
Stereoselective [4+3]-Cycloaddition of 2-Amino-β-nitrostyrenes with Azaoxyallyl Cations to Access Functionalized 1,4-Benzodiazepin-3-ones


Abstract
:
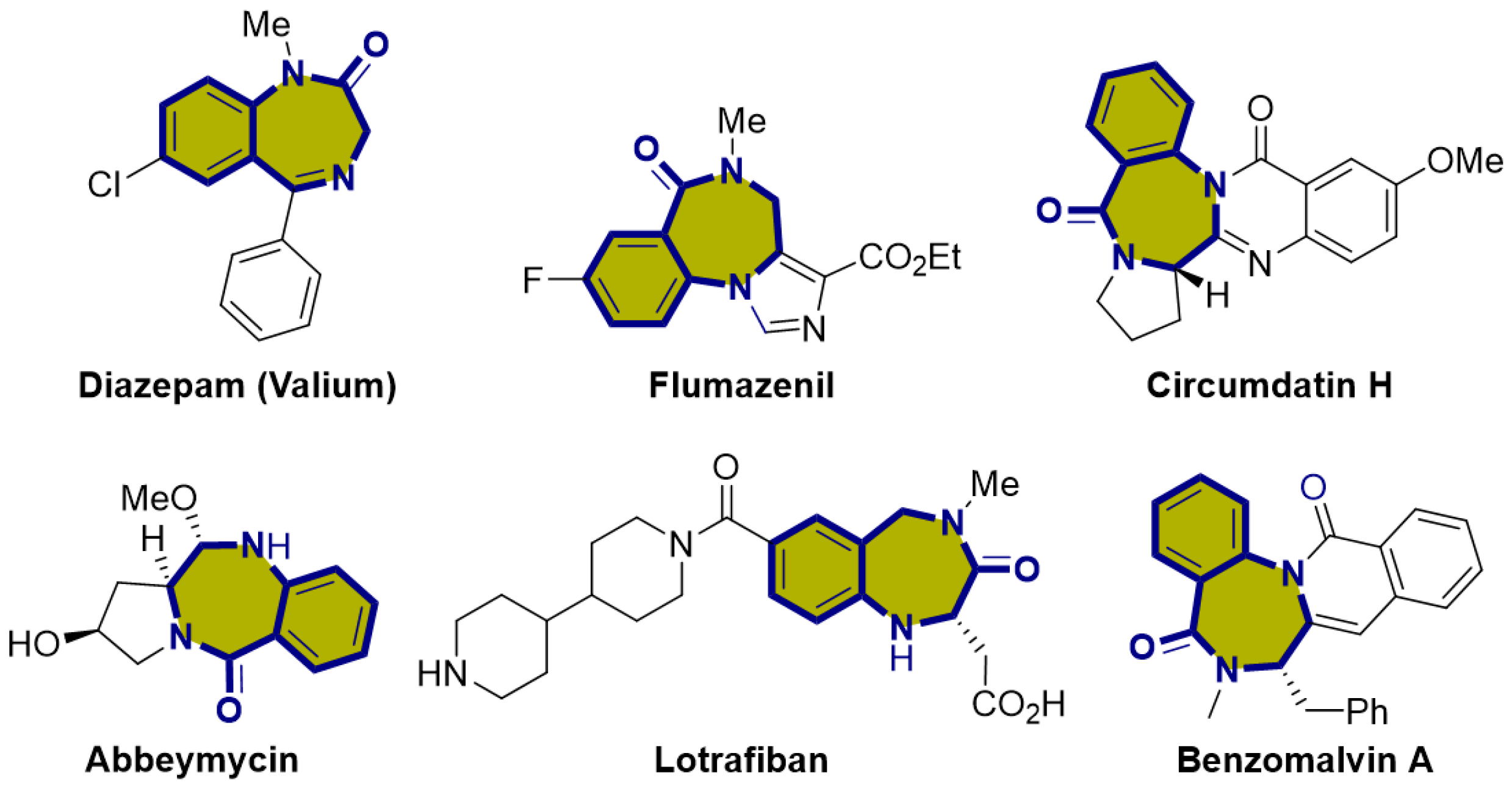
1. Introduction
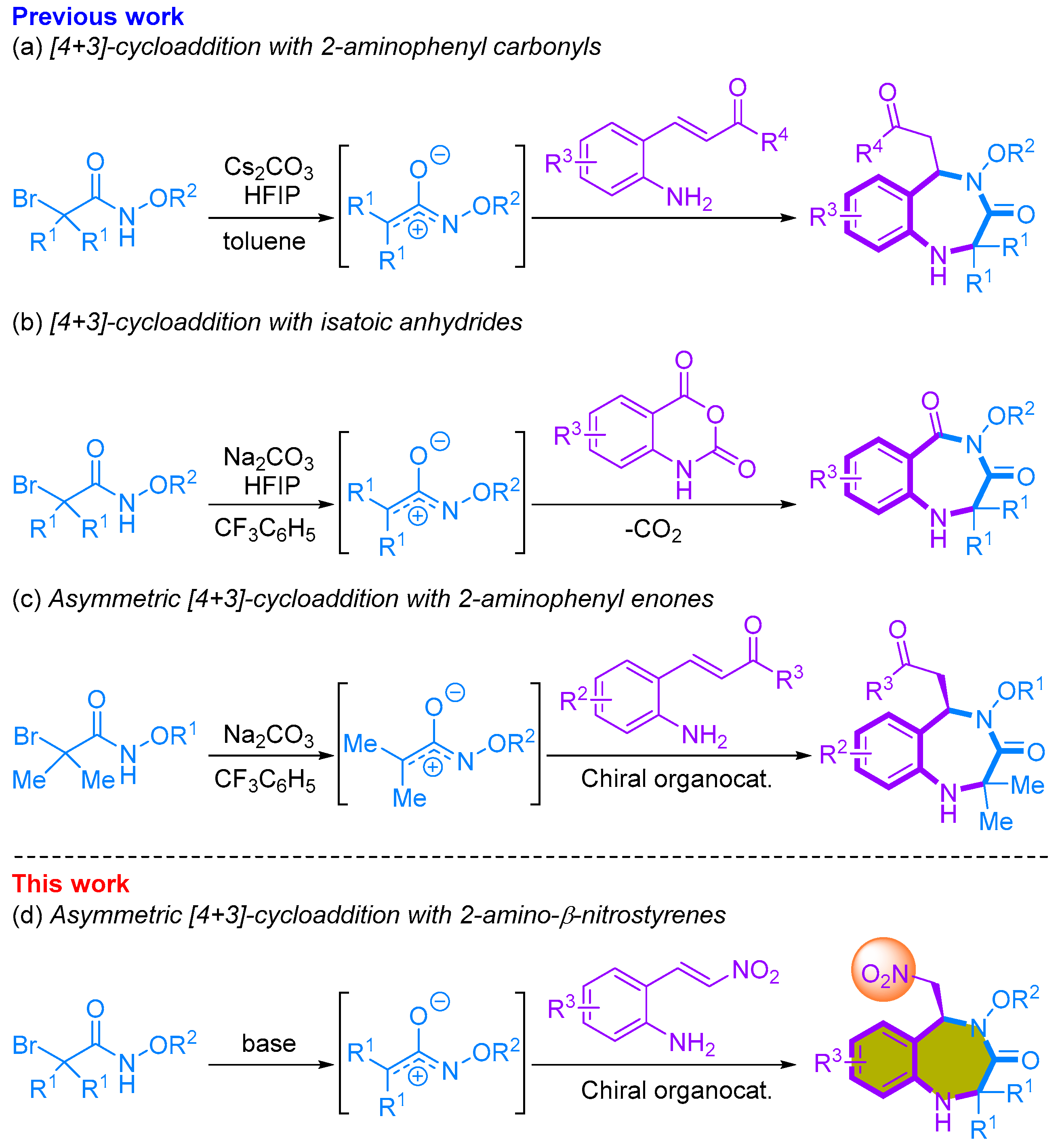
2. Results and Discussion
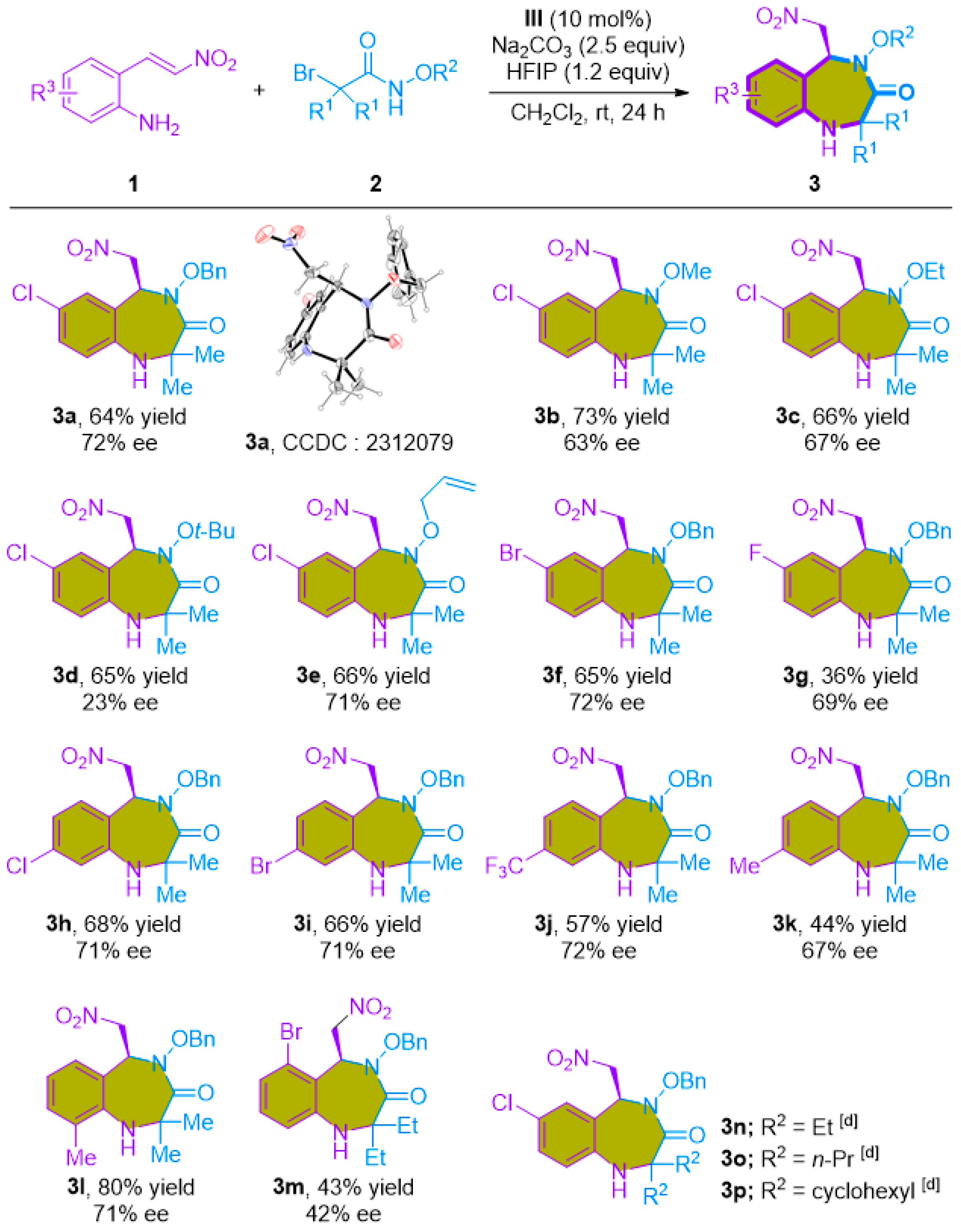
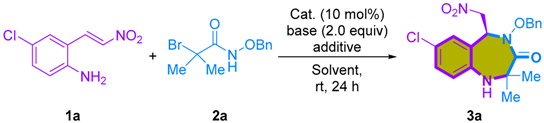
2.1. Non-Enantioselective [4+3]-Cycloaddition of 2-Amino-β-nitrostyrenes with Azaoxyallyl Cations
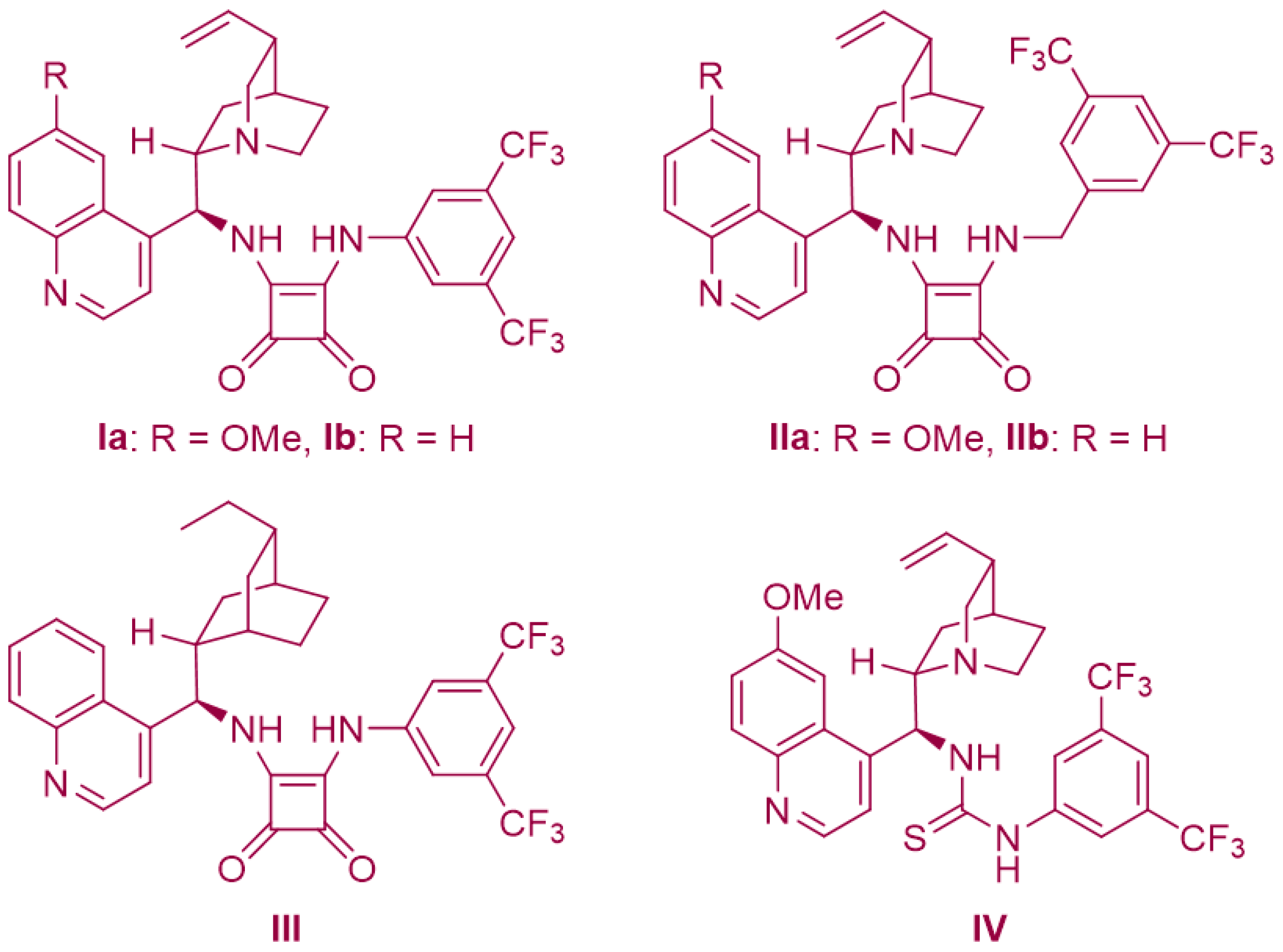
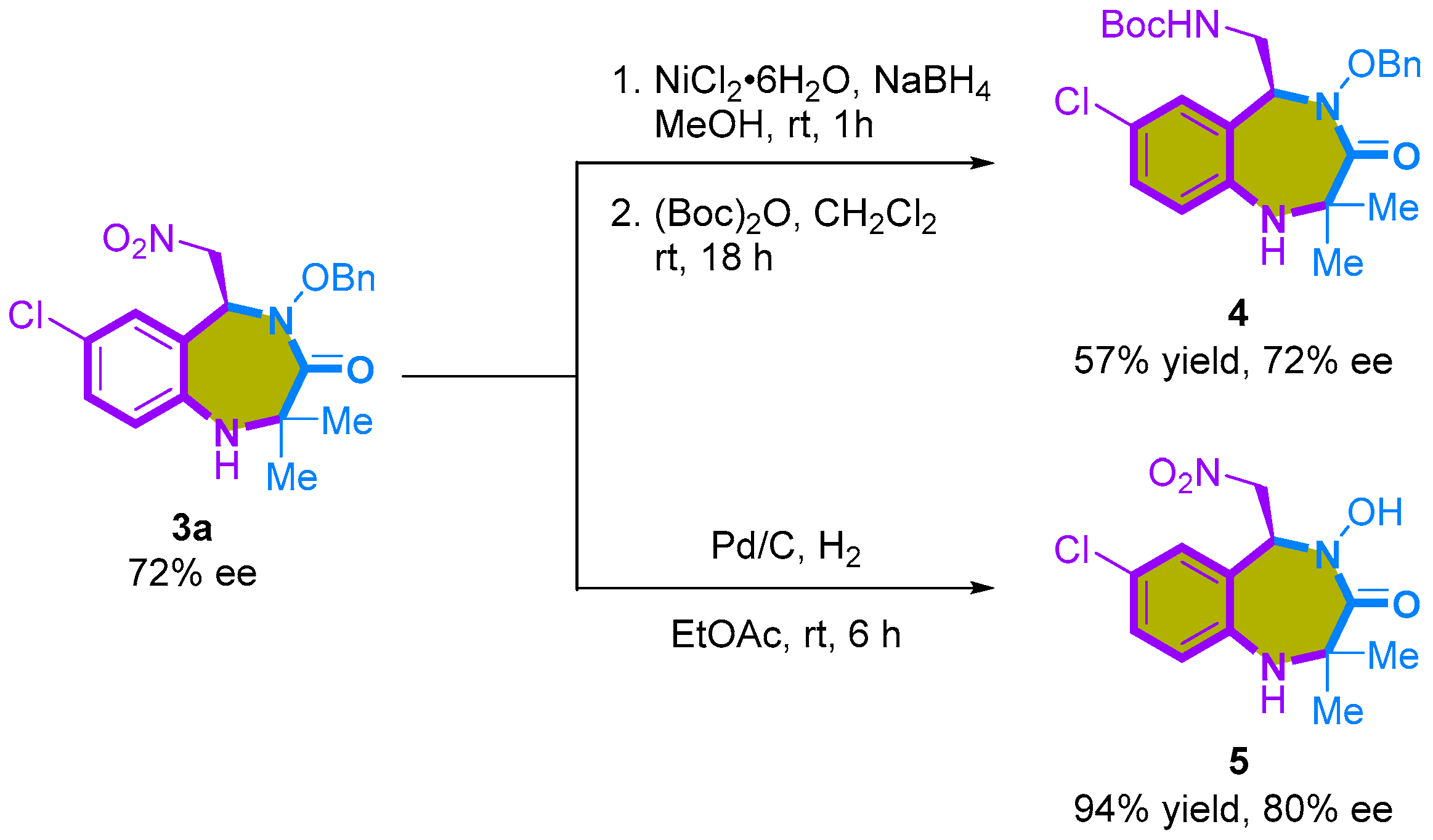
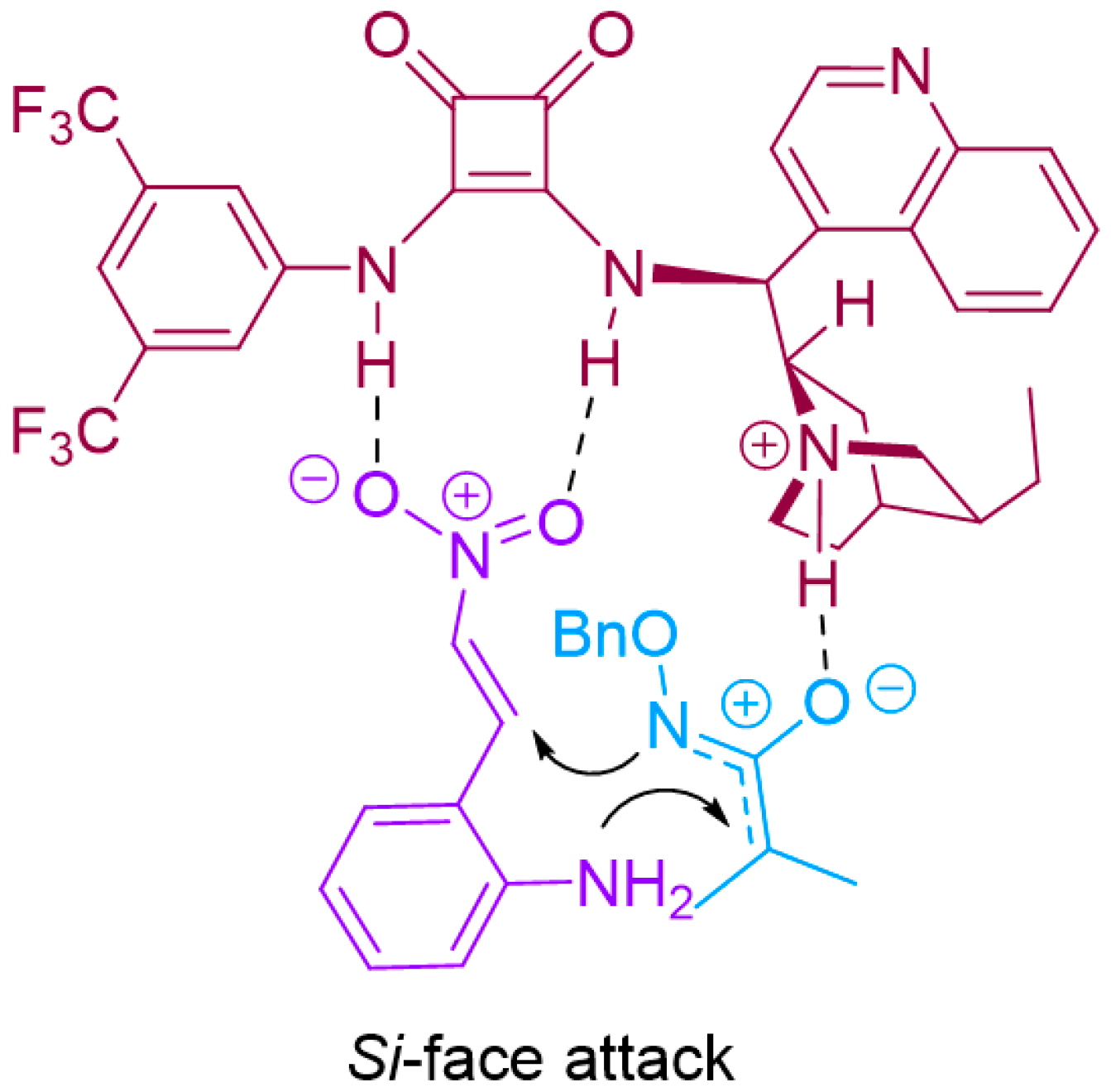
2.2. Enantioselective [4+3]-Cycloaddition of 2-Amino-β-nitrostyrenes with Azaoxyallyl Cations
3. Experimental
3.1. General Information
3.2. General Procedure I for the [4+3]-Cycloaddition of 2-Amino-β-nitrostyrenes with α-Bromohydroxamates
3.3. General Procedure II for the Asymmetric [4+3]-Cycloaddition of 2-Amino-β-nitrostyrenes with α-Bromohydroxamates
3.4. Procedure for a One-mmol Scale Synthesis of 3a
3.5. Procedure for a One-mmol Scale Synthesis of 3f
3.6. Procedure for Reduction of 3a
3.7. Procedure for Debenzylation of 3a
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Garg, M.; Chauhan, M.; Singh, P.K.; Alex, J.M.; Kumar, R. Pyrazoloquinazolines: Synthetic strategies and bioactivities. Eur. J. Med. Chem. 2015, 97, 444–461. [Google Scholar] [CrossRef] [PubMed]
- Vila, N.; Besada, P.; Costas, T.; Costas-Lago, M.C.; Terán, C. Phthalazin-1(2H)-one as a remarkable scaffold in drug discovery. Eur. J. Med. Chem. 2015, 97, 462–482. [Google Scholar] [CrossRef]
- Horton, D.A.; Bourne, G.T.; Smythe, M.L. The Combinatorial Synthesis of Bicyclic Privileged Structures or Privileged Substructures. Chem. Rev. 2003, 103, 893–930. [Google Scholar] [CrossRef] [PubMed]
- Arora, N.; Dhiman, P.; Kumar, S.; Singh, G.; Monga, V. Recent advances in synthesis and medicinal chemistry of benzodiazepines. Bioorg. Chem. 2020, 97, 103668. [Google Scholar] [CrossRef]
- Magi, E.; Fattorusso, C.; Persico, M.; Corvino, A.; Esposito, G.; Fiorino, F.; Luciano, P.; Perissutti, E.; Santagada, V.; Severino, B.; et al. New Insights into the Structure−Activity Relationship and Neuroprotective Profile of Benzodiazepinone Derivatives of Neurounina-1 as Modulators of the Na+/Ca2+ Exchanger Isoforms. J. Med. Chem. 2021, 64, 17901–17919. [Google Scholar] [CrossRef] [PubMed]
- Vézina-Dawod, S.; Perreault, M.; Guay, L.-D.; Gerber, N.; Gobeil, S.; Biron, E. Synthesis and biological evaluation of novel 1,4-benzodiazepin-3-one derivatives as potential antitumor agents against prostate cancer. Bioorg. Med. Chem. 2021, 45, 116314. [Google Scholar] [CrossRef] [PubMed]
- Alam, M.I.; Quasimi, H.; Kumar, A.; Alam, A.; Bhagat, S.; Alam, M.S.; Khan, G.A.; Dhulap, A.; Ansari, M.A. Protective effects of novel diazepinone derivatives in snake venom induced sterile inflammation in experimental animals. Eur. J. Pharmacol. 2022, 928, 175095. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Alam, M.S.; Hamid, H.; Chugh, V.; Tikla, T.; Kaul, R.; Dhulap, A.; Sharma, S.K. Design and synthesis of anti–inflammatory 1,2,3–triazolylpyrrolobenzodiazepinone derivatives and impact of molecular structure on COX–2 selective targeting. J. Mol. Struct. 2023, 1272, 134151. [Google Scholar] [CrossRef]
- Lee, S.; Love, M.S.; Modukuri, R.; Chatterjee, A.K.; Huerta, L.; Lawson, A.P.; McNamara, C.W.; Mead, J.R.; Hedstrom, L.; Cuny, G.D. Structure-activity relationship of BMS906024 derivatives for Cryptosporidium parvum growth inhibition. Bioorg. Med. Chem. Lett. 2023, 90, 129328. [Google Scholar] [CrossRef]
- Calcaterra, N.E.; Barrow, J.C. Classics in Chemical Neuroscience: Diazepam (Valium). ACS Chem. Neurosci. 2014, 5, 253–260. [Google Scholar] [CrossRef]
- Liu, F.; Craft, R.M.; Morris, S.A.; Carroll, R.C. Lotrafiban: An oral platelet glycoprotein IIb/IIIa blocker. Expert Opin. Investig. Drugs 2000, 9, 2673–2687. [Google Scholar] [CrossRef] [PubMed]
- Scarborough, R.M.; Gretler, D.D. Platelet glycoprotein IIb-IIIa antagonists as prototypical integrin blockers: Novel parenteral and potential oral antithrombotic agents. J. Med. Chem. 2000, 43, 3453–3473. [Google Scholar] [CrossRef] [PubMed]
- Saranya, P.V.; Neetha, M.; Radhika, S.; Anilkumar, G. An overview of palladium-catalyzed synthesis of seven-membered heterocycles. J. Heterocycl. Chem. 2021, 58, 673–684. [Google Scholar] [CrossRef]
- Sasiambarrena, L.D.; Barri, I.A.; Fraga, G.G.; Bravo, R.D.; Ponzinibbio, A. Facile synthesis of 4-substituted 1,2,4,5-tetrahydro-1,4-benzodiazepin-3-ones by reductive cyclization of 2-chloro-N-(2-nitrobenzyl)acetamides. Tetrahedron Lett. 2019, 60, 264–267. [Google Scholar] [CrossRef]
- Ma, X.; Zhang, X.; Awad, J.M.; Xie, G.; Qiu, W.; Zhang, W. One-pot synthesis of tetrahydro-pyrrolobenzodiazepines and tetrahydro-pyrrolobenzodiazepinones through sequential 1,3-dipolar cycloaddition/N-alkylation (N-acylation)/Staudinger/aza-Wittig reactions. Green Chem. 2019, 21, 4489–4494. [Google Scholar] [CrossRef]
- Velasco-Rubio, Á.; Varela, J.; Saá, C. Pd-Catalyzed allylic C–H activation to seven-membered N,O-heterocycles. Chem. Commun. 2021, 57, 10915–10918. [Google Scholar] [CrossRef] [PubMed]
- Ganesh, P.S.K.P.; Muthuraja, P.; Gopinath, P. Rh(III) Catalyzed Redox-Neutral C-H Activation/[5 + 2] Annulation of Aroyl Hydrazides and Sulfoxonium Ylides: Synthesis of Benzodiazepinones. Org. Lett. 2023, 25, 8361–8366. [Google Scholar] [CrossRef] [PubMed]
- Illuminati, G.; Mandolini, L. Ring closure reactions of bifunctional chain molecules. Acc. Chem. Res. 1981, 14, 95–102. [Google Scholar] [CrossRef]
- Moyano, A.; Rios, R. Asymmetric Organocatalytic Cyclization and Cycloaddition Reactions. Chem. Rev. 2011, 111, 4703–4832. [Google Scholar] [CrossRef]
- Kwon, Y.I.; Choi, S.; Jang, H.S.; Kim, S.-G. Rapid Access to Hindered α-Amino Acid Derivatives and Benzodiazepin-3-ones from Aza-Oxyallyl Cations. Org. Lett. 2020, 22, 1420–1425. [Google Scholar] [CrossRef]
- Kim, E.; Lee, C.Y.; Kim, S.-G. HFIP-Mediated Decarboxylative [4+3]-Annulation of Azaoxyallyl Cations with Isatoic Anhydride. Adv. Synth. Catal. 2020, 362, 3594–3603. [Google Scholar] [CrossRef]
- Jang, H.S.; Kwon, Y.I.; Kim, S.-G. Facile Synthesis of Functionalized 1,4-Benzodiazepine-3-one-5-acetates via [4+3]-Annulation of Azaoxyallyl Cations with 2-Aminophenyl α,β-Unsaturated Esters. Bull. Korean Chem. Soc. 2020, 41, 727–734. [Google Scholar] [CrossRef]
- Lee, C.Y.; Kwon, Y.I.; Jang, H.S.; Lee, S.; Chun, Y.L.; Jung, J.; Kim, S.-G. Organocatalytic Enantioselective [4+3]-Cycloadditions of Azaoxyallyl Cations with 2-Aminophenyl Enones. Adv. Synth. Catal. 2021, 363, 4197–4203. [Google Scholar] [CrossRef]
- Kim, Y.; Han, J.W.; Kim, S.-G. Organocatalytic Enantioselective Synthesis of Polycyclic Benzosultams from 2-Amino-β-nitrostyrenes with Cyclic N-Sulfonyl Ketimines. Org. Lett. 2024, 24, 1472–1477. [Google Scholar] [CrossRef]
- Khaksar, S. Fluorinated alcohols: A magic medium for the synthesis of heterocyclic compounds. J. Fluor. Chem. 2015, 172, 51–61. [Google Scholar] [CrossRef]
- Shuklov, I.A.; Dubrovina, N.V.; Börner, A. Fluorinated Alcohols as Solvents, Cosolvents and Additives in Homogeneous Catalysis. Synthesis 2007, 2007, 2925–2943. [Google Scholar] [CrossRef]
- Chauhan, P.; Mahajan, S.; Kaya, U.; Hack, D.; Enders, D. Bifunctional Amine-Squaramides: Powerful Hydrogen-Bonding Organocatalysts for Asymmetric Domino/Cascade Reactions. Adv. Synth. Catal. 2015, 357, 253–281. [Google Scholar] [CrossRef]
- Fang, X.; Wang, C.-J. Recent advances in asymmetric organocatalysis mediated by bifunctional amine–thioureas bearing multiple hydrogen-bonding donors. Chem. Commun. 2015, 51, 1185–1197. [Google Scholar] [CrossRef]
- Zhao, B.-L.; Li, J.-H.; Du, D.-M. Squaramide-Catalyzed Asymmetric Reactions. Chem. Rec. 2017, 17, 994–1018. [Google Scholar] [CrossRef] [PubMed]
- Parvin, T.; Yadav, R.; Choudhury, L.H. Recent applications of thiourea-based organocatalysts in asymmetric multicomponent reactions (AMCRs). Org. Biomol. Chem. 2020, 18, 5513–5532. [Google Scholar] [CrossRef]
- CCDC 2312079 (3a) Contains the Supplementary Crystallographic Data for This Paper. These Data Can Be Obtained Free of Charge from the Cambridge Crystallographic Data Centre. Available online: www.ccdc.cam.ac.uk/data_request/cif (accessed on 5 December 2023).
- Jeffrey, C.S.; Barnes, K.; Eickhoff, J.; Carson, C. Generation and Reactivity of Aza-Oxyallyl Cationic Intermediates: Aza-[4 + 3] Cycloaddition Reactions for Heterocycle Synthesis. J. Am. Chem. Soc. 2011, 133, 7688–7691. [Google Scholar] [CrossRef] [PubMed]
- DiPoto, M.C.; Wu, J. Synthesis of 2-Aminoimidazolones and Imidazolones by (3 + 2) Annulation of Azaoxyallyl Cations. Org. Lett. 2018, 20, 499–501. [Google Scholar] [CrossRef] [PubMed]
- Murata, Y.; Takeuchi, K.; Nishikata, T. The synthetic protocol for α-bromocarbonyl compounds via brominations. Tetrahedron 2019, 75, 2726–2736. [Google Scholar] [CrossRef]
- Malerich, J.P.; Hagihara, K.; Rawal, V.H. Chiral Squaramide Derivatives are Excellent Hydrogen Bond Donor Catalysts. J. Am. Chem. Soc. 2008, 130, 14416–14417. [Google Scholar] [CrossRef]
- Yang, W.; Du, D.M. Highly enantioselective Michael addition of nitroalkanes to chalcones using chiral squaramides as hydrogen bonding organocatalysts. Org. Lett. 2010, 12, 5450–5453. [Google Scholar] [CrossRef]
- Zhu, Y.; Malerich, J.P.; Rawal, V.H. Squaramide-catalyzed enantioselective Michael addition of diphenyl phosphite to nitroalkenes. Angew. Chem. Int. Ed. 2010, 49, 153–156. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||||
|---|---|---|---|---|---|
| Entry | Base | Additive | Solvent | Time (h) | Yield (%) b |
| 1 | Cs2CO3 | HFIP (5 equiv) | CF3C6H5 | 24 | 18 |
| 2 | Cs2CO3 | HFIP (5 equiv) | CH2Cl2 | 2 | 55 |
| 3 | Cs2CO3 | - | CH2Cl2/HFIP | 2 | 85 |
| 4 c | Cs2CO3 | - | CH2Cl2/HFIP | 1 | 91 |
| 5 c | Cs2CO3 | - | CHCl3/HFIP | 1 | 90 |
| 6 c | Cs2CO3 | - | CH3CN/HFIP | 2 | 35 |
| 7 | Cs2CO3 | - | toluene/HFIP | 2 | 86 |
| 8 | Cs2CO3 | - | THF/HFIP | 1 | 69 |
| 9 c,d | Cs2CO3 | - | CH2Cl2/HFIP | 1 | 81 |
| 10 c | K2CO3 | - | CH2Cl2/HFIP | 1 | 62 |
| 11 c | Na2CO3 | - | CH2Cl2/HFIP | 2 | 34 |
| 12 c | Et3N | - | CH2Cl2/HFIP | 1 | 84 |
| 13 c | DABCO | - | CH2Cl2/HFIP | 2 | 64 |
| 14 c | DMAP | - | CH2Cl2/HFIP | 1 | 76 |
| 15 c | DBU | - | CH2Cl2/HFIP | 2 | 83 |
 | |||||
|---|---|---|---|---|---|
| Entry | Cat. | Base | Solvent | Yield (%) b | Ee c |
| 1 | Ia | Na2CO3 | CF3C6H5 | 14 | 48 |
| 2 | Ia | K2CO3 | CF3C6H5 | 11 | 60 |
| 3 | Ia | Cs2CO3 | CF3C6H5 | 19 | 6 |
| 4 | Ia | Et3N | CF3C6H5 | 18 | 6 |
| 5 | Ia | DIPEA | CF3C6H5 | 27 | 6 |
| 6 | Ia | DABCO | CF3C6H5 | 10 | 22 |
| 7 | Ia | DMAP | CF3C6H5 | 11 | 4 |
| 8 | Ia | Na2CO3 | Toluene | 24 | 70 |
| 9 | Ia | Na2CO3 | CH2Cl2 | 37 | 78 |
| 10 | Ia | Na2CO3 | CHCl3 | 35 | 76 |
| 11 | Ia | Na2CO3 | ClCH2CH2Cl | 34 | 74 |
| 12 | Ia | Na2CO3 | THF | 15 | 4 |
| 13 | Ia | Na2CO3 | CH3CN | 33 | 2 |
| 14 | Ib | Na2CO3 | CH2Cl2 | 37 | 78 |
| 15 | IIa | Na2CO3 | CH2Cl2 | 23 | 60 |
| 16 | IIb | Na2CO3 | CH2Cl2 | 19 | 56 |
| 17 | III | Na2CO3 | CH2Cl2 | 38 | 80 |
| 18 | IV | Na2CO3 | CH2Cl2 | 72 | 2 |
| 19 d | III | Na2CO3 | CH2Cl2 | 73 | 58 |
| 20 e | III | Na2CO3 | CH2Cl2 | 63 | 64 |
| 21 f | III | Na2CO3 | CH2Cl2 | 61 | 72 |
| 22 f,g | III | Na2CO3 | CH2Cl2 | 64 | 72 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, Y.; Kim, S.-G. Stereoselective [4+3]-Cycloaddition of 2-Amino-β-nitrostyrenes with Azaoxyallyl Cations to Access Functionalized 1,4-Benzodiazepin-3-ones. Molecules 2024, 29, 1221. https://doi.org/10.3390/molecules29061221
Kim Y, Kim S-G. Stereoselective [4+3]-Cycloaddition of 2-Amino-β-nitrostyrenes with Azaoxyallyl Cations to Access Functionalized 1,4-Benzodiazepin-3-ones. Molecules. 2024; 29(6):1221. https://doi.org/10.3390/molecules29061221
Chicago/Turabian StyleKim, Yoseop, and Sung-Gon Kim. 2024. "Stereoselective [4+3]-Cycloaddition of 2-Amino-β-nitrostyrenes with Azaoxyallyl Cations to Access Functionalized 1,4-Benzodiazepin-3-ones" Molecules 29, no. 6: 1221. https://doi.org/10.3390/molecules29061221