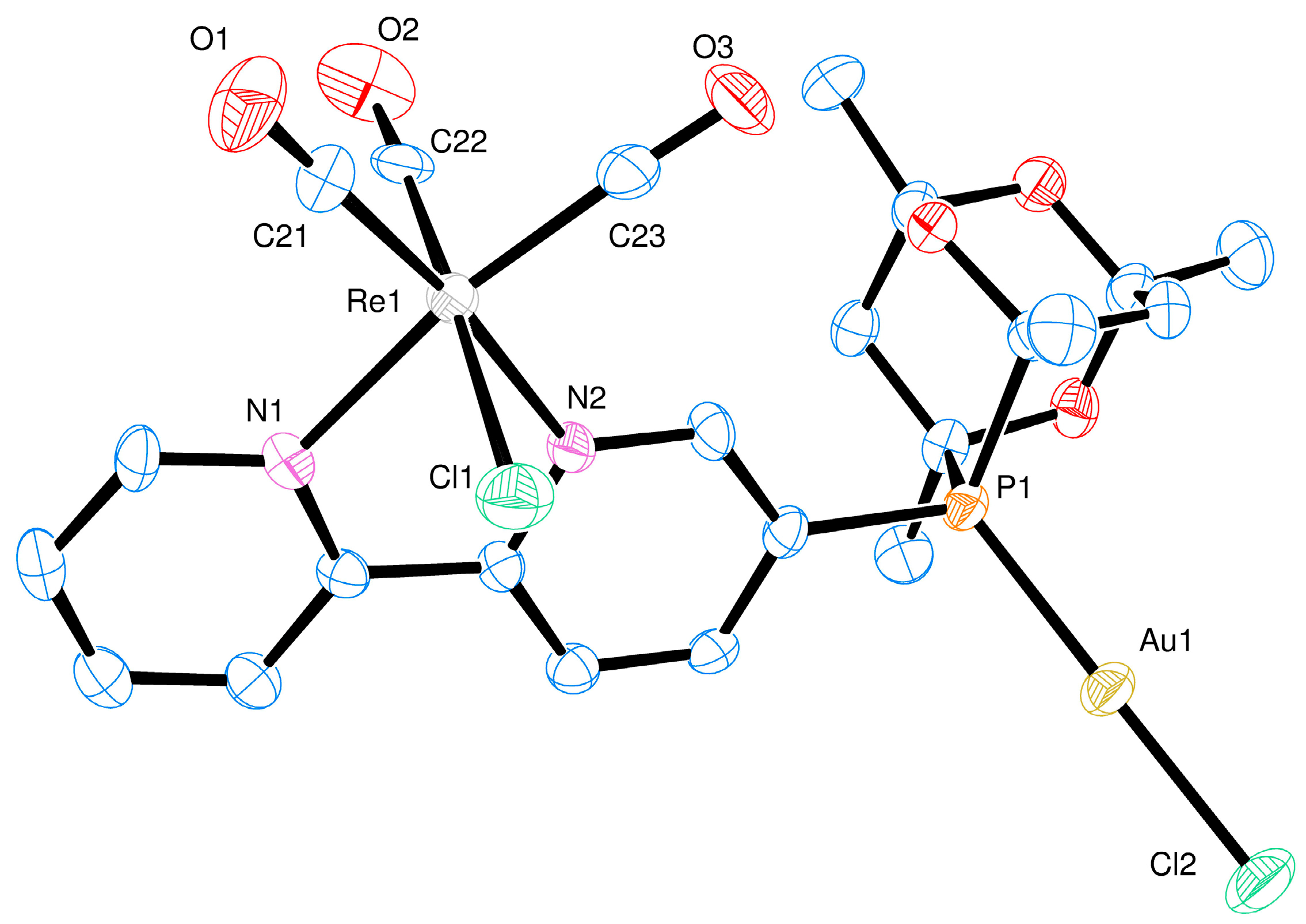
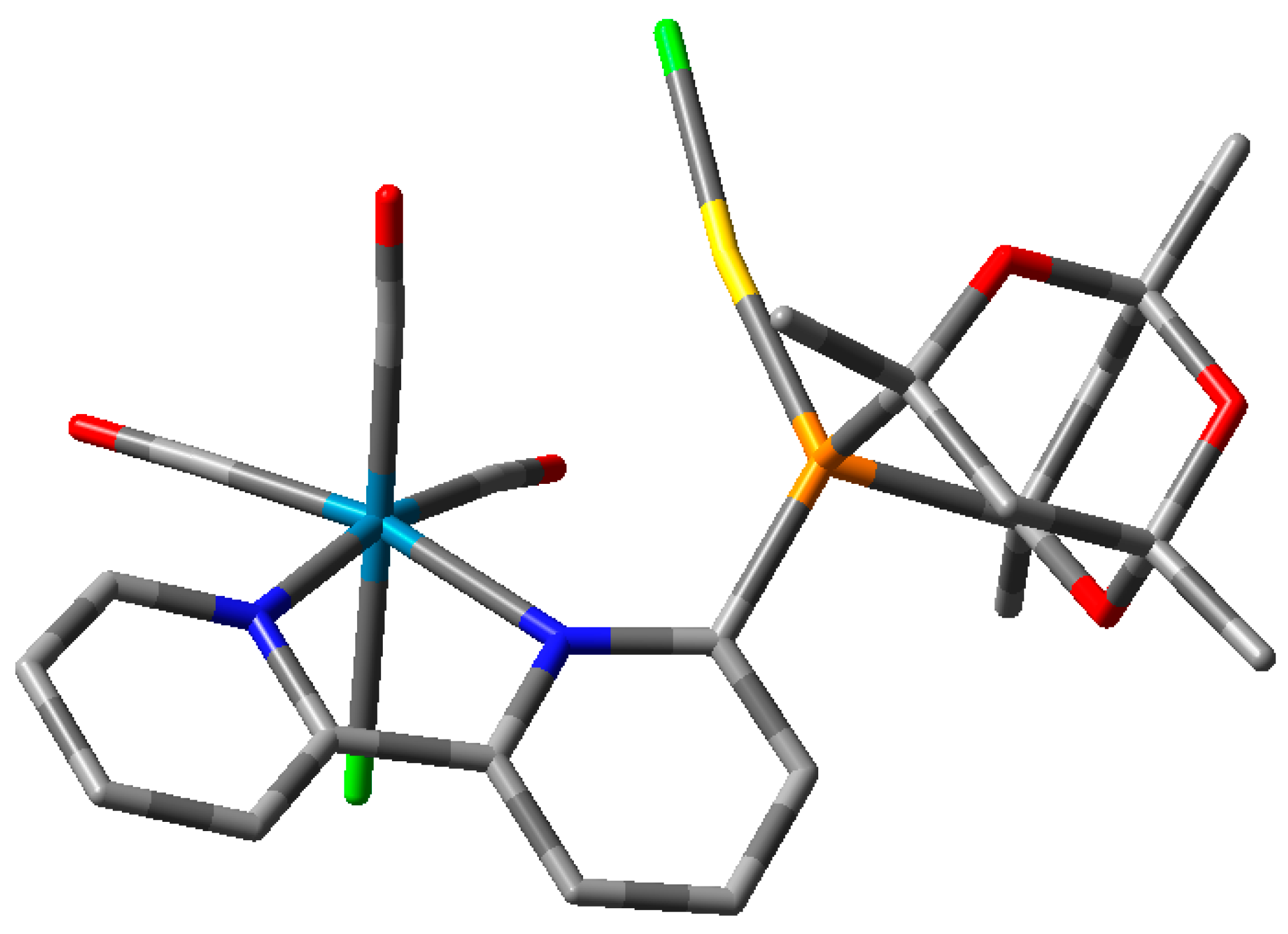
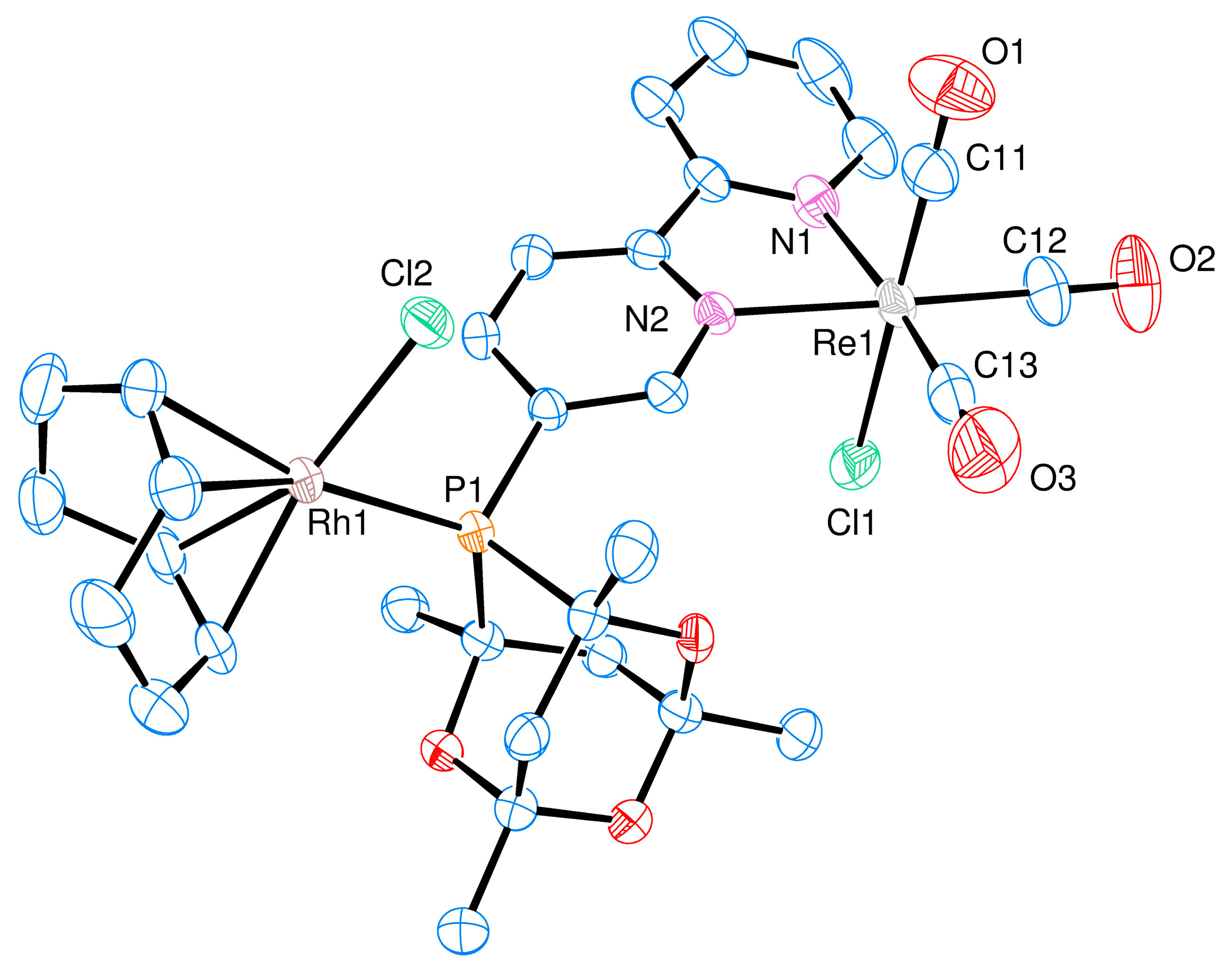
2. Results and Discussion
Ligand Synthesis
3. Materials and Methods
3.1. Synthesis of CgP-5bpy (5L)
3.2. Synthesis of CgP-6bpy (6L)
3.3. fac-[Re(κ2-N,N′-5L)(CO)3Cl], 5LRe
3.4. fac-[Re(κ2-N,N′-6L)(CO)3Cl], 6LRe
3.5. fac-[Re(κ2-N,N′-Re,κ-P-Au-5L)(CO)3Cl(AuCl)], 5LRe,Au
3.6. fac-[Re(κ2-N,N′-Re,κ-P-Au-6L)(CO)3Cl(AuCl)], 6LRe,Au
3.7. fac-[{Re(κ2-N,N′-Re,κ-P-Ag-5L)(CO)3Cl}2(Ag)]OTf, 5LRe,Ag
3.8. [Au(κ-P-6L)Cl]
3.9. [Ag(κ-P-6L)2]BF4
3.10. fac-[Re(κ2-N,N′-Re,κ-P-Rh-5L)(CO)3Cl(Rh(COD)Cl)], 5LRe,Rh
3.11. [Rh(κ-P-6L)(COD)Cl)]
3.12. Reaction of [Rh(κ-P-6L)(COD)Cl)] with [Re(CO)5Cl]
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Yuan, L.; Zhang, L.; Li, X.-X.; Liu, J.; Liu, J.-J.; Dong, L.-Z.; Li, D.-S.; Li, S.-L.; Lan, Y.-Q. Uncovering the synergistic photocatalytic behavior of bimetallic molecular catalysts. Chin. Chem. Lett. 2023, 34, 107146. [Google Scholar] [CrossRef]
- Stevens, M.A.; Colebatch, A.L. Cooperative approaches in catalytic hydrogenation and dehydrogenation. Chem. Soc. Rev. 2022, 51, 1881–1898. [Google Scholar] [CrossRef]
- Navarro, N.; Moreno, J.J.; Pérez-Jiménez, M.; Campos, J. Small molecule activation with bimetallic systems: A landscape of cooperative reactivity. Chem. Commun. 2022, 58, 11220–11235. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.-W.; Zhang, X.; Velasco, L.; Karnahl, M.; Li, Z.; Luo, Z.-M.; Huang, Y.; Yu, J.; Hu, W.; Zhang, X.; et al. Precious-Metal-Free CO2 Photoreduction Boosted by Dynamic Coordinative Interaction between Pyridine-Tethered Cu(I) Sensitizers and a Co(II) Catalyst. J. Am. Chem. Soc. Au 2023, 3, 1984–1997. [Google Scholar] [CrossRef]
- Cesari, C.; Berti, B.; Calcagno, F.; Lucarelli, C.; Garavelli, M.; Mazzoni, R.; Rivalta, I.; Zacchini, S. Bimetallic Co–M (M = Cu, Ag, and Au) Carbonyl Complexes Supported by N-Heterocyclic Carbene Ligands: Synthesis, Structures, Computational Investigation, and Catalysis for Ammonia Borane Dehydrogenation. Organometallics 2021, 40, 2724–2735. [Google Scholar] [CrossRef]
- Inagaki, A.; Akita, M. Visible-light promoted bimetallic catalysis. Coord. Chem. Rev. 2010, 254, 1220–1239. [Google Scholar] [CrossRef]
- Fickenscher, Z.; Hey-Hawkins, E. Added complexity!—Mechanistic aspects of heterobimetallic complexes for application in homogeneous catalysis. Molecules 2023, 28, 4233. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, A.; Singh, A.; Kamboj, R.C. Heterobimetallic complexes as promising catlysts. Chem. Sci. Rev. Lett. 2016, 5, 170. [Google Scholar]
- Luengo, A.; Fernández-Moreira, V.; Marzo, I.; Concepción Gimeno, M. Trackable Metallodrugs Combining Luminescent Re(I) and Bioactive Au(I) Fragments. Inorg. Chem. 2017, 56, 15159–15170. [Google Scholar] [CrossRef] [PubMed]
- Luengo, A.; Redrado, M.; Marzo, I.; Fernández-Moreira, V.; Concepción Gimeno, M. Luminescent Re(I)/Au(I) Species As Selective Anticancer Agents for HeLa Cells. Inorg. Chem. 2020, 59, 8960–8970. [Google Scholar] [CrossRef]
- Huang, Z.; Wilson, J.J. Therapeutic and Diagnostic Applications of Multimetallic Rhenium(I) Tricarbonyl Complexes. Eur. J. Inorg. Chem. 2021, 2021, 1312–1324. [Google Scholar] [CrossRef]
- Quental, L.; Raposinho, P.; Mendes, F.; Santos, I.; Navarro-Ranninger, C.; Alvarez-Valdes, A.; Huang, H.; Chao, H.; Rubbiani, R.; Gasser, G.; et al. Combining imaging and anticancer properties with new heterobimetallic Pt(II)/M(I) (M = Re, 99mTc) complexes. Dalton Trans. 2017, 46, 14523–14536. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Gallardo, J.; Elie, B.T.; Sanaú, M.; Contel, M. Versatile synthesis of cationic N-heterocyclic carbene–gold(I) complexes containing a second ancillary ligand. Design of heterobimetallic ruthenium–gold anticancer agents. Chem. Commun. 2016, 52, 3155–3158. [Google Scholar] [CrossRef]
- Elie, B.T.; Hubbard, K.; Layek, B.; Yang, W.S.; Prabha, S.; Ramos, J.W.; Contel, M. Auranofin-Based Analogues Are Effective Against Clear Cell Renal Carcinoma In Vivo and Display No Significant Systemic Toxicity. ACS Pharmacol. Transl. Sci. 2020, 3, 644–654. [Google Scholar] [CrossRef]
- Zheng, Y.; Zhang, D.-Y.; Zhang, H.; Cao, J.-J.; Tan, C.-P.; Ji, L.-N.; Mao, Z.-W. Photodamaging of Mitochondrial DNA to Overcome Cisplatin Resistance by a RuII–PtII Bimetallic Complex. Chem.–Eur. J. 2018, 24, 18971–18980. [Google Scholar] [CrossRef] [PubMed]
- Mauro, M. Phosphorescent multinuclear complexes for optoelectronics: Tuning of the excited-state dynamics. Chem. Commun. 2021, 57, 5857–5870. [Google Scholar] [CrossRef] [PubMed]
- Salassa, G.; Coenen, M.J.J.; Wezenberg, S.J.; Hendriksen, B.L.M.; Speller, S.; Elemans, J.A.A.W.; Kleij, A.W. Extremely Strong Self-Assembly of a Bimetallic Salen Complex Visualized at the Single-Molecule Level. J. Am. Chem. Soc. 2012, 134, 7186–7192. [Google Scholar] [CrossRef]
- Li, Z.; Lau, M.-T.; Li, J.; Qiu, F.; Meng, Z.; Wong, W.-Y. Seeded-growth self-assembled polymerization of a ferrocene-bearing palladium(II)-terpyridyl bimetallic complex. Chem. Commun. 2022, 58, 9878–9881. [Google Scholar] [CrossRef]
- Bonfiglio, A.; Hsiao, P.-W.; Chen, Y.; Gourlaouen, C.; Marchand, Q.; César, V.; Bellemin-Laponnaz, S.; Wang, Y.-X.; Lu, C.-W.; Daniel, C.; et al. Highly Emissive Red Heterobimetallic IrIII/MI (MI = CuI and AuI) Complexes for Efficient Light-Emitting Electrochemical Cells. Chem. Mater. 2022, 34, 1756–1769. [Google Scholar] [CrossRef]
- Termühlen, S.; Wilm, L.F.B.; Dutschke, P.D.; Hepp, A.; Hahn, F.E. Synthesis of Heterobimetallic Complexes through Chemoselective 2,4-Metalation of a Thiazolium Salt. Organometallics 2021, 40, 1565–1570. [Google Scholar] [CrossRef]
- Coles, N.T.; Gasperini, D.; Provis-Evans, C.B.; Mahon, M.F.; Webster, R.L. Heterobimetallic Complexes of 1,1-Diphosphineamide Ligands. Organometallics 2021, 40, 148–155. [Google Scholar] [CrossRef]
- Cherepakhin, V.; Hellman, A.; La, Z.; Mallikarjun Sharada, S.; Williams, T.J. Heterobimetallic complexes of IrM (M = FeII, CoII, and NiII) core and bridging 2-(diphenylphosphino)pyridine: Electronic structure and electrochemical behavior. Dalton Trans. 2020, 49, 10509–10515. [Google Scholar] [CrossRef]
- Fickenscher, Z.B.G.; Lönnecke, P.; Müller, A.K.; Hollóczki, O.; Kirchner, B.; Hey-Hawkins, E. Synergistic catalysis in heterobimetallic complexes for homogeneous carbon dioxide hydrogenation. Molecules 2023, 28, 2574. [Google Scholar] [CrossRef]
- Kumar, S.; Patra, D.K.; Rit, A. HeterobimetallicComplexes Bridged by an Unsymmetrical Bis(NHC) Ligand: Study of Enhanced Catalytic Activity in Tandem Transformations and Understanding of Cooperativity between the Metal Centers. Chem. Eur. J. 2023, 29, e202302180. [Google Scholar] [CrossRef]
- Sue, T.; Sunada, Y.; Nagashima, H. Zirconium(IV) Tris(phosphinoamide) Complexes as a Tripodal-Type Metalloligand: A Route to Zr–M (M = Cu, Mo, Pt) Heterodimetallic Complexes. Eur. J. Inorg. Chem. 2007, 46, 2897–2908. [Google Scholar] [CrossRef]
- Ye, J.; Cammarota, R.C.; Xie, J.; Vollmer, M.V.; Truhlar, D.G.; Cramer, C.J.; Lu, C.C.; Gagliardi, L. Rationalizing the Reactivity of Bimetallic Molecular Catalysts for CO2 Hydrogenation. ACS Catal. 2018, 8, 4955–4968. [Google Scholar] [CrossRef]
- Liu, S.; Motta, A.; Mouat, A.R.; Delferro, M.; Marks, T.J. Very Large Cooperative Effects in Heterobimetallic Titanium-Chromium Catalysts for Ethylene Polymerization/Copolymerization. J. Am. Chem. Soc. 2014, 136, 10460–10469. [Google Scholar] [CrossRef] [PubMed]
- Bayer, L.; Birenheide, B.S.; Krämer, F.; Lebedkin, S.; Breher, F. Heterobimetallic Gold/Ruthenium Complexes Synthesized via Post-functionalization and Applied in Dual Photoredox Gold Catalysis. Chem. Eur. J. 2022, 28, e202201856. [Google Scholar] [CrossRef] [PubMed]
- Zippel, C.; Israil, R.; Schüssler, L.; Hassan, Z.; Schneider, E.K.; Weis, P.; Nieger, M.; Bizzarri, C.; Kappes, M.M.; Riehn, C.; et al. Metal-to-Metal Distance Modulated Au(I)/Ru(II) Cyclophanyl Complexes: Cooperative Effects in Photoredox Catalysis. Chem. Eur. J. 2021, 27, 15188–15201. [Google Scholar] [CrossRef] [PubMed]
- Knoll, D.M.; Zippel, C.; Hassan, Z.; Nieger, M.; Weis, P.; Kappes, M.M.; Bräse, S. A highly stable, Au/Ru heterobimetallic photoredox catalyst with a [2.2]paracyclophane backbone. Dalton Trans. 2019, 48, 17704–17708. [Google Scholar] [CrossRef]
- Dietl, M.C.; Vethacke, V.; Keshavarzi, A.; Mulks, F.F.; Rominger, F.; Rudolph, M.; Mkhalid, I.A.I.; Hashmi, A.S.K. Synthesis of Heterobimetallic Gold(I) Palladium(II) Bis(acyclic diaminocarbene) Complexes via the Isonitrile Route. Organometallics 2022, 41, 802. [Google Scholar] [CrossRef]
- Osawa, M.; Hoshino, M.; Wakatsuki, Y. A Light-Harvesting tert-Phosphane Ligand Bearing a Ruthenium(II) Polypyridyl Complex as Substituent. Angew. Chem. Int. Ed. 2001, 40, 3472–3474. [Google Scholar] [CrossRef]
- West, N.M.; Labinger, J.A.; Bercaw, J.E. Heterobimetallic Complexes of Rhenium and Zinc: Potential Catalysts for Homogeneous Syngas Conversion. Organometallics 2011, 30, 2690–2700. [Google Scholar] [CrossRef]
- He, X.; Cao, Y.; Lang, X.-D.; Wang, N.; He, L.-N. Integrative Photoreduction of CO2 with Subsequent Carbonylation: Photocatalysis for Reductive Functionalization of CO2. ChemSusChem 2018, 11, 3382–3387. [Google Scholar] [CrossRef]
- Monticelli, S.; Talbot, A.; Gotico, P.; Caillé, F.; Loreau, O.; Del Vecchio, A.; Malandain, A.; Sallustrau, A.; Leibl, W.; Aukauloo, A.; et al. Unlocking full and fast conversion in photocatalytic carbon dioxide reduction for applications in radio-carbonylation. Nat. Commun. 2023, 14, 4451. [Google Scholar] [CrossRef] [PubMed]
- Gotico, P.; Del Vecchio, A.; Audisio, D.; Quaranta, A.; Halime, Z.; Leibl, W.; Aukauloo, A. Visible-Light-Driven Reduction of CO2 to CO and Its Subsequent Valorization in Carbonylation Chemistry and 13C Isotope Labeling. ChemPhotoChem 2018, 2, 715–719. [Google Scholar] [CrossRef]
- Mikhel, I.S.; Garland, M.; Hopewell, J.; Mastroianni, S.; McMullin, C.L.; Orpen, A.G.; Pringle, P.G. Cage Phosphinites: Ligands for Efficient Nickel-Catalyzed Hydrocyanation of 3-Pentenenitrile. Organometallics 2011, 30, 974–985. [Google Scholar] [CrossRef]
- Downing, J.H.; Floure, J.; Heslop, K.; Haddow, M.F.; Hopewell, J.; Lusi, M.; Phetmung, H.; Orpen, A.G.; Pringle, P.G.; Pugh, R.I.; et al. General Routes to Alkyl Phosphatrioxaadamantane Ligands. Organometallics 2008, 27, 3216–3224. [Google Scholar] [CrossRef]
- Cunningham, T.J.; Elsegood, M.R.J.; Kelly, P.F.; Smith, M.B.; Staniland, P.M. Coordination Studies of a New Nonsymmetric Ditertiary Phosphane Bearing a Single Phosphaadamantane Cage. Eur. J. Inorg. Chem. 2008, 14, 2326–2335. [Google Scholar] [CrossRef]
- Baber, R.A.; Clarke, M.L.; Heslop, K.M.; Marr, A.C.; Orpen, A.G.; Pringle, P.G.; Ward, A.; Zambrano-Williams, D.E. Phenylphosphatrioxa-adamantanes: Bulky, robust, electron-poor ligands that give very efficient rhodium(I) hydroformylation catalysts. Dalton Trans. 2005, 6, 1079–1085. [Google Scholar] [CrossRef]
- Shuttleworth, T.A.; Miles-Hobbs, A.M.; Pringle, P.G.; Sparkes, H.A. 2-Pyridyl substituents enhance the activity of palladium–phospha-adamantane catalysts for the methoxycarbonylation of phenylacetylene. Dalton Trans. 2017, 46, 125–137. [Google Scholar] [CrossRef]
- Jankowski, P.; McMullin, C.L.; Gridnev, I.D.; Orpen, A.G.; Pringle, P.G. Is restricted M–P rotation a common feature of enantioselective monophos catalysts? An example of restricted Rh–P rotation in a secondary phosphine complex. Tetrahedron Asymmetry 2010, 21, 1206–1209. [Google Scholar] [CrossRef]
- Rotta-Loria, N.L.; Chisholm, A.J.; MacQueen, P.M.; McDonald, R.; Ferguson, M.J.; Stradiotto, M. Exploring the Influence of Phosphine Ligation on the Gold-Catalyzed Hydrohydrazination of Terminal Alkynes at Room Temperature. Organometallics 2017, 36, 2470–2475. [Google Scholar] [CrossRef]
- Kariuki, B.M.; Newman, P.D. Asymmetric Cationic Phosphines: Synthesis, Coordination Chemistry, and Reactivity. Inorg. Chem. 2018, 57, 9554–9563. [Google Scholar] [CrossRef]
- Farrugia, L.J. WinGX and ORTEP for Windows: An update. J. Appl. Cryst. 2012, 45, 849–854. [Google Scholar] [CrossRef]
- Lamola, J.L.; Adeyinka, A.S.; Malan, F.P.; Moshapo, P.T.; Holzapfela, C.W.; Maumela, M.C. Exploring steric and electronic parameters of biaryl phosphacycles. New J. Chem. 2022, 46, 4677–4686. [Google Scholar] [CrossRef]
- Heydová, R.; Gindensperger, E.; Romano, R.; Sýkora, J.; Vlček, A., Jr.; Záliš, S.; Daniel, C. Spin–Orbit Treatment of UV–vis Absorption Spectra and Photophysics of Rhenium(I) Carbonyl–Bpyridine Complexes: MS-CASPT2 and TD-DFT Analysis. J. Phys. Chem. 2012, 116, 11319–11329. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. Shelxt—Integrated space-group and crystal-structure determination. Acta Crystallogr. Sect. A 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with shelxl. Acta Crystallogr. Sect. C 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. (Eds.) Gaussian 09, Revision D.01; Gaussian, Inc.: Wallingford, CT, USA, 2013. [Google Scholar]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic-behavior. Phys. Rev. A 1998, 38, 3098–3100. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1998, 37, 785–789. [Google Scholar] [CrossRef] [PubMed]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parameterization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Platts, J.A.; Kariuki, B.M.; Newman, P.D. Welcoming Neighbour or Inhospitable Host? Selective Second Metal Binding in 5- and 6-Phospha-Substituted Bpy Ligands. Molecules 2024, 29, 1150. https://doi.org/10.3390/molecules29051150
Platts JA, Kariuki BM, Newman PD. Welcoming Neighbour or Inhospitable Host? Selective Second Metal Binding in 5- and 6-Phospha-Substituted Bpy Ligands. Molecules. 2024; 29(5):1150. https://doi.org/10.3390/molecules29051150
Chicago/Turabian StylePlatts, James A., Benson M. Kariuki, and Paul D. Newman. 2024. "Welcoming Neighbour or Inhospitable Host? Selective Second Metal Binding in 5- and 6-Phospha-Substituted Bpy Ligands" Molecules 29, no. 5: 1150. https://doi.org/10.3390/molecules29051150
APA StylePlatts, J. A., Kariuki, B. M., & Newman, P. D. (2024). Welcoming Neighbour or Inhospitable Host? Selective Second Metal Binding in 5- and 6-Phospha-Substituted Bpy Ligands. Molecules, 29(5), 1150. https://doi.org/10.3390/molecules29051150