
Selectivity Studies and Free Energy Calculations of AKT Inhibitors


Abstract
:1. Introduction
2. Results and Discussion
2.1. Multiple Sequence Alignment
2.2. Building Model Proteins
2.3. Docking Scores and Validation
2.3.1. Validation of Method
2.3.2. Prediction of the AKT3 Ligand Binding
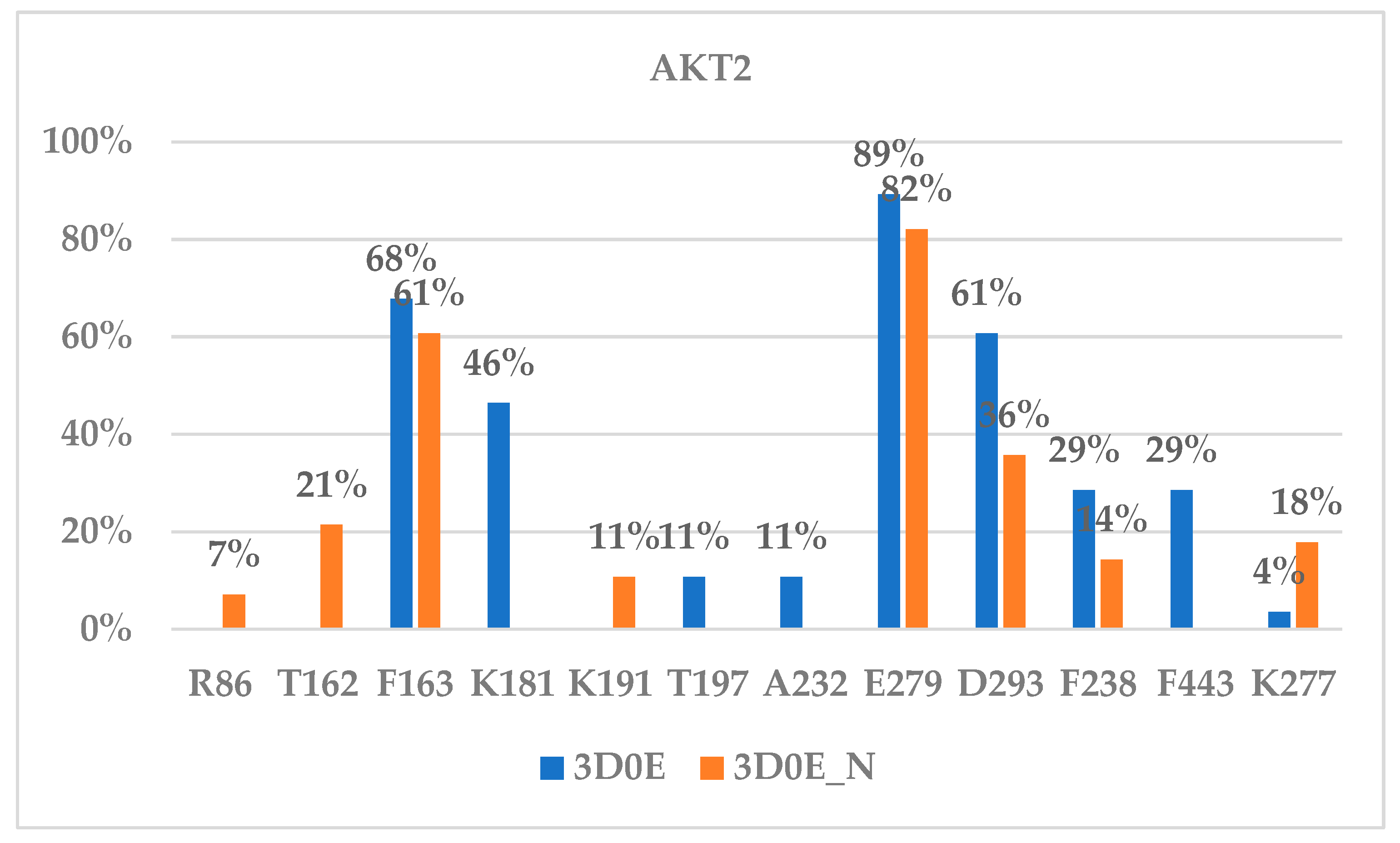
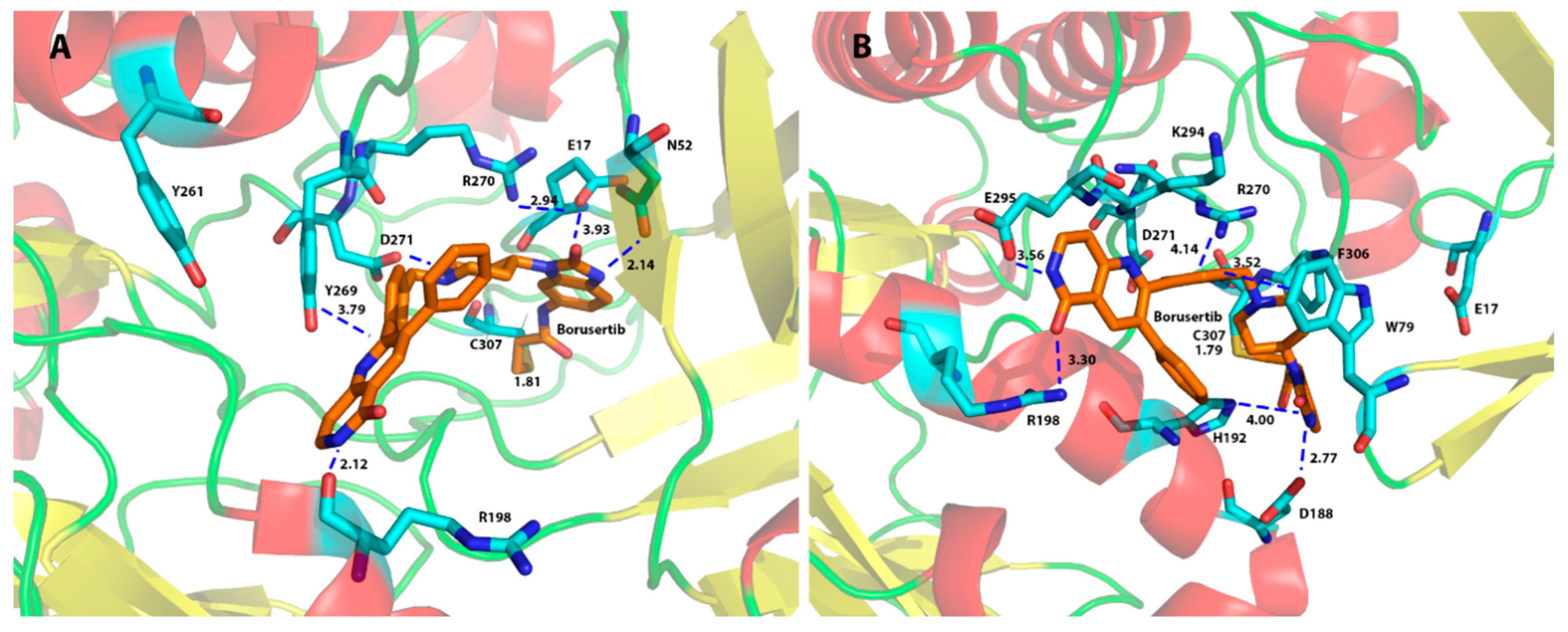
2.3.3. Binding Interaction of AKT Inhibitors
2.4. Binding Selectivity and Drug Design Perspectives
3. Materials and Methods
3.1. Multiple Sequence Alignment
3.2. Preparation of Protein Structures
3.3. Preparation of a Homology Model for AKT3
3.4. Preparation of Ligand Structures
3.5. Schrödinger Covalent Docking
3.6. The Calculation of MT-Based Free Energy
3.7. Protein–Ligand Interactions and Binding Affinity
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Huang, J.; Chen, L.; Wu, J.; Ai, D.; Zhang, J.Q.; Chen, T.G.; Wang, L. Targeting the PI3K/AKT/mTOR signaling pathway in the treatment of human diseases: Current status, trends, and solutions. J. Med. Chem. 2022, 65, 16033–16061. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.S.; Cui, W. Proliferation, survival and metabolism: The role of PI3K/AKT/mTOR signalling in pluripotency and cell fate determination. Development 2016, 143, 3050–3060. [Google Scholar] [CrossRef] [PubMed]
- Tan, A.C. Targeting the PI3K/AKT/mTOR pathway in non-small cell lung cancer (NSCLC). Thorac. Cancer 2020, 11, 511–518. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Liu, G.; Guo, J.; Su, Z. The PI3K/AKT pathway in obesity and type 2 diabetes. Int. J. Biol. Sci. 2018, 14, 1483–1496. [Google Scholar] [CrossRef]
- Santi, S.A.; Lee, H. The Akt isoforms are present at distinct subcellular locations. Am. J. Physiol. Cell Physiol. 2010, 298, C580–C591. [Google Scholar] [CrossRef]
- Cho, H.; Mu, J.; Kim, J.K.; Thorvaldsen, J.L.; Chu, Q.; Crenshaw, E.B., 3rd; Kaestner, K.H.; Bartolomei, M.S.; Shulman, G.I.; Birnbaum, M.J. Insulin resistance and a diabetes mellitus-like syndrome in mice lacking the protein kinase AKT2 (PKB beta). Science 2001, 292, 1728–1731. [Google Scholar] [CrossRef]
- DuBois, J.C.; Ray, A.K.; Gruber, R.C.; Zhang, Y.; Aflakpui, R.; Macian-Juan, F.; Shafit-Zagardo, B. AKT3-mediated protection against inflammatory demyelinating disease. Front. Immunol. 2019, 10, 1738. [Google Scholar] [CrossRef]
- Linnerth-Petrik, N.M.; Santry, L.A.; Petrik, J.J.; Wootton, S.K. Opposing functions of Akt isoforms in lung tumor initiation and progression. PLoS ONE 2014, 9, e94595. [Google Scholar] [CrossRef]
- Riggio, M.; Perrone, M.C.; Polo, M.L.; Rodriguez, M.J.; May, M.; Abba, M.; Lanari, C.; Novaro, V. AKT1 and AKT2 isoforms play distinct roles during breast cancer progression through the regulation of specific downstream proteins. Sci. Rep. 2017, 7, 44244. [Google Scholar] [CrossRef]
- Chu, N.; Viennet, T.; Bae, H.; Salguero, A.; Boeszoermenyi, A.; Arthanari, H.; Cole, P.A. The structural determinants of PH domain-mediated regulation of Akt revealed by segmental labeling. Elife 2020, 9, e59151. [Google Scholar] [CrossRef]
- Hanada, M.; Feng, J.; Hemmings, B.A. Structure, regulation and function of PKB/AKT--a major therapeutic target. Biochim. Biophys. Acta 2004, 1697, 3–16. [Google Scholar] [CrossRef]
- Szymonowicz, K.; Oeck, S.; Malewicz, N.M.; Jendrossek, V. New Insights into Protein Kinase B/Akt Signaling: Role of Localized Akt Activation and Compartment-Specific Target Proteins for the Cellular Radiation Response. Cancers 2018, 10, 78. [Google Scholar] [CrossRef]
- Yi, K.H.; Lauring, J. Recurrent AKT mutations in human cancers: Functional consequences and effects on drug sensitivity. Oncotarget 2016, 7, 4241–4251. [Google Scholar] [CrossRef]
- Turner, N.C.; Oliveira, M.; Howell, S.J.; Dalenc, F.; Cortes, J.; Gomez Moreno, H.L.; Hu, X.; Jhaveri, K.; Krivorotko, P.; Loibl, S.; et al. Capivasertib in Hormone Receptor-Positive Advanced Breast Cancer. N. Engl. J. Med. 2023, 388, 2058–2070. [Google Scholar] [CrossRef]
- Spencer, A.; Yoon, S.S.; Harrison, S.J.; Morris, S.R.; Smith, D.A.; Brigandi, R.A.; Gauvin, J.; Kumar, R.; Opalinska, J.B.; Chen, C. The novel AKT inhibitor afuresertib shows favorable safety, pharmacokinetics, and clinical activity in multiple myeloma. Blood 2014, 124, 2190–2195. [Google Scholar] [CrossRef]
- Shi, Z.; Wulfkuhle, J.; Nowicka, M.; Gallagher, R.I.; Saura, C.; Nuciforo, P.G.; Calvo, I.; Andersen, J.; Passos-Coelho, J.L.; Gil-Gil, M.J.; et al. Functional mapping of AKT signaling and biomarkers of response from the FAIRLANE trial of neoadjuvant ipatasertib plus paclitaxel for triple-negative breast cancer. Clin. Cancer Res. 2022, 28, 993–1003. [Google Scholar] [CrossRef]
- Ragon, B.K.; Odenike, O.; Baer, M.R.; Stock, W.; Borthakur, G.; Patel, K.; Han, L.; Chen, H.; Ma, H.; Joseph, L.; et al. Oral MEK 1/2 inhibitor trametinib in combination with AKT inhibitor GSK2141795 in patients with acute myeloid leukemia with RAS mutations: A phase II study. Clin. Lymphoma Myeloma Leuk. 2019, 19, 431–440.e413. [Google Scholar] [CrossRef]
- Levy, D.S.; Kahana, J.A.; Kumar, R. AKT inhibitor, GSK690693, induces growth inhibition and apoptosis in acute lymphoblastic leukemia cell lines. Blood 2009, 113, 1723–1729. [Google Scholar] [CrossRef]
- Weisner, J.; Gontla, R.; van der Westhuizen, L.; Oeck, S.; Ketzer, J.; Janning, P.; Richters, A.; Mühlenberg, T.; Fang, Z.; Taher, A.; et al. Covalent-allosteric kinase inhibitors. Angew. Chem. Int. Ed. 2015, 54, 10313–10316. [Google Scholar] [CrossRef]
- Weisner, J.; Landel, I.; Reintjes, C.; Uhlenbrock, N.; Trajkovic-Arsic, M.; Dienstbier, N.; Hardick, J.; Ladigan, S.; Lindemann, M.; Smith, S.; et al. Preclinical efficacy of covalent-allosteric AKT inhibitor borussertib in combination with trametinib in KRAS-mutant pancreatic and colorectal cancer. Cancer Res. 2019, 79, 2367–2378. [Google Scholar] [CrossRef]
- Hirai, H.; Sootome, H.; Nakatsuru, Y.; Miyama, K.; Taguchi, S.; Tsujioka, K.; Ueno, Y.; Hatch, H.; Majumder, P.K.; Pan, B.S.; et al. MK-2206, an allosteric Akt inhibitor, enhances antitumor efficacy by standard chemotherapeutic agents or molecular targeted drugs in vitro and in vivo. Mol. Cancer Ther. 2010, 9, 1956–1967. [Google Scholar] [CrossRef] [PubMed]
- Andrikopoulou, A.; Chatzinikolaou, S.; Panourgias, E.; Kaparelou, M.; Liontos, M.; Dimopoulos, M.A.; Zagouri, F. The emerging role of capivasertib in breast cancer. Breast 2022, 63, 157–167. [Google Scholar] [CrossRef] [PubMed]
- Che, J.; Dai, X.; Gao, J.; Sheng, H.; Zhan, W.; Lu, Y.; Li, D.; Gao, Z.; Jin, Z.; Chen, B.; et al. Discovery of N-((3S,4S)-4-(3,4-difluorophenyl)piperidin-3-yl)-2-fluoro-4-(1-methyl-1H-pyrazol-5-yl)benzamide (Hu7691), a potent and selective Akt inhibitor that enables decrease of cutaneous toxicity. J. Med. Chem. 2021, 64, 12163–12180. [Google Scholar] [CrossRef]
- Landel, I.; Quambusch, L.; Depta, L.; Rauh, D. Spotlight on AKT: Current therapeutic challenges. ACS Med. Chem. Lett. 2020, 11, 225–227. [Google Scholar] [CrossRef]
- Quambusch, L.; Landel, I.; Depta, L.; Weisner, J.; Uhlenbrock, N.; Müller, M.P.; Glanemann, F.; Althoff, K.; Siveke, J.T.; Rauh, D. Covalent-allosteric inhibitors to achieve Akt isoform-selectivity. Angew. Chem. Int. Ed. 2019, 58, 18823–18829. [Google Scholar] [CrossRef] [PubMed]
- Heerding, D.A.; Rhodes, N.; Leber, J.D.; Clark, T.J.; Keenan, R.M.; Lafrance, L.V.; Li, M.; Safonov, I.G.; Takata, D.T.; Venslavsky, J.W.; et al. Identification of 4-(2-(4-amino-1,2,5-oxadiazol-3-yl)-1-ethyl-7-{[(3S)-3-piperidinylmethyl]oxy}-1H-imidazo[4,5-c]pyridin-4-yl)-2-methyl-3-butyn-2-ol (GSK690693), a novel inhibitor of AKT kinase. J. Med. Chem. 2008, 51, 5663–5679. [Google Scholar] [CrossRef] [PubMed]
- AAD24196.1, AKT3 Homo Sapiens (Human) Sequence Was Taken from the GenBank. Available online: https://www.ncbi.nlm.nih.gov/gene/?term=AAD24196.1 (accessed on 5 June 2023).
- The Clustal Omega World-Wide Web. Available online: https://www.ebi.ac.uk/jdispatcher/msa/clustalo (accessed on 5 June 2023).
- MOE. The Molecular Operating Environment (MOE); Chemical Computing Group Inc.: Montreal, QC, Canada, 2022; Version 2022.02. [Google Scholar]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Varadi, M.; Anyango, S.; Deshpande, M.; Nair, S.; Natassia, C.; Yordanova, G.; Yuan, D.; Stroe, O.; Wood, G.; Laydon, A.; et al. AlphaFold protein structure database: Massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 2022, 50, D439–D444. [Google Scholar] [CrossRef]
- Colovos, C.; Yeates, T.O. Verification of protein structures: Patterns of nonbonded atomic interactions. Protein Sci. 1993, 2, 1511–1519. [Google Scholar] [CrossRef]
- Quambusch, L.; Depta, L.; Landel, I.; Lubeck, M.; Kirschner, T.; Nabert, J.; Uhlenbrock, N.; Weisner, J.; Kostka, M.; Levy, L.M.; et al. Cellular model system to dissect the isoform-selectivity of Akt inhibitors. Nat. Commun. 2021, 12, 5297. [Google Scholar] [CrossRef]
- Auguin, D.; Barthe, P.; Augé-Sénégas, M.T.; Stern, M.H.; Noguchi, M.; Roumestand, C. Solution structure and backbone dynamics of the pleckstrin homology domain of the human protein kinase B (PKB/Akt). Interaction with inositol phosphates. J. Biomol. NMR 2004, 28, 137–155. [Google Scholar] [CrossRef]
- Zheng, Z.; Merz, K.M., Jr. Development of the knowledge-based and empirical combined scoring algorithm (KECSA) to score protein–ligand interactions. J. Chem. Inf. Model. 2013, 53, 1073–1083. [Google Scholar] [CrossRef]
- Zheng, Z.; Borbulevych, O.Y.; Liu, H.; Deng, J.; Martin, R.I.; Westerhoff, L.M. MovableType software for fast free energy-based virtual screening: Protocol development, deployment, validation, and assessment. J. Chem. Inf. Model. 2020, 60, 5437–5456. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Liu, Z.; Liu, H.; Westerhoff, L.M.; Zheng, Z. Free energy calculations using the Movable Type method with molecular dynamics driven protein-ligand sampling. J. Chem. Inf. Model. 2022, 62, 5645–5665. [Google Scholar] [CrossRef]
- Sabbah, D.A.; Vennerstrom, J.L.; Zhong, H.A. Binding selectivity studies of phosphoinositide 3-kinases using free energy calculations. J. Chem. Inf. Model. 2012, 52, 3213–3224. [Google Scholar] [CrossRef]
- Zhong, H.A.; Santos, E.M.; Vasileiou, C.; Zheng, Z.; Geiger, J.H.; Borhan, B.; Merz, K.M., Jr. Free-energy-based protein design: Re-engineering cellular retinoic acid binding protein II assisted by the Moveable-Type approach. J. Am. Chem. Soc. 2018, 140, 3483–3486. [Google Scholar] [CrossRef] [PubMed]
- Al Hasan, M.; Sabirianov, M.; Redwine, G.; Goettsch, K.; Yang, S.X.; Zhong, H.A. Binding and selectivity studies of phosphatidylinositol 3-kinase (PI3K) inhibitors. J. Mol. Graph. Model. 2023, 121, 108433. [Google Scholar] [CrossRef] [PubMed]
- Zhong, H.A.; Almahmoud, S. Docking and selectivity studies of covalently bound Janus kinase 3 inhibitors. Int. J. Mol. Sci. 2023, 24, 6023. [Google Scholar] [CrossRef]
- Zhong, H.; Carlson, H.A. Computational studies and peptidomimetic design for the human p53-MDM2 complex. Proteins 2005, 58, 222–234. [Google Scholar] [CrossRef]
- Schrödinger Suite 2022-1 Protein Preparation Wizard, Maestro, Protein Grid Generation, Glide, Macromodel, Epik and Covalent Dock (CovDock); Schrödinger, LLC: New York, NY, USA, 2022.
- Lapierre, J.M.; Eathiraj, S.; Vensel, D.; Liu, Y.; Bull, C.O.; Cornell-Kennon, S.; Iimura, S.; Kelleher, E.W.; Kizer, D.E.; Koerner, S.; et al. Discovery of 3-(3-(4-(1-Aminocyclobutyl)phenyl)-5-phenyl-3H-imidazo[4,5-b]pyridin-2-yl)pyridin-2-amine (ARQ 092): An orally bioavailable, selective, and potent allosteric AKT inhibitor. J. Med. Chem. 2016, 59, 6455–6469. [Google Scholar] [CrossRef]
- Yu, X.; Xu, J.; Cahuzac, K.M.; Xie, L.; Shen, Y.; Chen, X.; Liu, J.; Parsons, R.E.; Jin, J. Novel allosteric inhibitor-derived AKT proteolysis targeting chimeras (PROTACs) enable potent and selective AKT degradation in KRAS/BRAF mutant cells. J. Med. Chem. 2022, 65, 14237–14260. [Google Scholar] [CrossRef] [PubMed]
- Xu, F.; Zhang, X.; Chen, Z.; He, S.; Guo, J.; Yu, L.; Wang, Y.; Hou, C.; Ai-Furas, H.; Zheng, Z.; et al. Discovery of isoform-selective Akt3 degraders overcoming osimertinib-induced resistance in non-small cell lung cancer cells. J. Med. Chem. 2022, 65, 14032–14048. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.; Wu, J.; Wang, L.; Ji, X.; Wu, Y.; Miao, L.; Chen, D.; Zhang, L.; Wu, Y.; Feng, H.; et al. Discovery of clinical candidate NTQ1062 as a potent and bioavailable Akt inhibitor for the treatment of human tumors. J. Med. Chem. 2022, 65, 8144–8168. [Google Scholar] [CrossRef] [PubMed]
- Cantley, L.C.; Hopkins, B.; Goncalves, M.; Mukherjee, S. Combination therapy for PI3K-associated disease or disorder. United States Patent Application US 15/733,852, 5 December 2019. [Google Scholar]
- Xu, J.; Yu, X.; Martin, T.C.; Bansal, A.; Cheung, K.; Lubin, A.; Stratikopoulos, E.; Cahuzac, K.M.; Wang, L.; Xie, L.; et al. AKT Degradation selectively inhibits the growth of PI3K/PTEN pathway-mutant cancers with wild-type KRAS and BRAF by destabilizing Aurora kinase B. Cancer Discov. 2021, 11, 3064–3089. [Google Scholar] [CrossRef]
- You, I.; Erickson, E.C.; Donovan, K.A.; Eleuteri, N.A.; Fischer, E.S.; Gray, N.S.; Toker, A. Discovery of an AKT degrader with prolonged inhibition of downstream signaling. Cell Chem. Biol. 2020, 27, 66–73.e7. [Google Scholar] [CrossRef]
- Page, N.; Wappett, M.; O’Dowd, C.R.; O’Rourke, M.; Gavory, G.; Zhang, L.; Rountree, J.S.S.; Jordan, L.; Barker, O.; Gibson, H.; et al. Identification and development of a subtype-selective allosteric AKT inhibitor suitable for clinical development. Sci. Rep. 2022, 12, 15715. [Google Scholar] [CrossRef]







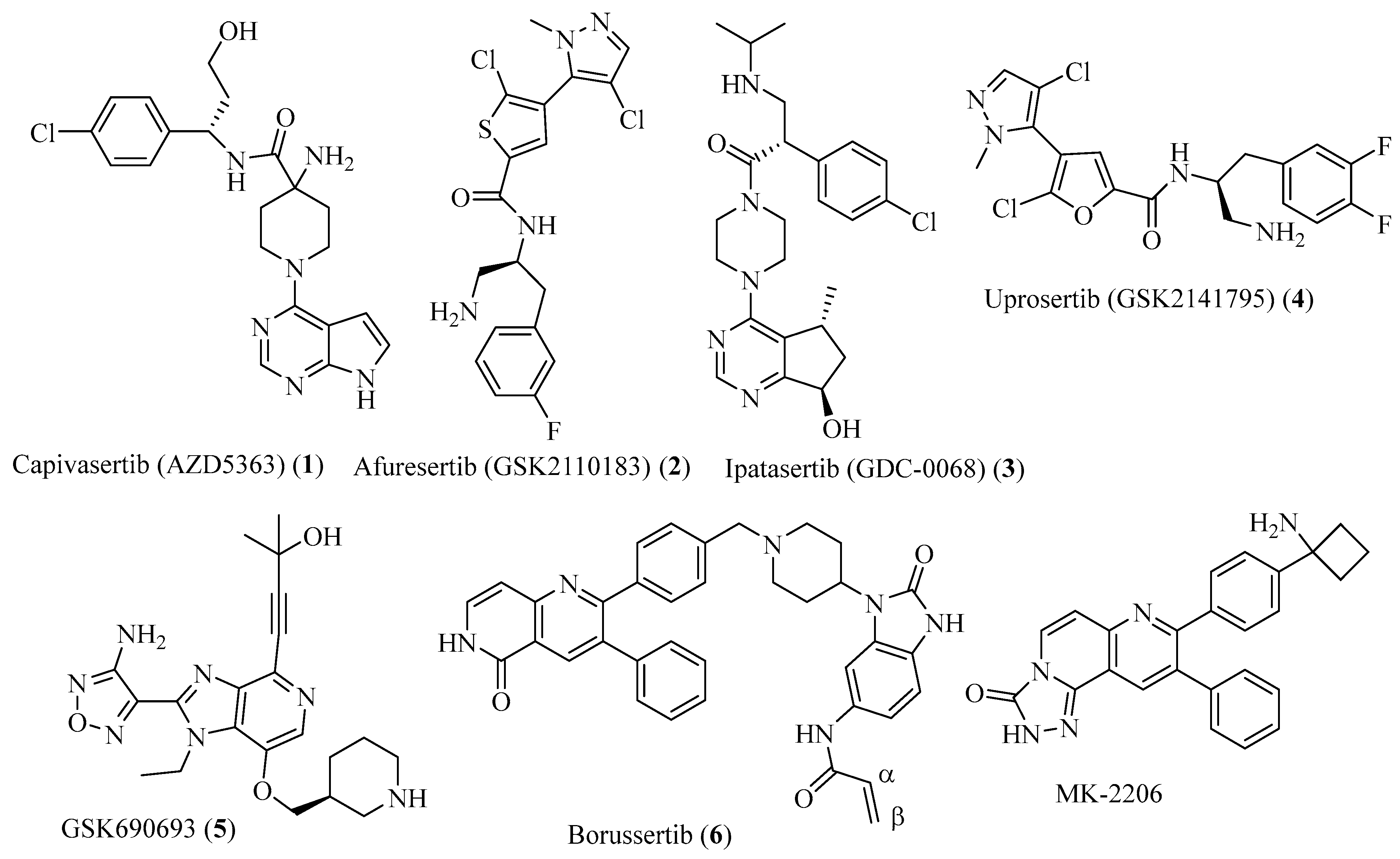{kind=link}
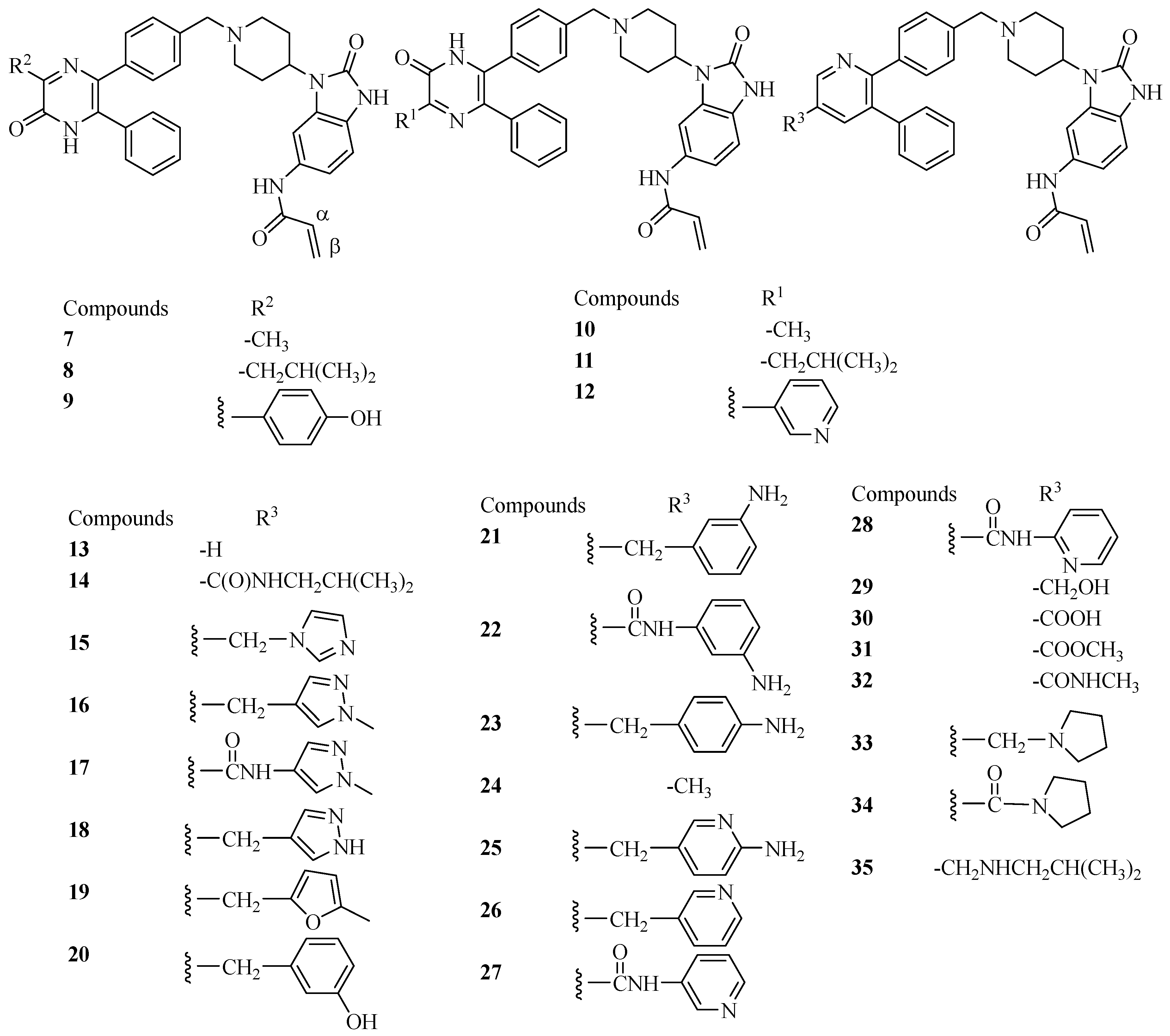{kind=link}
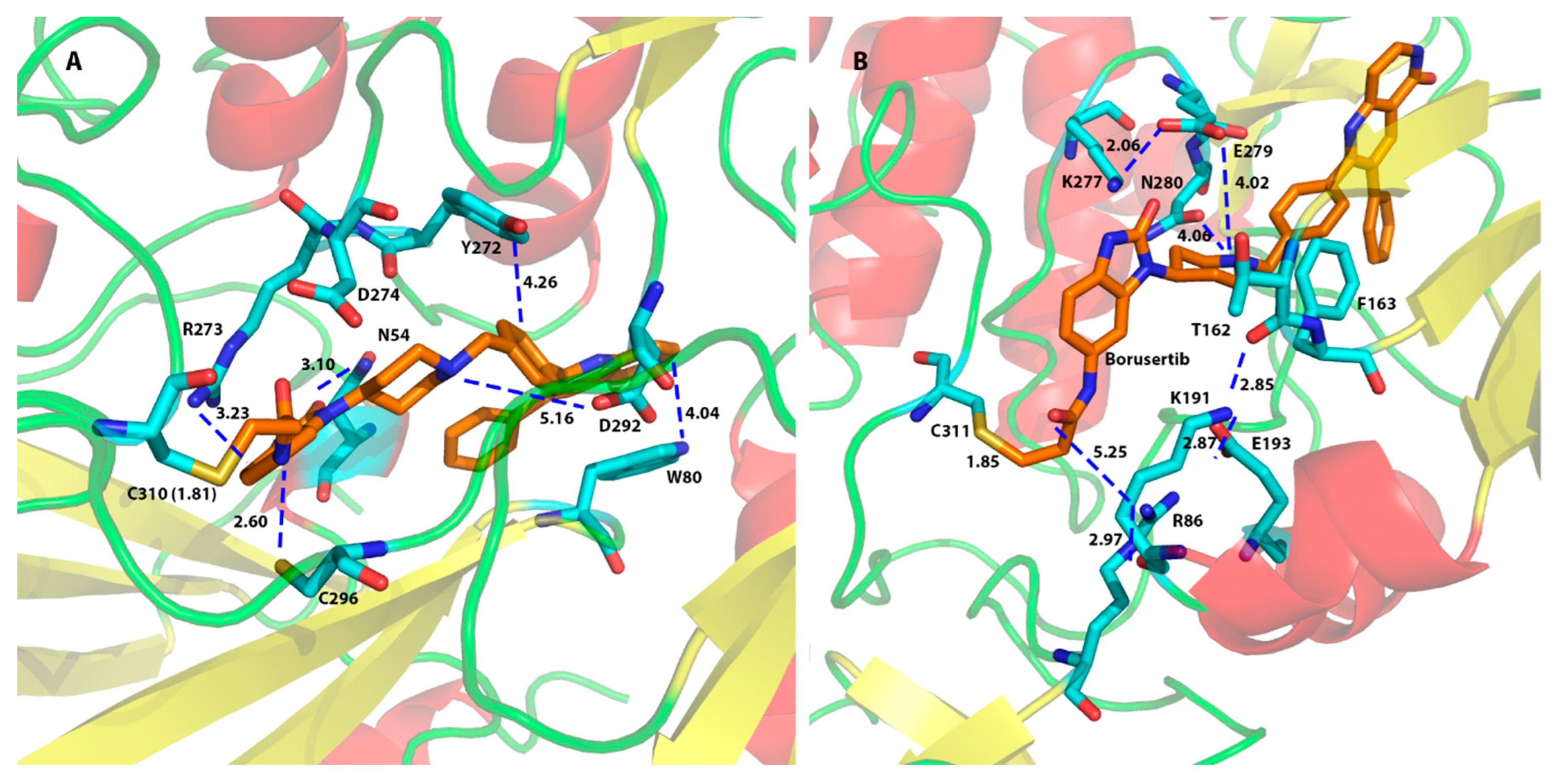{kind=link}
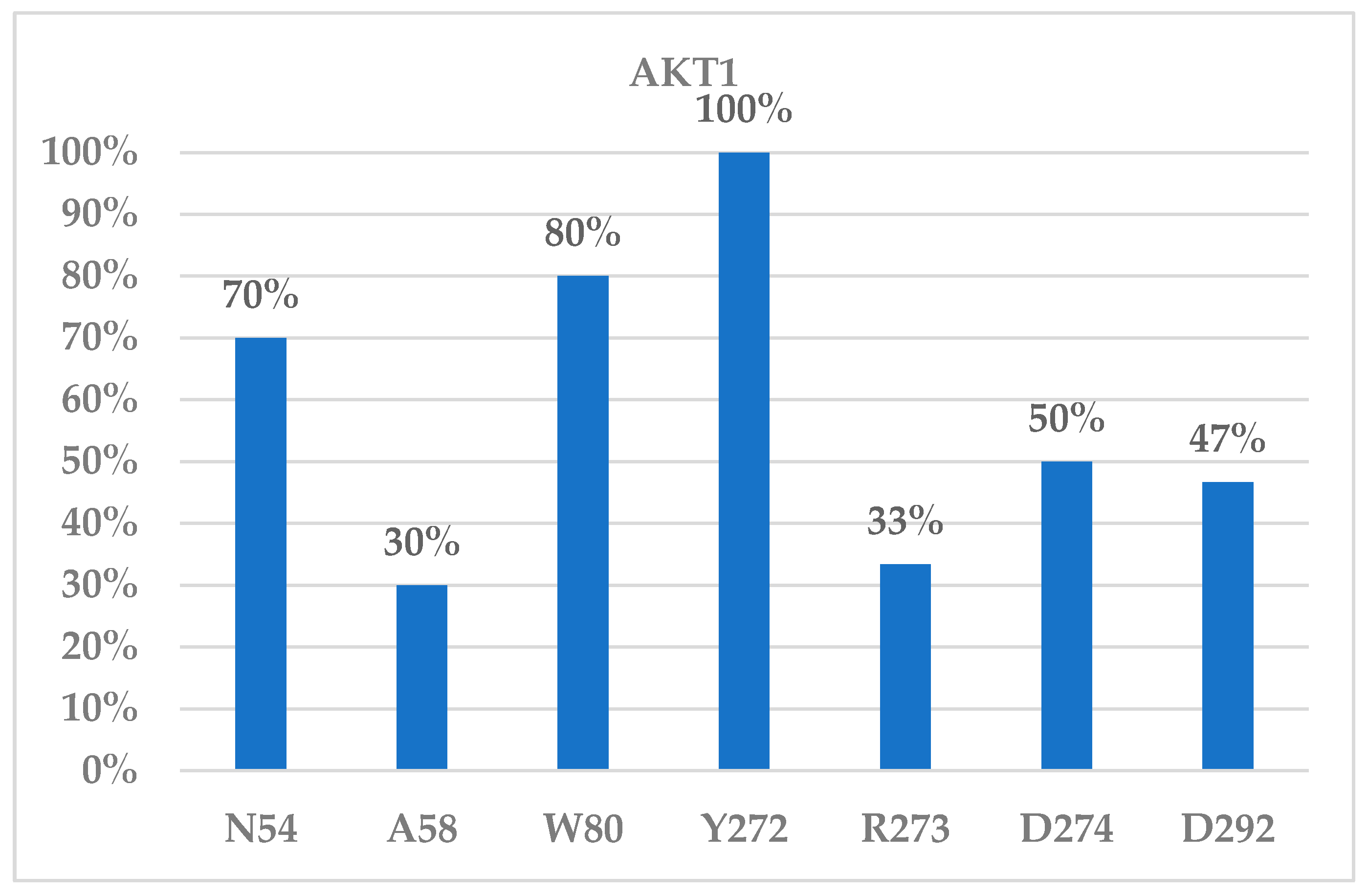{kind=link}
{kind=link}
{kind=link}
| Subtypes | Residues | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 6S9W/AKT1 | N54 | W80 | T160 | F161 | K179 | R200 | Y272 | R273 | D274 | E278 | D292 | E298 | C310 |
| 3D0E/AKT2 | N54 | W80 | T162 | F163 | K181 | F238 | Y273 | R274 | D275 | E279 | D293 | E299 | C311 |
| HM/AF/AKT3 | N53 | W79 | T158 | F159 | K177 | R198 | Y269 | R270 | D271 | E275 | D289 | E295 | C307 |
| Compd. | Ki (nM) | ∆Gexp | 6S9W | ΔΔG (6S9W) | 6S9W_L | ΔΔG (6S9W_L) | MT | ∆∆G (MT) |
|---|---|---|---|---|---|---|---|---|
| 6 | 2 | −11.87 | −12.94 | 1.07 | −14.695 | 2.83 | −14.62 | 2.75 |
| 7 | 71 | −9.75 | −11.88 | 2.13 | −14.31 | 4.56 | −12.75 | 3.00 |
| 8 | 1243 | −8.06 | −13.85 | 5.80 | −14.08 | 6.02 | −14.24 | 6.18 |
| 10 | 59 | −9.86 | −11.71 | 1.85 | −14.58 | 4.72 | −13.10 | 3.24 |
| 11 | 1432 | −7.97 | −12.75 | 4.78 | −14.58 | 6.61 | −14.88 | 6.91 |
| 13 | 6 | −11.22 | −12.03 | 0.81 | −15.56 | 4.35 | −11.71 | 0.50 |
| 15 | 9 | −10.98 | −12.49 | 1.51 | −13.88 | 2.90 | −12.34 | 1.36 |
| 17 | 8 | −11.05 | −12.28 | 1.23 | −9.99 | 1.06 | −15.22 | 4.17 |
| 22 | 6 | −11.22 | −12.65 | 1.43 | −7.65 | 3.56 | −16.64 | 5.43 |
| 27 | 9 | −10.98 | −12.11 | 1.13 | −9.13 | 1.85 | −16.11 | 5.14 |
| mean | 2.18 | 3.85 | 3.87 | |||||
| stdev | 1.70 | 1.75 | 2.08 |
| Compd. | IC50 (nM) | ∆Gexp | 3D0E | ∆∆G (3D0E) | 3D0E_N | ∆∆G (3D0E_N) | MT (3D0E) | ΔΔG (MT, 3D0E) | MT (3D0E_N) | ΔΔG (MT, 3D0E_N) |
|---|---|---|---|---|---|---|---|---|---|---|
| 6 | 14 | −10.71 | −10.49 | 0.22 | −9.00 | 1.71 | −8.94 | 1.77 | −11.19 | 0.47 |
| 7 | 1569 | −7.92 | −8.60 | 0.68 | −8.05 | 0.13 | −9.96 | 2.04 | −9.75 | 1.83 |
| 10 | 13,030 | −6.66 | −8.37 | 1.71 | −8.22 | 1.55 | −8.84 | 2.17 | −10.27 | 3.60 |
| 11 | 119 | −9.45 | −9.66 | 0.21 | −8.38 | 1.07 | −13.38 | 3.93 | −11.73 | 2.28 |
| 12 | 5033 | −7.23 | −7.14 | 0.09 | −6.39 | 0.84 | −11.16 | 3.93 | −12.10 | 4.87 |
| 13 | 599 | −8.49 | −9.12 | 0.63 | −9.17 | 0.69 | −12.61 | 4.12 | −11.09 | 2.60 |
| 14 | 94 | −9.59 | −6.31 | 3.28 | −7.53 | 2.06 | −11.21 | 1.63 | −12.12 | 2.54 |
| 15 | 76 | −9.71 | −9.09 | 0.62 | −9.89 | 0.18 | −11.46 | 1.75 | −11.58 | 1.87 |
| 16 | 251 | −9.00 | −10.97 | 1.96 | −11.53 | 2.53 | −9.43 | 0.43 | −11.53 | 2.53 |
| 17 | 53 | −9.92 | −7.75 | 2.17 | −7.60 | 2.32 | −12.27 | 2.35 | −10.61 | 0.69 |
| 18 | 23 | −10.42 | −10.31 | 0.11 | −9.66 | 0.76 | −9.61 | 0.81 | −10.92 | 0.50 |
| 19 | 1400 | −7.99 | −7.77 | 0.21 | −8.33 | 0.34 | −10.67 | 2.69 | −10.87 | 2.89 |
| 20 | 682 | −8.41 | −8.21 | 0.20 | −8.65 | 0.24 | −11.41 | 3.00 | −11.30 | 2.89 |
| 21 | 133 | −9.38 | −8.80 | 0.58 | −7.75 | 1.63 | −12.29 | 2.91 | −11.88 | 2.50 |
| 22 | 39 | −10.11 | −8.84 | 1.26 | −8.50 | 1.61 | −10.24 | 0.14 | −12.29 | 2.18 |
| 23 | 511 | −8.58 | −4.57 | 4.01 | −9.37 | 0.78 | −9.58 | 1.00 | −13.67 | 5.09 |
| 24 | 150 | −9.31 | −9.77 | 0.47 | −8.38 | 0.93 | −11.42 | 2.11 | −10.10 | 0.79 |
| 25 | 175 | −9.22 | −8.29 | 0.93 | −7.30 | 1.92 | −10.73 | 1.51 | −11.30 | 2.08 |
| 26 | 99 | −9.55 | −8.85 | 0.71 | −10.22 | 0.66 | −10.76 | 1.21 | −10.87 | 1.31 |
| 27 | 55 | −9.90 | −8.75 | 1.15 | −8.32 | 1.58 | −10.80 | 0.90 | −12.09 | 2.19 |
| 28 | 151 | −9.30 | −8.24 | 1.06 | −8.31 | 0.99 | −10.88 | 1.58 | −12.35 | 3.05 |
| 29 | 961 | −8.21 | −11.45 | 3.24 | −9.84 | 1.63 | −13.52 | 5.31 | −11.13 | 2.92 |
| 30 | 248 | −9.01 | −6.92 | 2.09 | −8.44 | 0.57 | −14.50 | 5.49 | −10.85 | 1.84 |
| 31 | 116 | −9.46 | −9.54 | 0.08 | −8.20 | 1.26 | −12.61 | 3.15 | −9.90 | 0.44 |
| 32 | 108 | −9.50 | −9.10 | 0.41 | −7.47 | 2.03 | −13.04 | 3.53 | −10.14 | 0.63 |
| 33 | 479 | −8.62 | −9.01 | 0.39 | −9.09 | 0.47 | −12.42 | 3.80 | −13.29 | 4.67 |
| 34 | 2068 | −7.75 | −7.61 | 0.14 | −8.16 | 0.41 | −9.65 | 1.90 | −11.12 | 3.36 |
| 35 | 4214 | −7.33 | −7.24 | 0.09 | −9.16 | 1.83 | −8.18 | 0.85 | −11.43 | 4.10 |
| mean | 1.03 | 1.17 | 2.36 | 2.38 | ||||||
| stdev | 1.08 | 0.69 | 1.40 | 1.32 |
| Compd. | Ki (nM) | ∆Gexp | 3D0E | ∆∆G (3D0E) | 3D0E_N | ∆∆G (3D0E_N) | MT (3D0E) | ΔΔG (MT, 3D0E) | MT (3D0E_N) | ΔΔG (MT, 3D0E_N) |
|---|---|---|---|---|---|---|---|---|---|---|
| 6 | 30 | −10.26 | −10.49 | 0.23 | −9.00 | 1.26 | −8.94 | 1.32 | −11.19 | 0.92 |
| 11 | 57 | −9.88 | −9.66 | 0.22 | −8.38 | 1.50 | −13.38 | 3.49 | −11.73 | 1.85 |
| 14 | 4 | −11.46 | −6.31 | 5.15 | −7.53 | 3.93 | −11.21 | 0.24 | −12.12 | 0.67 |
| 15 | 11 | −10.86 | −9.09 | 1.77 | −9.89 | 0.97 | −11.46 | 0.61 | −11.58 | 0.72 |
| 17 | 40 | −10.09 | −7.75 | 2.34 | −7.60 | 2.49 | −12.27 | 2.18 | −10.61 | 0.52 |
| 18 | 11 | −10.86 | −10.31 | 0.55 | −9.66 | 1.20 | −9.61 | 1.25 | −10.92 | 0.06 |
| 22 | 11 | −10.86 | −8.84 | 2.01 | −8.50 | 2.36 | −10.24 | 0.61 | −12.29 | 1.43 |
| 25 | 95 | −9.58 | −8.29 | 1.29 | −7.30 | 2.28 | −10.73 | 1.15 | −11.30 | 1.72 |
| 26 | 21 | −10.47 | −8.85 | 1.63 | −10.22 | 0.26 | −10.76 | 0.29 | −10.87 | 0.40 |
| 27 | 11 | −10.86 | −8.75 | 2.11 | −8.32 | 2.53 | −10.80 | 0.05 | −12.09 | 1.23 |
| mean | 1.73 | 1.88 | 1.12 | 0.95 | ||||||
| stdev | 1.43 | 1.05 | 1.05 | 0.59 |
| Compd. | IC50 | ∆Gexp | HM | ∆∆G (HM) | AF | ∆∆G (AF) | MT (AF) | ∆∆G (MT, AF) |
|---|---|---|---|---|---|---|---|---|
| 6 | 431 | −8.68 | −10.05 | 1.36 | −5.98 | 2.70 | −9.27 | 0.59 |
| 11 | 16,316 | −6.53 | −9.14 | 2.61 | −6.22 | 0.31 | −10.36 | 3.83 |
| 12 | 1277 | −8.04 | −10.74 | 2.70 | −8.02 | 0.02 | −11.39 | 3.35 |
| 13 | 6627 | −7.06 | −9.52 | 2.46 | −7.21 | 0.14 | −10.00 | 2.94 |
| 14 | 1564 | −7.92 | −10.25 | 2.33 | −6.80 | 1.12 | −10.43 | 2.51 |
| 15 | 1890 | −7.81 | −9.86 | 2.05 | −6.56 | 1.24 | −9.20 | 1.40 |
| 16 | 607 | −8.48 | −10.49 | 2.01 | −6.67 | 1.81 | −10.76 | 2.28 |
| 17 | 3543 | −7.44 | −10.79 | 3.36 | −6.88 | 0.56 | −11.46 | 4.03 |
| 18 | 356 | −8.80 | −11.11 | 2.31 | −6.25 | 2.55 | −9.91 | 1.11 |
| 19 | 4356 | −7.31 | −10.45 | 3.14 | −6.79 | 0.52 | −9.81 | 2.50 |
| 20 | 5339 | −7.19 | −10.70 | 3.51 | −7.42 | 0.23 | −10.30 | 3.11 |
| 21 | 2108 | −7.74 | −10.86 | 3.12 | −6.99 | 0.75 | −10.86 | 3.12 |
| 22 | 5266 | −7.20 | −10.37 | 3.17 | −7.54 | 0.33 | −11.29 | 4.09 |
| 23 | 1509 | −7.94 | −10.82 | 2.88 | −6.93 | 1.01 | −9.81 | 1.87 |
| 24 | 7509 | −6.99 | −9.35 | 2.36 | −5.44 | 1.55 | −8.59 | 1.60 |
| 25 | 187 | −9.18 | −11.17 | 1.99 | −7.15 | 2.03 | −10.66 | 1.49 |
| 26 | 272 | −8.96 | −10.29 | 1.33 | −6.64 | 2.31 | −10.12 | 1.17 |
| 27 | 3049 | −7.52 | −10.54 | 3.01 | −6.99 | 0.53 | −10.16 | 2.63 |
| 28 | 6303 | −7.09 | −11.08 | 3.99 | −6.35 | 0.74 | −8.99 | 1.89 |
| 30 | 6446 | −7.08 | −9.89 | 2.81 | −5.67 | 1.41 | −7.42 | 0.34 |
| 31 | 3396 | −7.46 | −9.93 | 2.47 | −5.67 | 1.80 | −6.98 | 0.48 |
| 32 | 5752 | −7.15 | −10.15 | 3.00 | −6.36 | 0.79 | −9.13 | 1.98 |
| 35 | 8374 | −6.93 | −10.72 | 3.80 | −6.85 | 0.07 | −9.62 | 2.69 |
| mean | 2.69 | 1.07 | 2.22 | |||||
| stdev | 0.69 | 0.82 | 1.10 |
| Compd. | Ki | ∆Gexp | HM | ∆∆G (HM) | AF | ∆∆G (AF) | MT (AF) | ∆∆G (MT, AF) |
|---|---|---|---|---|---|---|---|---|
| 6 | 46.00 | −10.01 | −10.05 | 0.04 | −5.98 | 4.03 | −9.27 | 0.74 |
| 18 | 29.00 | −10.28 | −11.11 | 0.83 | −6.25 | 4.04 | −9.91 | 0.38 |
| 25 | 53.00 | −9.92 | −10.29 | 0.36 | −7.15 | 2.78 | −10.66 | 0.74 |
| 26 | 64.00 | −9.81 | −10.54 | 0.72 | −6.64 | 3.17 | −10.12 | 0.31 |
| mean | 0.49 | 3.50 | 0.54 | |||||
| stdev | 0.36 | 0.63 | 0.23 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhong, H.A.; Goodwin, D.T. Selectivity Studies and Free Energy Calculations of AKT Inhibitors. Molecules 2024, 29, 1233. https://doi.org/10.3390/molecules29061233
Zhong HA, Goodwin DT. Selectivity Studies and Free Energy Calculations of AKT Inhibitors. Molecules. 2024; 29(6):1233. https://doi.org/10.3390/molecules29061233
Chicago/Turabian StyleZhong, Haizhen A., and David T. Goodwin. 2024. "Selectivity Studies and Free Energy Calculations of AKT Inhibitors" Molecules 29, no. 6: 1233. https://doi.org/10.3390/molecules29061233
APA StyleZhong, H. A., & Goodwin, D. T. (2024). Selectivity Studies and Free Energy Calculations of AKT Inhibitors. Molecules, 29(6), 1233. https://doi.org/10.3390/molecules29061233