
Study on the Relationship between Particulate Methane Monooxygenase and Methanobactin on Gold-Nanoparticles-Modified Electrodes

Abstract
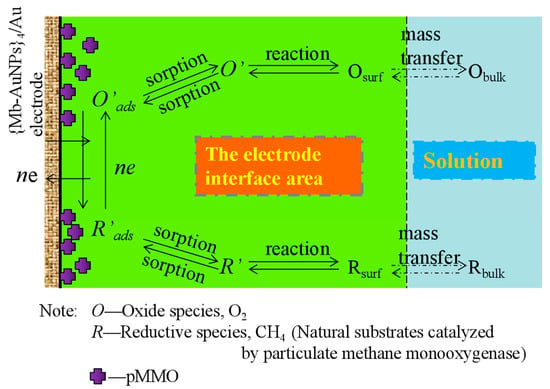
:
1. Introduction
2. Results and Discussion
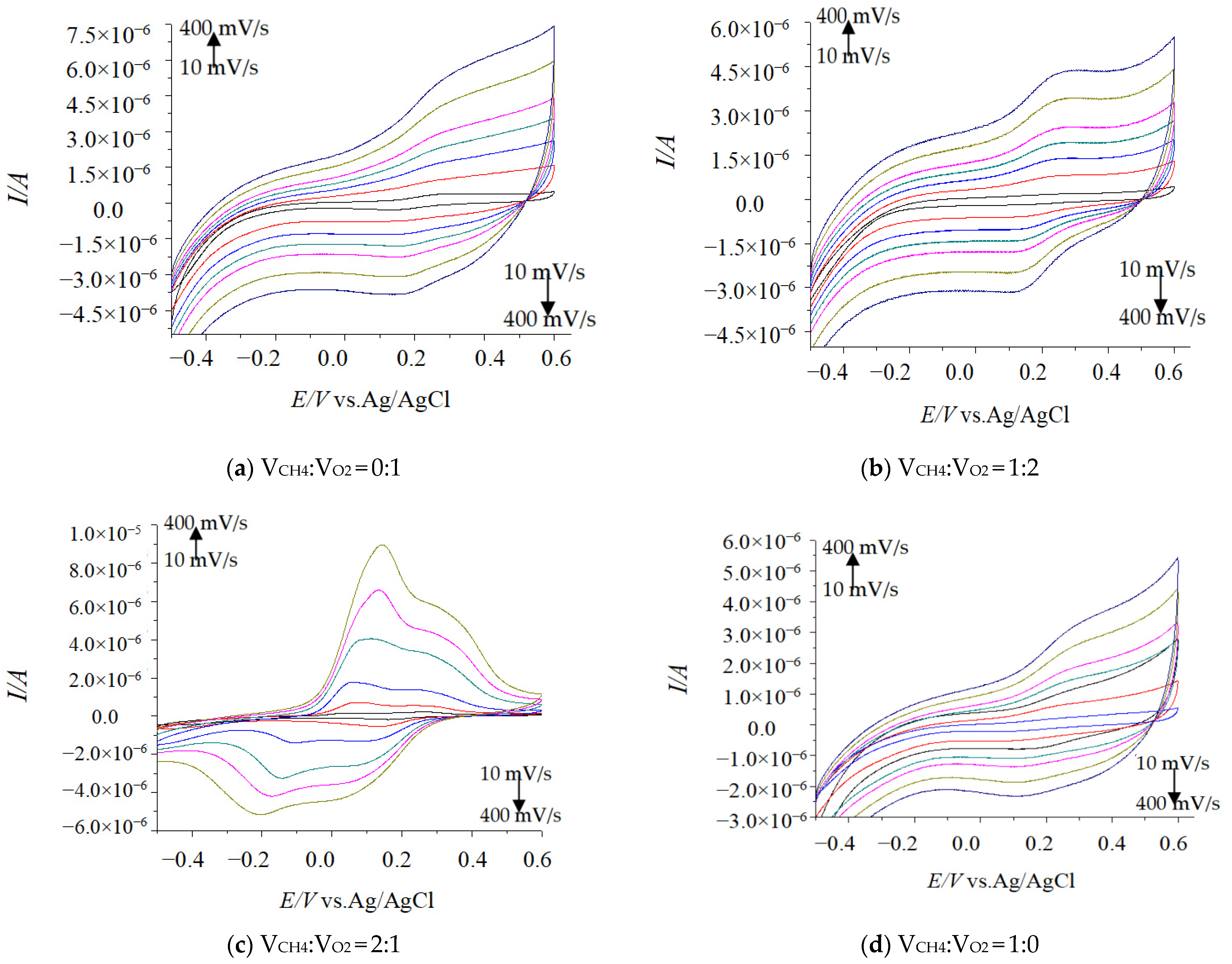
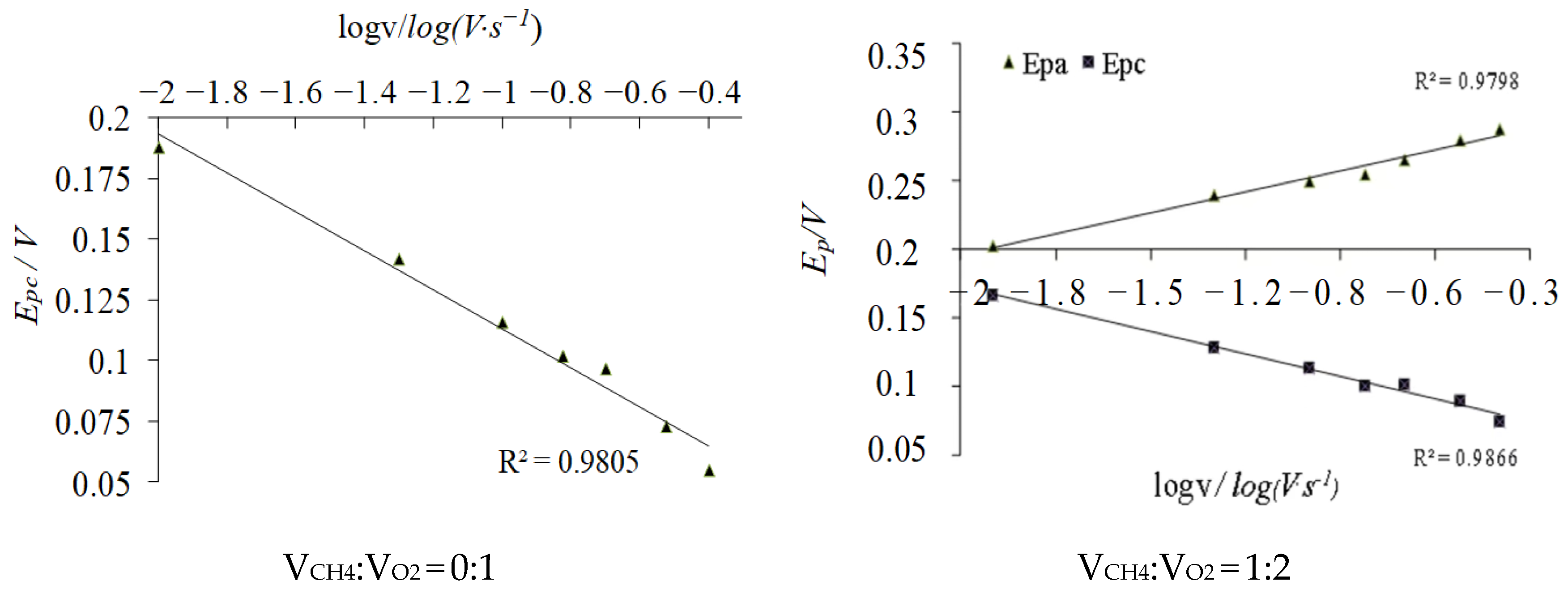
2.1. Impact Tests of Different CV Scanning Speed
2.2. Effect of Dissolved CH4 and O2 Volumes Ratio on Electrochemical Kinetics Parameters
2.2.1. Transferred Electron Number (n)
2.2.2. Electron Transfer Coefficient (α)
2.2.3. Electron Transfer Rate Constant (ks)
2.3. Analysis of the Kinetic Behavior of pMMO Bioelectrocatalysis
3. Materials and Methods
3.1. Chemicals
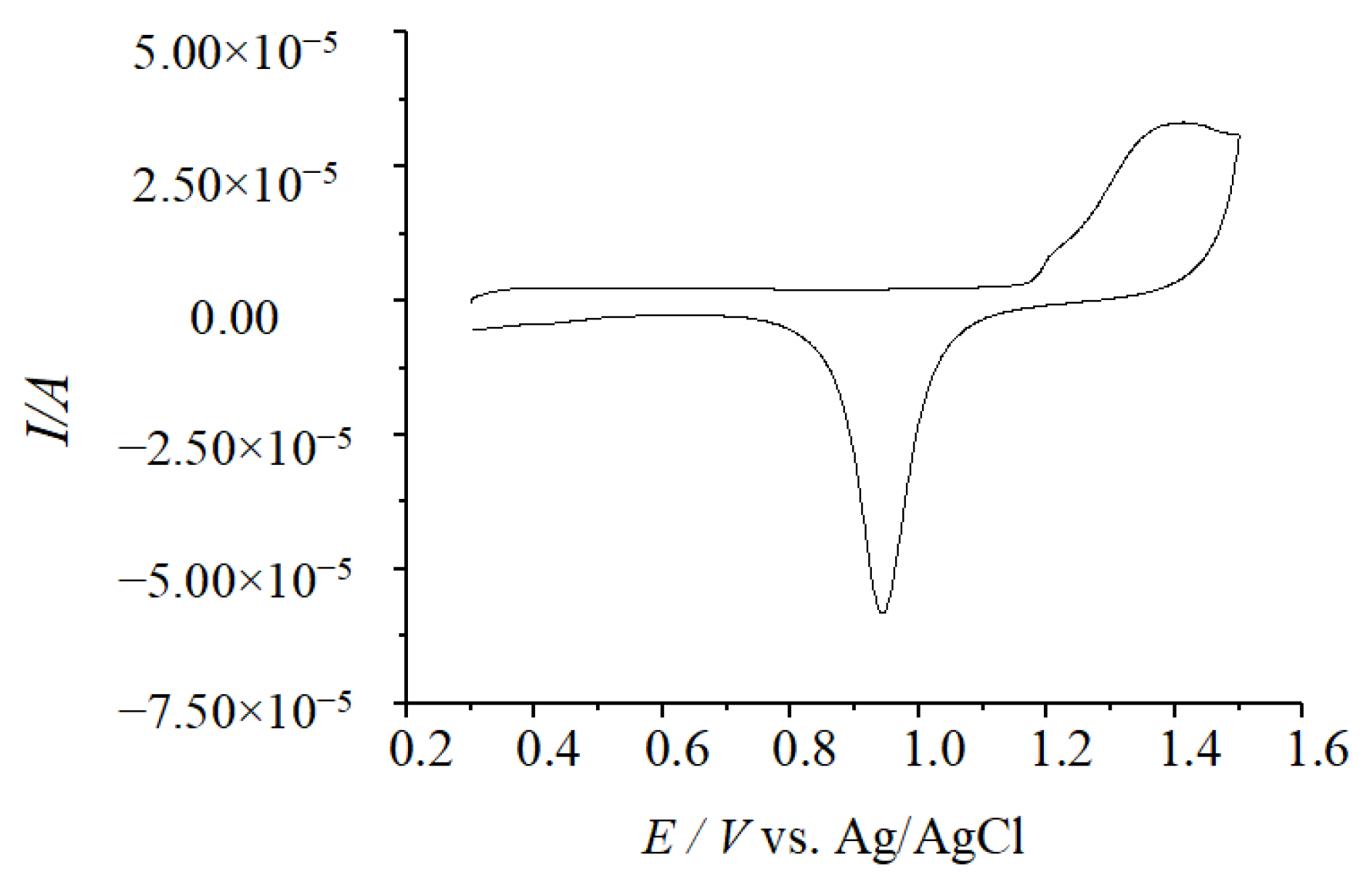
3.2. Culture Preparation and Isolation and Purification of pMMO
3.2.1. Fermentation Culture
3.2.2. Purification of pMMO-Containing Endosomes
3.2.3. pMMO Specific Activity Measurement
3.3. Gold Disk Electrode Pretreatment
3.4. Preparation of Pure AuNPs Sols by Chemical Method
3.5. Preparation of AuNPs Modified with Mb (Mb-AuNPs)
3.6. Preparation of AuNPs with Mixed Modifications of Mb and Mercapto Compounds
3.7. Preparation of {Mb-AuNPs}4/Au Disk Electrodes
3.8. Kinetic Behavior and Analysis of pMMO Bioelectrocatalysis
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Anthony, C. The Biochemistry of Methylotrophs; Academic Press: London, UK, 1982. [Google Scholar]
- Bédard, C.; Knowles, R. Physiology, biochemistry, and specific inhibitors of CH4, NH4+, and CO oxidation by methanotrophs and nitrifiers. Microbiol. Rev. 1989, 53, 68–84. [Google Scholar] [CrossRef] [PubMed]
- Lipscomb, J.D. Biochemistry of the Soluble Methane Monooxygenase. Annu. Rev. Microbiol. 2003, 48, 371–399. [Google Scholar] [CrossRef] [PubMed]
- Yılmaz, G.E.; Saylan, Y.; Göktürk, I.; Yılmaz, F.; Denizli, A. Selective Amplification of Plasmonic Sensor Signal for Cortisol Detection Using Gold Nanoparticles. Biosensors 2022, 12, 482. [Google Scholar] [CrossRef]
- Gokturk, I.; Bakhshpour, M.; Cimen, D.; Yilmaz, F.; Bereli, N.; Denizli, A. SPR Signal Enhancement with Silver Nanoparticle-Assisted Plasmonic Sensor for Selective Adenosine Detection. IEEE Sens. J. 2022, 22, 14862–14869. [Google Scholar] [CrossRef]
- Choi, D.W.; Semrau, J.D.; Antholine, W.E.; Hartsel, S.C.; Anderson, R.C.; Carey, J.N.; Dreis, A.M.; Kenseth, E.M.; Renstrom, J.M.; Scardino, L.L.; et al. Oxidase, superoxide dismutase, and hydrogen peroxide reductase activities of methanobactin from types I and II methanotrophs. J. Inorg. Biochem. 2008, 102, 1571–1580. [Google Scholar] [CrossRef] [PubMed]
- Laviron, E. Use of linear potential sweep voltammetry and of ac voltammetry for the study of the surface electrochemical reaction of strongly adsorbed systems and of redox modified electrodes. Electroanal. Chem. 1979, 100, 263–270. [Google Scholar] [CrossRef]
- Bi, Y. Electron Transfer and Fluorescence Spectroelectrochemistry of Biomolecules at Biomimetic Interfaces. Ph.D. Thesis, Huazhong University of Science and Technology, Wuhan, China, 2006. [Google Scholar]
- Janet, D.C. Electrochemistry of Soluble Methane Monooxygenase on a Modified Gold Electrode Implications for Chemical Sensing in Natural Waters. Ph.D. Thesis, University of California, Berkeley, CA, USA, 2001. [Google Scholar]
- Xin, J.Y.; Cui, J.R.; Hu, X.X.; Li, S.B.; Xia, C.G.; Zhu, L.M.; Wang, Y.Q. Particulate methane monooxygenase from Methylosinus trichosporium is a copper-containing enzyme. Biochem. Biophys. Res. Commun. 2002, 295, 1800–1806. [Google Scholar] [CrossRef] [PubMed]
- Xin, J.Y.; Zhang, L.X.; Chen, D.D. Colorimetric detection of melamine based on methanobactin-mediated synthesis of gold nanoparticles. Food Chem. 2015, 174, 473–479. [Google Scholar] [CrossRef]
- Xin, J.; Dou, B.; Wang, Z.; Wang, Y.; Xia, C.; Liu, Z. Direct Electrochemistry of Methanobactin Functionalized Gold Nanoparticles on Au Electrode. J. Nanosci. Nanotechnol. 2018, 18, 4805–4813. [Google Scholar] [CrossRef]
- Huang, L.; Chen, Y.T.; Li, Y.X.; Yu, L.S. Application of Chiral Ionic Liquid-Modified Gold Nanoparticles in the Chiral Recognition of Amino Acid Enantiomers. J. Anal. Sci. 2016, 70, 1649–1654. [Google Scholar] [CrossRef]
- Khlebtsov, N.G. Determination of Size and Concentration of Gold Nanoparticles from Extinction Spectra. Anal. Chem. 2008, 80, 6620–6625. [Google Scholar] [CrossRef] [PubMed]
- Haiss, W.; Thanh, N.T.K.; Aveyard, J.; Fernig, D.G. Determination of Size and Concentration of Gold Nanoparticles from UV-Vis Spectra. Anal. Chem. 2007, 79, 4215–4221. [Google Scholar] [CrossRef]
- Thorum, M.; Anderson, C.; Hatch, J. Direct, Electrocatalytic Oxygen Reduction by Laccase on Anthracene-2-methanethiol-Modified Gold. J. Phys. Chem. Lett. 2010, 1, 2251. [Google Scholar] [CrossRef]
- Xue, Q.; Bian, C.; Tong, J. Micro Biosensor Antibody Immobilization Method Based on Mixed Self-assembly-monolayers Wrapped Nano-particle. Chin. J. Anal. Chem. 2011, 39, 804–808. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| The Volume Ratio of Dissolved CH4 and Dissolved O2 (VCH4:VO2) | 0:1 | 1:2 | 2:1 | 1:0 |
|---|---|---|---|---|
| Actual calculated transferred electron number | 0.947 | 1.57 | 1.81 | 1.019 |
| Approximately considered transferred electron number | 1 | 2 | 2 | 1 |
| The Volume Ratio of Dissolved CH4 and Dissolved O2 (VCH4:VO2) | 0:1 | 1:2 | 2:1 | 1:0 |
|---|---|---|---|---|
| α | 0.7058 | 0.59 | 0.47 | 0.74 |
| The Volume Ratio of Dissolved CH4 and Dissolved O2 (VCH4:VO2) | 0:1 | 1:2 | 2:1 | 1:0 |
|---|---|---|---|---|
| ks(s−1) | 0.5571 | 0.3886 | 0.113 | 0.5459 |
| The Volume Ratio of Dissolved CH4 and Dissolved O2 (VCH4:VO2) | The Kinetic Behavior and Parameters of pMMO Bioelectrocatalysis |
|---|---|
| 0:1 | The pMMO-catalyzed redox reaction was a single-electron transport process; the electron transfer coefficient was 0.7058, and the electron transfer rate constant was 0.5571 s−1. |
| 1:2 | The pMMO-catalyzed redox reaction was a two-electron transport process; the electron transfer coefficient was 0.59, and the electron transfer rate constant was 0.3886 s−1. |
| 2:1 | The pMMO-catalyzed redox reaction was a single-electron transport process; the electron transfer coefficient was 0.47, and the electron transfer rate constant was 0.113 s−1. |
| 1:0 | The pMMO-catalyzed redox reaction was a single-electron transport process; the electron transfer coefficient was 0.47, and the electron transfer rate constant was 0.5459 s−1. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dou, B.; Li, M.; Sun, L.; Xin, J.; Xia, C. Study on the Relationship between Particulate Methane Monooxygenase and Methanobactin on Gold-Nanoparticles-Modified Electrodes. Molecules 2024, 29, 1270. https://doi.org/10.3390/molecules29061270
Dou B, Li M, Sun L, Xin J, Xia C. Study on the Relationship between Particulate Methane Monooxygenase and Methanobactin on Gold-Nanoparticles-Modified Electrodes. Molecules. 2024; 29(6):1270. https://doi.org/10.3390/molecules29061270
Chicago/Turabian StyleDou, Boxin, Mingyu Li, Lirui Sun, Jiaying Xin, and Chungu Xia. 2024. "Study on the Relationship between Particulate Methane Monooxygenase and Methanobactin on Gold-Nanoparticles-Modified Electrodes" Molecules 29, no. 6: 1270. https://doi.org/10.3390/molecules29061270