
Discovery of Novel Noncovalent KRAS G12D Inhibitors through Structure-Based Virtual Screening and Molecular Dynamics Simulations


Abstract
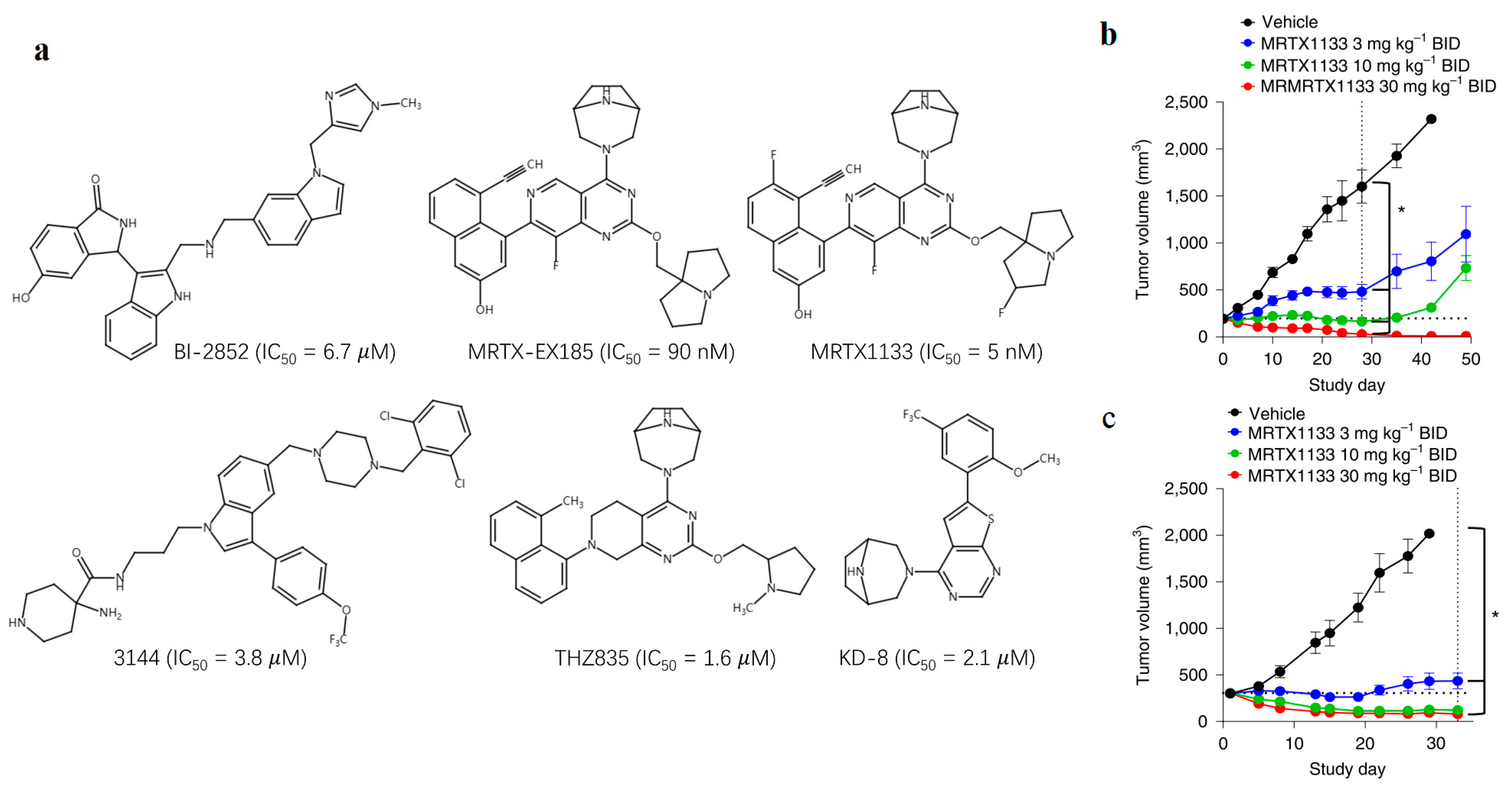
1. Introduction
2. Results and Discussion
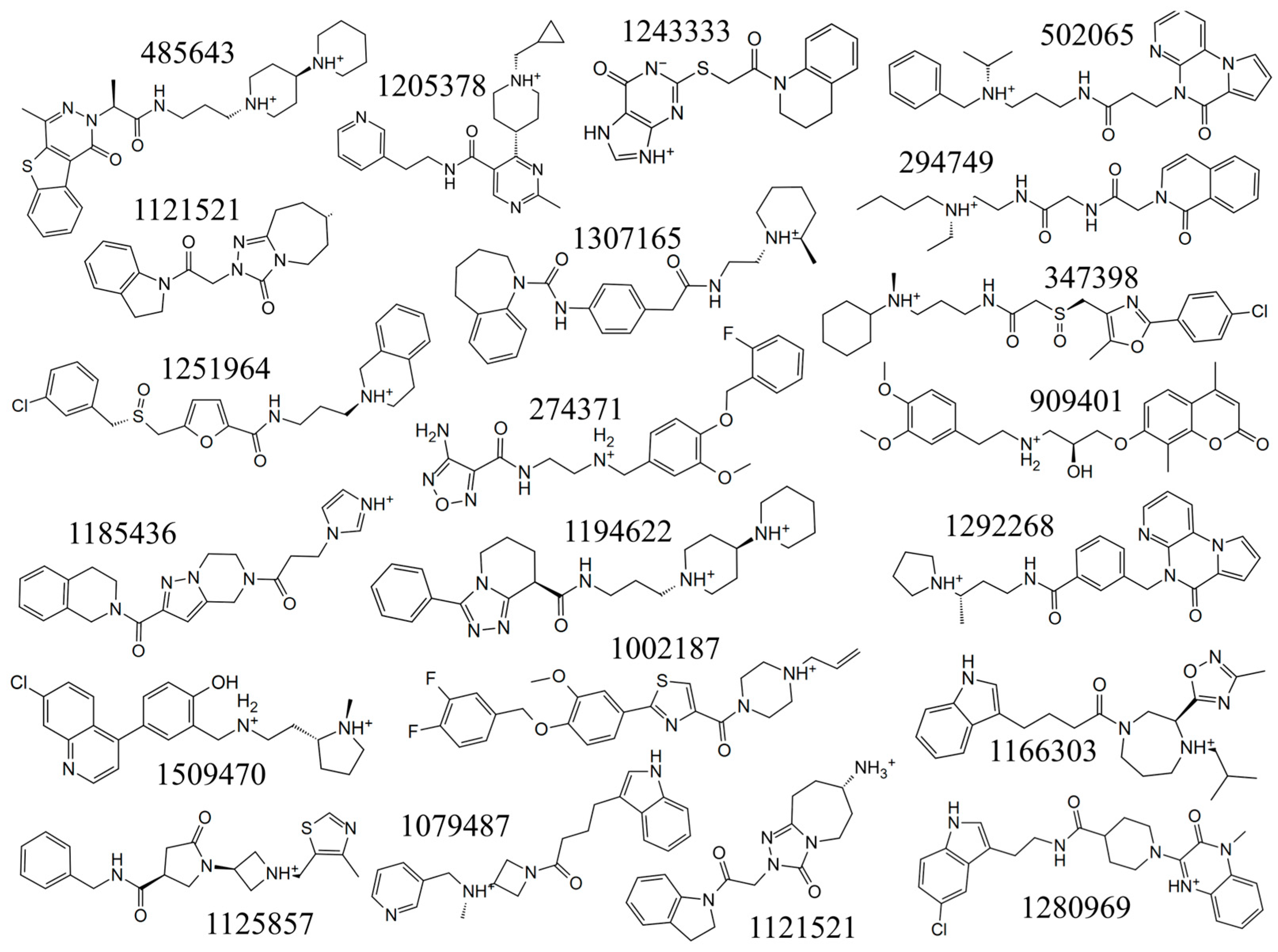
2.1. Virtual Screening of Compounds against KRASG12D
2.2. Compounds with ADME Properties against KRASG12D
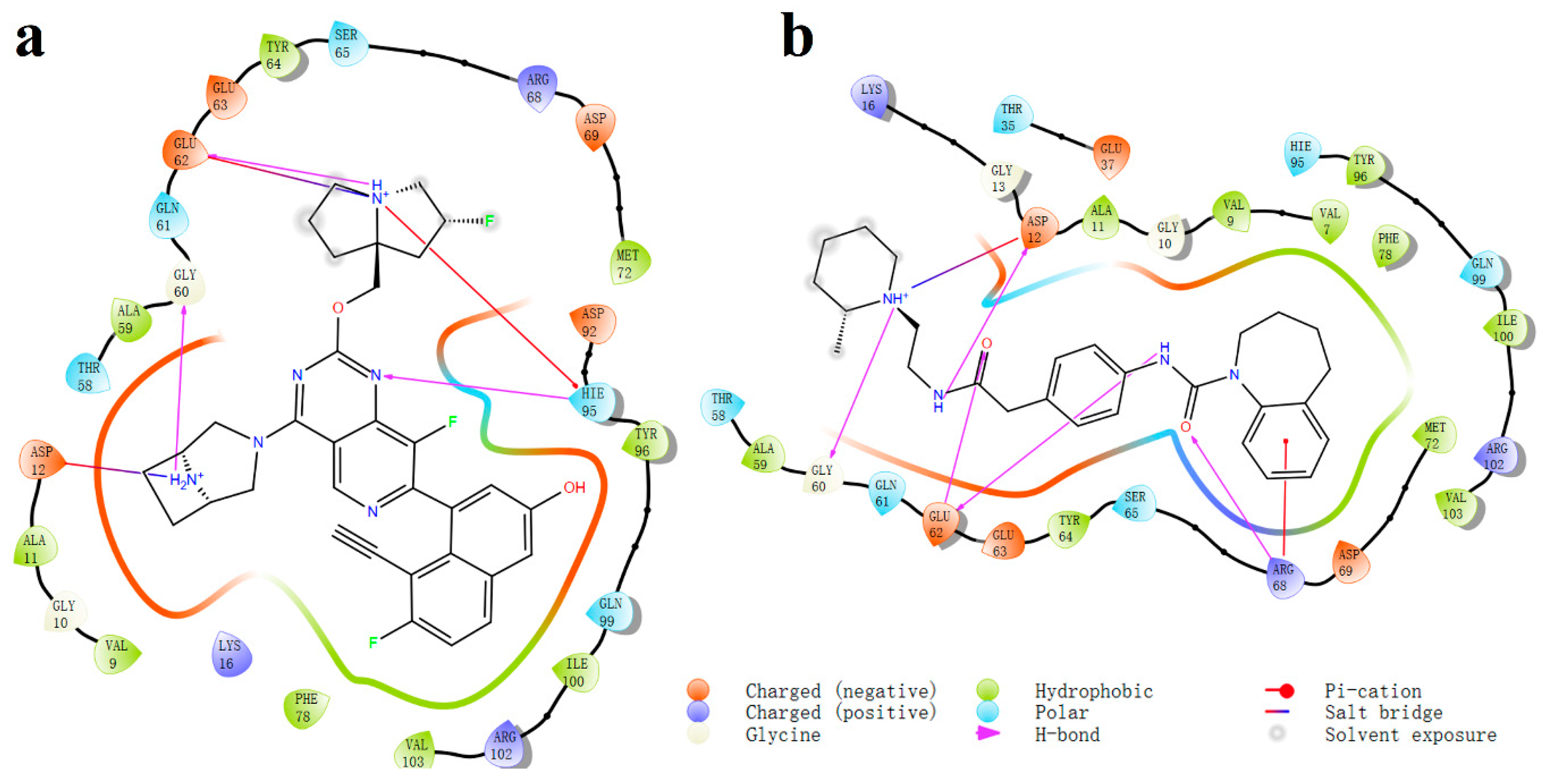
2.3. MD Simulation of Ligand-Protein Complex
3. Method
3.1. Preparation of Receptor and Ligands
3.2. Structure-Based Virtual Screening
3.3. ADME Properties Prediction
3.4. Molecular Dynamic (MD) Simulations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tejpar, S.; Celik, I.; Schlichting, M.; Sartorius, U.; Bokemeyer, C.; Van Cutsem, E. Association of KRAS G13D tumor mutations with outcome in patients with metastatic colorectal cancer treated with first-line chemotherapy with or without cetuximab. J. Clin. Oncol. 2012, 30, 3570–3577. [Google Scholar] [CrossRef]
- Kwan, A.K.; Piazza, G.A.; Keeton, A.B.; Leite, C.A. The path to the clinic: A comprehensive review on direct KRASG12C inhibitors. J. Exp. Clin. Cancer Res. 2022, 41, 27. [Google Scholar] [CrossRef] [PubMed]
- Ostrem, J.M.; Shokat, K.M. Targeting KRAS G12C with Covalent Inhibitors. Annu. Rev. Cancer Biol. 2022, 6, 49–64. [Google Scholar] [CrossRef]
- Bröker, J.; Waterson, A.G.; Smethurst, C.; Kessler, D.; Böttcher, J.; Mayer, M.; Gmaschitz, G.; Phan, J.; Little, A.; Abbott, J.R.; et al. Fragment Optimization of Reversible Binding to the Switch II Pocket on KRAS Leads to a Potent, In Vivo Active KRASG12C Inhibitor. J. Med. Chem. 2022, 65, 14614–14629. [Google Scholar] [CrossRef] [PubMed]
- Cox, A.D.; Fesik, S.W.; Kimmelman, A.C.; Luo, J.; Der, C.J. Drugging the undruggable RAS: Mission Possible? Nat. Rev. Drug Discov. 2014, 13, 828–851. [Google Scholar] [CrossRef]
- Vatansever, S.; Erman, B.; Gümüş, Z.H. Oncogenic G12D mutation alters local conformations and dynamics of K-Ras. Sci. Rep. 2019, 9, 11730. [Google Scholar] [CrossRef] [PubMed]
- Mao, Z.; Xiao, H.; Shen, P.; Yang, Y.; Xue, J.; Yang, Y.; Shang, Y.; Zhang, L.; Li, X.; Zhang, Y.; et al. KRAS(G12D) can be targeted by potent inhibitors via formation of salt bridge. Cell Discov. 2022, 8, 5. [Google Scholar] [CrossRef]
- Wang, X.; Allen, S.; Blake, J.F.; Bowcut, V.; Briere, D.M.; Calinisan, A.; Dahlke, J.R.; Fell, J.B.; Fischer, J.P.; Gunn, R.J.; et al. Identification of MRTX1133, a Noncovalent, Potent, and Selective KRAS G12D Inhibitor. J. Med. Chem. 2022, 65, 3123–3133. [Google Scholar] [CrossRef] [PubMed]
- Hallin, J.; Bowcut, V.; Calinisan, A.; Briere, D.M.; Hargis, L.; Engstrom, L.D.; Laguer, J.; Medwid, J.; Vanderpool, D.; Lifset, E.; et al. Anti-tumor efficacy of a potent and selective non-covalent KRASG12D inhibitor. Nat. Med. 2022, 28, 2171–2182. [Google Scholar] [CrossRef]
- Kessler, D.; Gmachl, M.; Mantoulidis, A.; Martin, L.J.; Zoephel, A.; Mayer, M.; Gollner, A.; Covini, D.; Fischer, S.; Gerstberger, T.; et al. Drugging an undruggable pocket on KRAS. Proc. Natl. Acad. Sci. USA 2019, 116, 15823–15829. [Google Scholar] [CrossRef]
- Kessler, D.; Bergner, A.; Böttcher, J.; Fischer, G.; Döbel, S.; Hinkel, M.; Müllauer, B.; Weiss-Puxbaum, A.; McConnell, D.B. Drugging all RAS isoforms with one pocket. Futur. Med. Chem. 2020, 12, 1911–1923. [Google Scholar] [CrossRef]
- Vasta, J.D.; Peacock, D.M.; Zheng, Q.; Walker, J.A.; Zhang, Z.; Zimprich, C.A.; Thomas, M.R.; Beck, M.T.; Binkowski, B.F.; Corona, C.R.; et al. KRAS is vulnerable to reversible switch-II pocket engagement in cells. Nat. Chem. Biol. 2022, 18, 596–604. [Google Scholar] [CrossRef]
- Welsch, M.E.; Kaplan, A.; Chambers, J.M.; Stokes, M.E.; Bos, P.H.; Zask, A.; Zhang, Y.; Sanchez-Martin, M.; Badgley, M.A.; Huang, C.S.; et al. Multivalent Small-Molecule Pan-RAS Inhibitors. Cell 2017, 168, 878–889.e29. [Google Scholar] [CrossRef]
- Kemp, S.B.; Cheng, N.; Markosyan, N.; Sor, R.; Kim, I.-K.; Hallin, J.; Shoush, J.; Quinones, L.; Brown, N.V.; Bassett, J.B.; et al. Efficacy of a Small-Molecule Inhibitor of KrasG12D in Immunocompetent Models of Pancreatic Cancer. Cancer Discov. 2023, 13, 298–311. [Google Scholar] [CrossRef]
- Zhou, C.; Li, W.; Song, Z.; Zhang, Y.; Huang, D.; Yang, Z.; Zhou, M.; Mao, R.; Huang, C.; Li, X.; et al. LBA33 A first-in-human phase I study of a novel KRAS G12D inhibitor HRS-4642 in patients with advanced solid tumors harboring KRAS G12D mutation. Ann. Oncol. 2023, 34, S1273. [Google Scholar] [CrossRef]
- Nagashima, T.; Inamura, K.; Nishizono, Y.; Suzuki, A.; Tanaka, H.; Yoshinari, T.; Yamanaka, Y. ASP3082, a First-in-class novel KRAS G12D degrader, exhibits remarkable anti-tumor activity in KRAS G12D mutated cancer models. Eur. J. Cancer 2022, 174, S30. [Google Scholar] [CrossRef]
- Tolcher, A.W.; Park, W.; Wang, J.S.; Spira, A.I.; Janne, P.A.; Lee, H.-J.; Gill, S.; LoRusso, P.; Herzberg, B.; Goldman, J.W.; et al. Trial in progress: A phase 1, first-in-human, open-label, multicenter, dose-escalation and dose-expansion study of ASP3082 in patients with previously treated advanced solid tumors and KRAS G12D mutations. J. Clin. Oncol. 2023, 41, TPS764. [Google Scholar] [CrossRef]
- Taylor, P. AstraZeneca Joins KRAS Push in Cancer with Chinese Deal. 2023. Available online: https://pharmaphorum.com/news/astrazeneca-joins-kras-push-cancer-chinese-deal (accessed on 1 September 2022).
- Li, L.; Liu, J.; Yang, Z.; Zhao, H.; Deng, B.; Ren, Y.; Mai, R.; Huang, J.; Chen, J. Discovery of Thieno[2,3-d]pyrimidine-based KRAS G12D inhibitors as potential anticancer agents via combinato-rial virtual screening. Eur. J. Med. Chem. 2022, 233, 114243. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhang, H.; Li, J.; Niu, M.-M.; Zhou, Y.; Qu, Y. Discovery of potent and noncovalent KRASG12D inhibitors: Structure-based virtual screening and biological evaluation. Front. Pharmacol. 2022, 13, 1094887. [Google Scholar] [CrossRef]
- Lin, J.; Sahakian, D.C.; de Morais, S.M.F.; Xu, J.J.; Polzer, R.J.; Winter, S.M. The Role of Absorption, Distribution, Metabolism, Excretion and Toxicity in Drug Discovery. Curr. Top. Med. Chem. 2003, 3, 1125–1154. [Google Scholar] [CrossRef] [PubMed]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef] [PubMed]
- Hou, T.; Wang, J.; Li, Y.; Wang, W. Assessing the Performance of the MM/PBSA and MM/GBSA Methods. 1. The Accuracy of Binding Free Energy Calculations Based on Molecular Dynamics Simulations. J. Chem. Inf. Model. 2011, 51, 69–82. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Docking Scores (kcal/mol) | Compound | Docking Scores (kcal/mol) |
|---|---|---|---|
| MRTX1133 | −12.18 | 274371 | −8.72 |
| 485643 | −10.11 | 1121521 | −8.49 |
| 1002187 | −9.71 | 347398 | −8.48 |
| 1166303 | −9.31 | 1125857 | −8.48 |
| 1243333 | −9.19 | 1251964 | −8.45 |
| 502065 | −9.05 | 1509470 | −8.35 |
| 294749 | −9.04 | 1185436 | −8.21 |
| 1079487 | −9.02 | 909401 | −8.11 |
| 1307165 | −9.00 | 1205378 | −8.11 |
| 1280969 | −8.96 | 1292268 | −8.04 |
| 1194622 | −8.87 |
| Compound | Molecular Weight | logP (o/w) a | logS b | Caco c | PMDCK d | Percent Human Oral Absorption e |
|---|---|---|---|---|---|---|
| MRTX1133 | 600.64 | 5.59 | −6.77 | 51.67 | 84.37 | 64.45 |
| 485643 | 495.68 | 3.10 | −2.26 | 110.38 | 119.74 | 81.66 |
| 1002187 | 485.55 | 4.74 | −4.69 | 839.28 | 2372.93 | 100.00 |
| 1166303 | 423.56 | 2.77 | −3.05 | 243.47 | 157.30 | 85.87 |
| 1243333 | 341.39 | 2.26 | −4.37 | 318.84 | 238.58 | 84.97 |
| 502065 | 445.56 | 3.79 | −4.57 | 187.40 | 145.95 | 89.84 |
| 294749 | 386.49 | 1.61 | −2.38 | 53.46 | 69.44 | 67.30 |
| 1079487 | 362.47 | 3.43 | −3.41 | 151.03 | 139.69 | 86.00 |
| 1307165 | 448.61 | 3.56 | −3.40 | 557.12 | 501.26 | 96.95 |
| 1280969 | 463.97 | 4.36 | −6.60 | 715.90 | 1315.61 | 100.00 |
| 1194622 | 450.63 | 3.33 | −4.48 | 60.07 | 47.50 | 78.29 |
| 274371 | 415.42 | 2.57 | −4.48 | 25.02 | 15.63 | 67.02 |
| 1121521 | 327.39 | 1.57 | −3.15 | 103.17 | 46.99 | 72.18 |
| 347398 | 466.04 | 3.48 | −2.98 | 9.42 | 676.19 | 64.75 |
| 1125857 | 384.50 | 2.81 | −4.32 | 722.39 | 1305.50 | 94.58 |
| 1251964 | 471.01 | 4.47 | −4.53 | 15.08 | 629.79 | 74.21 |
| 1509470 | 395.93 | 4.06 | −4.24 | 119.25 | 149.77 | 87.89 |
| 1185436 | 404.47 | 2.42 | −4.05 | 442.24 | 348.33 | 88.44 |
| 909401 | 427.50 | 3.61 | −5.02 | 167.70 | 79.44 | 87.88 |
| 1205378 | 379.50 | 3.35 | −4.60 | 392.49 | 199.16 | 92.96 |
| 1292268 | 443.55 | 4.06 | −5.38 | 329.09 | 164.63 | 95.78 |
| Compound | MMGBSA ΔG (kcal/mol) | Compound | MMGBSA ΔG (kcal/mol) |
|---|---|---|---|
| MRTX1133 | −60.58 | 1292268 | −20.39 |
| 1307165 | −41.94 | 1185436 | −18.34 |
| 1194622 | −31.71 | 1002187 | −17.84 |
| 1166303 | −30.47 | 1121521 | −16.01 |
| 502065 | −27.81 | 1509470 | −15.02 |
| 1243333 | −26.11 | 1280969 | −13.39 |
| 909401 | −25.33 | 1079487 | −10.02 |
| 294749 | −24.63 | 1125857 | −5.42 |
| 485643 | −22.59 | 1205378 | −1.68 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Du, Z.; Tu, G.; Gong, Y.; Fu, X.; Wu, Q.; Long, G. Discovery of Novel Noncovalent KRAS G12D Inhibitors through Structure-Based Virtual Screening and Molecular Dynamics Simulations. Molecules 2024, 29, 1229. https://doi.org/10.3390/molecules29061229
Du Z, Tu G, Gong Y, Fu X, Wu Q, Long G. Discovery of Novel Noncovalent KRAS G12D Inhibitors through Structure-Based Virtual Screening and Molecular Dynamics Simulations. Molecules. 2024; 29(6):1229. https://doi.org/10.3390/molecules29061229
Chicago/Turabian StyleDu, Zhenya, Gao Tu, Yaguo Gong, Xiangzheng Fu, Qibiao Wu, and Guankui Long. 2024. "Discovery of Novel Noncovalent KRAS G12D Inhibitors through Structure-Based Virtual Screening and Molecular Dynamics Simulations" Molecules 29, no. 6: 1229. https://doi.org/10.3390/molecules29061229
APA StyleDu, Z., Tu, G., Gong, Y., Fu, X., Wu, Q., & Long, G. (2024). Discovery of Novel Noncovalent KRAS G12D Inhibitors through Structure-Based Virtual Screening and Molecular Dynamics Simulations. Molecules, 29(6), 1229. https://doi.org/10.3390/molecules29061229