
Identification of Novel Antimicrobial Compounds Targeting Mycobacterium tuberculosis S-Adenosyl-L-Homocysteine Hydrolase Using Dual Hierarchical In Silico Structure-Based Drug Screening
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
2.1. In Silico SBDS Pathway
2.2. Dual Hierarchical In Silico SBDS of MtSAHH Using Dual Pathways
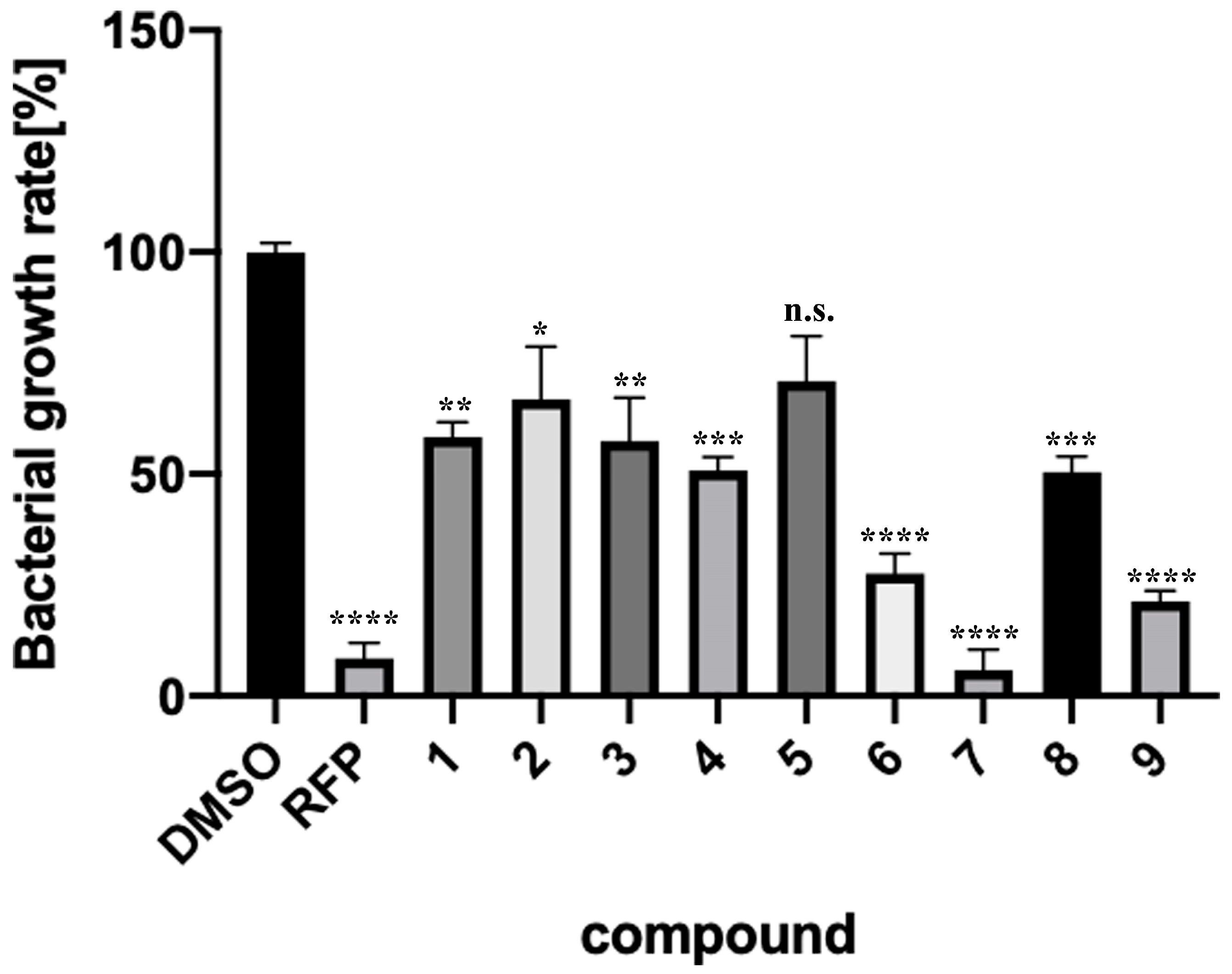
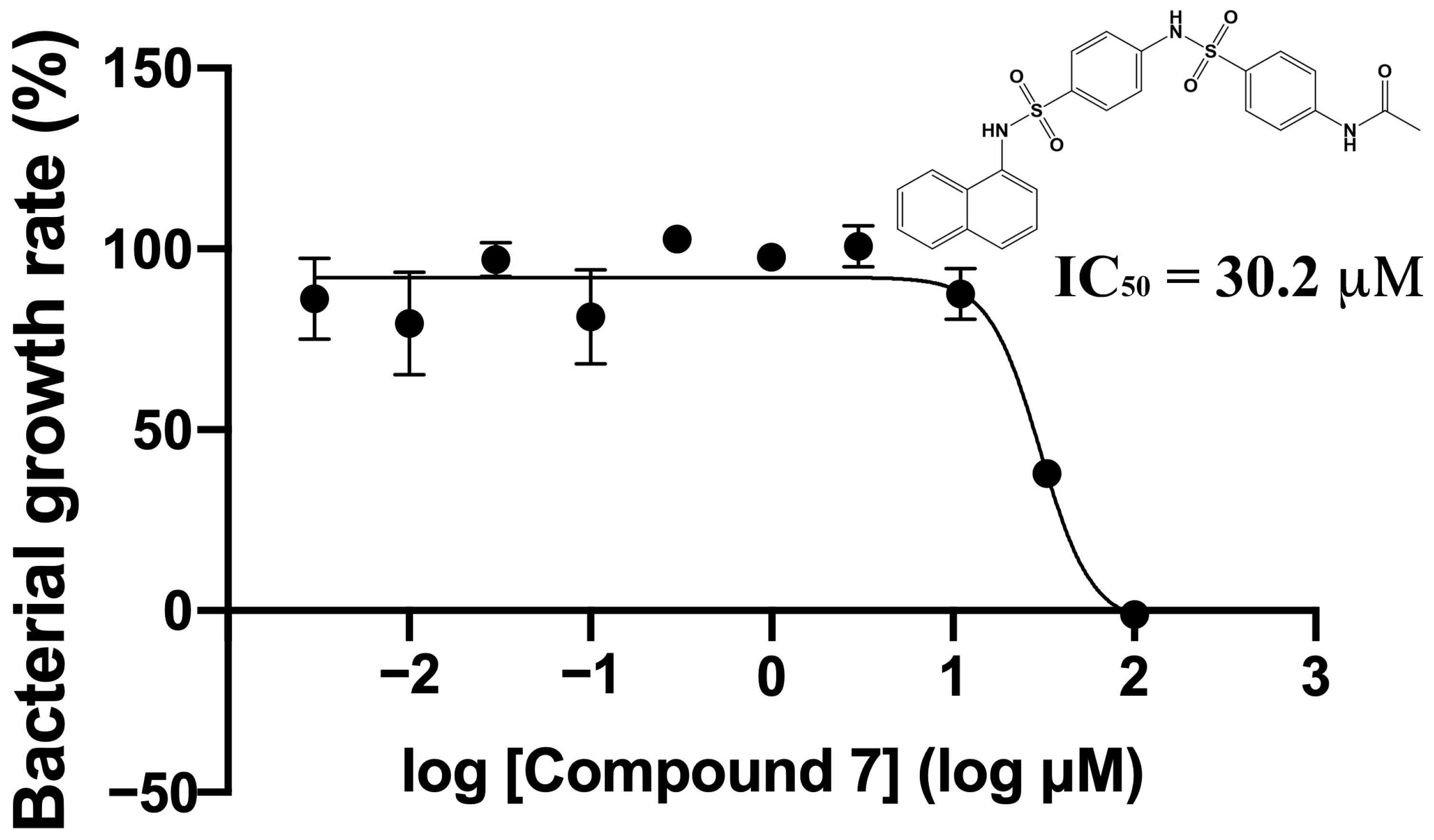
2.3. Verification of Antibacterial Effects of Compounds against M. smegmatis
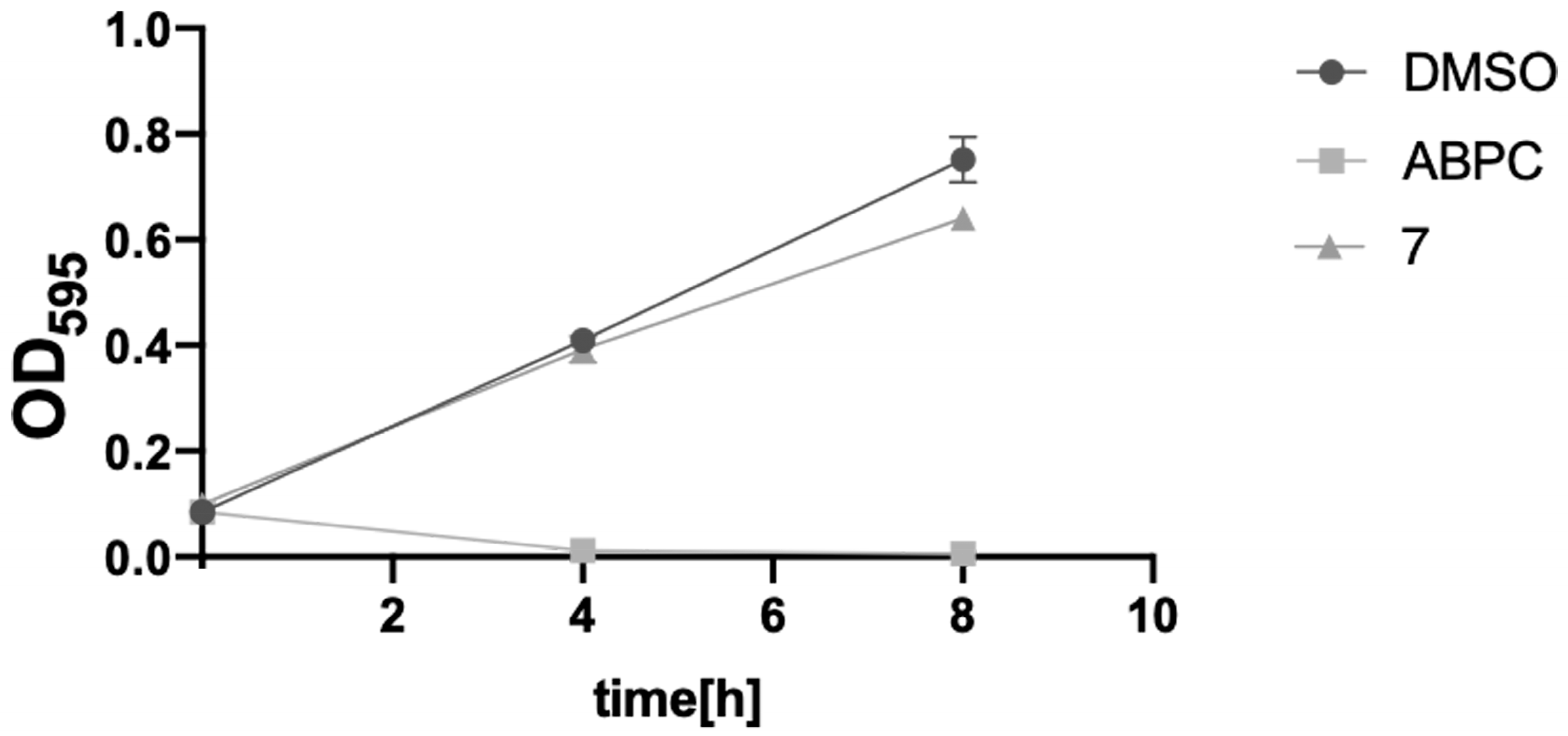
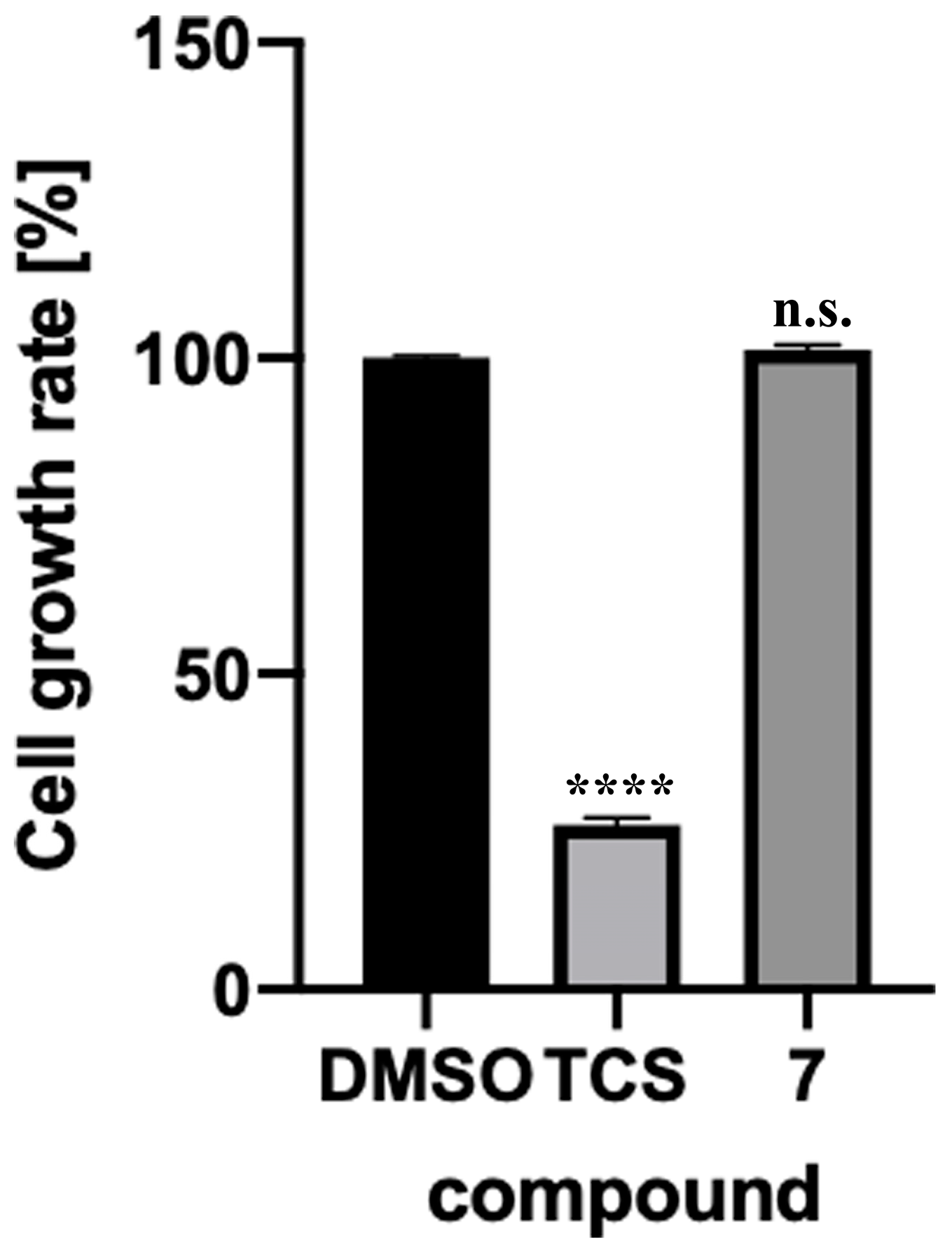
2.4. Toxicity Verification for Escherichia coli and Mammalian Cells
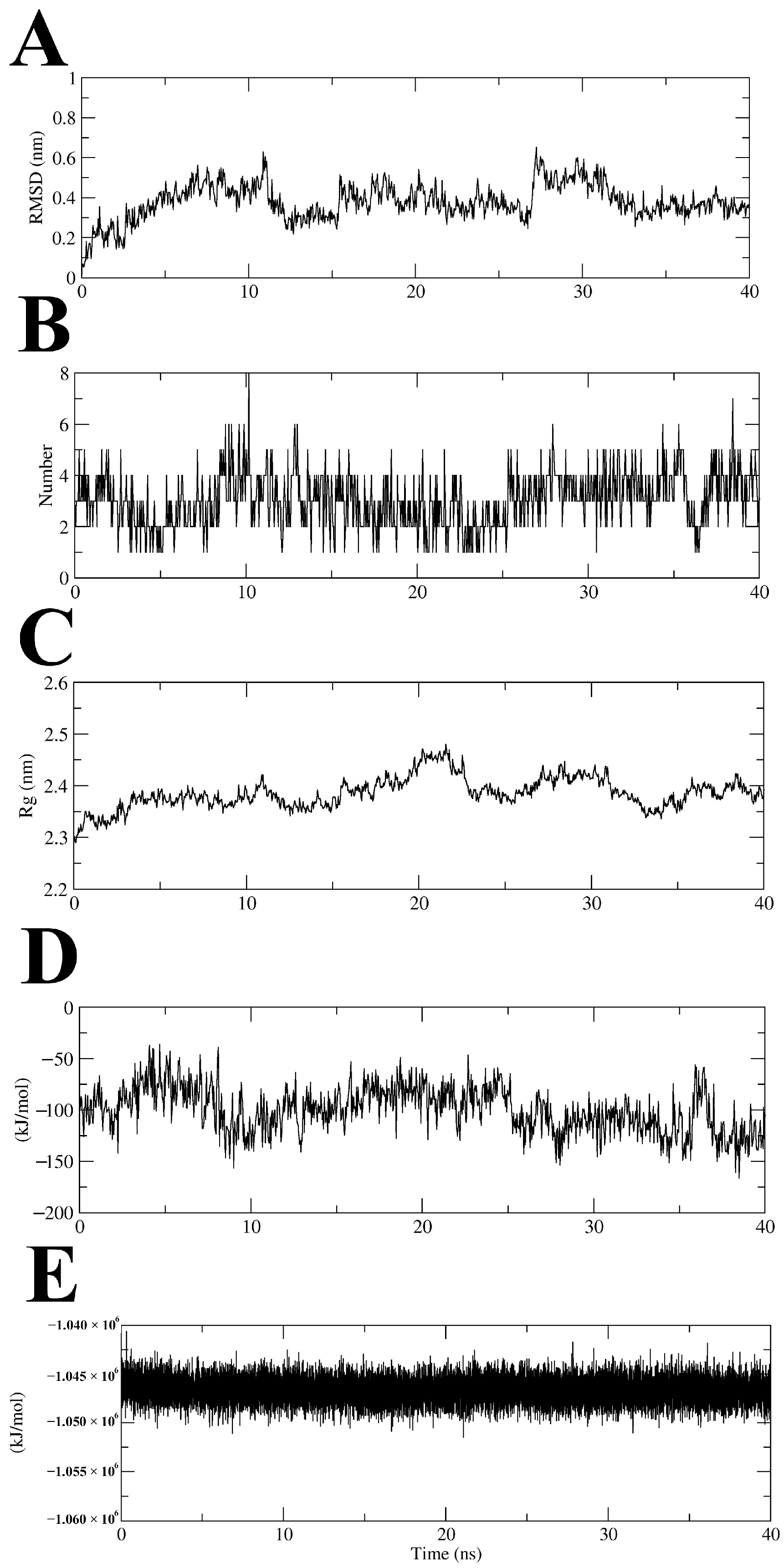
2.5. Molecular Dynamics Simulation of MtSAHH–Compound 7 Complex
2.6. Binding Mode Analysis of MtSAHH–Compound 7 Complex
2.7. ADME/Tox Prediction for Compound 7
3. Materials and Methods
3.1. Target Protein
3.2. Compound Structure Library
3.3. Dual Hierarchical In Silico SBDS
- Ntotal: Total number of compounds;
- Nact: Total number of active compounds;
- Nx%: Number of compounds in the top x% of scores;
- Nx%act: Number of active compounds in the top x% of scores.
3.4. Preparation of Compounds
3.5. Growth Inhibition Assay against Mycobacterium
3.6. Evaluation of Growth Activity against Gram-Negative Bacteria
3.7. Toxicity Assays for Human Cells
3.8. MD Simulation
3.9. In Silico Estimation of ADME Properties and Toxicities
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Global Tuberculosis Report 2023. 2023. Available online: https://iris.who.int/ (accessed on 1 December 2023).
- Stillo, J.; Frick, M.; Galarza, J.; Kondratyuk, S.; Makone, A.; McKenna, L.; Vandevelde, W.; Winarni, P.; Agbassi, P. Addressing the needs of people with extensively drug-resistant TB through pre-approval access to drugs and research. Public Health Action 2023, 13, 126. [Google Scholar] [CrossRef]
- Corrales, R.M.; Leiba, J.; Cohen-Gonsaud, M.; Molle, V.; Kremer, L. Mycobacterium tuberculosis S-adenosyl-L-homocysteine hydrolase is negatively regulated by Ser/Thr phosphorylation. Biochem. Biophys. Res. Commun. 2013, 430, 858–864. [Google Scholar] [CrossRef]
- Jones, G.; Willett, P.; Glen, R.C.; Leach, A.R.; Taylor, R. Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol. 1997, 267, 727–748. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Allen, W.J.; Balius, T.E.; Mukherjee, S.; Brozell, S.R.; Moustakas, D.T.; Lang, P.T.; Case, D.A.; Kuntz, I.D.; Rizzo, R.C. DOCK 6: Impact of new features and current docking performance. J. Comput. Chem. 2015, 36, 1132–1156. [Google Scholar] [CrossRef]
- Kawamoto, S.; Hori, C.; Taniguchi, H.; Okubo, S.; Aoki, S. Identification of novel antimicrobial compounds targeting Mycobacterium tuberculosis shikimate kinase using in silico hierarchical structure-based drug screening. Tuberculosis 2023, 141, 102362. [Google Scholar] [CrossRef] [PubMed]
- Mysinger, M.M.; Carchia, M.; Irwin, J.J.; Shoichet, B.K. Directory of useful decoys, enhanced (DUD-E): Better ligands and decoys for better benchmarking. J. Med. Chem. 2012, 55, 6582–6594. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, P.C.D.; Skillman, A.G.; Nicholls, A. Comparison of shape-matching and docking as virtual screening tools. J. Med. Chem. 2007, 50, 74–82. [Google Scholar] [CrossRef] [PubMed]
- Lagarde, N.; Goldwaser, E.; Pencheva, T.; Jereva, D.; Pajeva, I.; Rey, J.; Tuffery, P.; Villoutreix, B.O.; Miteva, M.A. A Free Web-Based Protocol to Assist Structure-Based Virtual Screening Experiments. Int. J. Mol. Sci. 2019, 20, 4648. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A. Lead- and drug-like compounds: The rule-of-five revolution. Drug Discov. Today Technol. 2004, 1, 337–341. [Google Scholar] [CrossRef]
- Wang, Y.; Cheng, T.; Bryant, S.H. PubChem BioAssay: A Decade’s Development toward Open High-Throughput Screening Data Sharing. SLAS Discov. 2017, 22, 655–666. [Google Scholar] [CrossRef]
- Namasivayam, S.; Maiga, M.; Yuan, W.; Thovarai, V.; Costa, D.L.; Mittereder, L.R.; Wipperman, M.F.; Glickman, M.S.; Dzutsev, A.; Trinchieri, G.; et al. Longitudinal profiling reveals a persistent intestinal dysbiosis triggered by conventional anti-tuberculosis therapy. Microbiome 2017, 5, 71. [Google Scholar] [CrossRef]
- Kuriki, K.; Taira, J.; Kuroki, M.; Sakamoto, H.; Aoki, S. Computer-assisted screening of mycobacterial growth inhibitors: Exclusion of frequent hitters with the assistance of the multiple target screening method. Int. J. Mycobacteriol. 2021, 10, 307–311. [Google Scholar] [CrossRef]
- Miller, B.R.; McGee, T.D.; Swails, J.M.; Homeyer, N.; Gohlke, H.; Roitberg, A.E. MMPBSA.py: An efficient program for end-state free energy calculations. J. Chem. Theory Comput. 2012, 8, 3314–3321. [Google Scholar] [CrossRef]
- Valdés-Tresanco, M.S.; Valdés-Tresanco, M.E.; Valiente, P.A.; Moreno, E. Gmx_MMPBSA: A New Tool to Perform End-State Free Energy Calculations with GROMACS. J. Chem. Theory Comput. 2021, 17, 6281–6291. [Google Scholar] [CrossRef]
- Adasme, M.F.; Linnemann, K.L.; Bolz, S.N.; Kaiser, F.; Salentin, S.; Haupt, V.J.; Schroeder, M. PLIP 2021: Expanding the scope of the protein–ligand interaction profiler to DNA and RNA. Nucleic Acids Res. 2021, 49, W530–W534. [Google Scholar] [CrossRef] [PubMed]
- Reddy, M.C.M.; Kuppan, G.; Shetty, N.D.; Owen, J.L.; Ioerger, T.R.; Sacchettini, J.C. Crystal structures of Mycobacterium tuberculosis S-adenosyl-L-homocysteine hydrolase in ternary complex with substrate and inhibitors. Protein Sci. 2008, 17, 2134–2144. [Google Scholar] [CrossRef] [PubMed]
- Daina, A.; Zoete, V. A BOILED-Egg To Predict Gastrointestinal Absorption and Brain Penetration of Small Molecules. ChemMedChem 2016, 11, 1117–1121. [Google Scholar] [CrossRef] [PubMed]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, P.; Eckert, A.O.; Schrey, A.K.; Preissner, R. ProTox-II: A webserver for the prediction of toxicity of chemicals. Nucleic Acids Res. 2018, 46, W257–W263. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28. Available online: http://www.rcsb.org/structure/3CE6 (accessed on 23 January 2024). [CrossRef] [PubMed]
- Vilar, S.; Cozza, G.; Moro, S. Medicinal chemistry and the molecular operating environment (MOE): Application of QSAR and molecular docking to drug discovery. Curr. Top. Med. Chem. 2008, 8, 1555–1572. Available online: https://www.ingentaconnect.com/content/ben/ctmc/2008/00000008/00000018/art00002 (accessed on 23 January 2024). [CrossRef] [PubMed]
- Li, Q.; Shah, S. Structure-based virtual screening. In Protein Bioinformatics; Methods in Molecular Biology; Wu, C., Arighi, C., Ross, K., Eds.; Humana Press: New York, NY, USA, 2017; Volume 1558, pp. 111–124. [Google Scholar] [CrossRef] [PubMed]
- Hendrix, D.K.; Kuntz, I.D. Surface Solid Angle-Based Site Points for Molecular Docking. 1998. Available online: http://psb.stanford.edu/psb-online/proceedings/psb98/hendrix.pdf (accessed on 19 January 2024).
- Lipinski, C.A. Drug-like properties and the causes of poor solubility and poor permeability. J. Pharmacol. Toxicol. Methods 2000, 44, 235–249. [Google Scholar] [CrossRef] [PubMed]
- Taira, J.; Murakami, K.; Monobe, K.; Kuriki, K.; Fujita, M.; Ochi, Y.; Sakamoto, H.; Aoki, S. Identification of novel inhibitors for mycobacterial polyketide synthase 13 via in silico drug screening assisted by the parallel compound screening with genetic algorithm-based programs. J. Antibiot. 2022, 75, 552–558. [Google Scholar] [CrossRef]
- Jo, S.; Kim, T.; Iyer, V.G.; Im, W. CHARMM-GUI: A web-based graphical user interface for CHARMM. J. Comput. Chem. 2008, 29, 1859–1865. [Google Scholar] [CrossRef]
- Brooks, B.R.; Brooks, C.L., III; MacKerell, A.D., Jr.; Nilsson, L.; Petrella, R.J.; Roux, B.; Won, Y.; Archontis, G.; Bartels, C.; Boresch, S.; et al. CHARMM: The biomolecular simulation program. J. Comput. Chem. 2009, 30, 1545–1614. [Google Scholar] [CrossRef]
- Lee, J.; Cheng, X.; Swails, J.M.; Yeom, M.S.; Eastman, P.K.; Lemkul, J.A.; Wei, S.; Buckner, J.; Jeong, J.C.; Qi, Y.; et al. CHARMM-GUI input generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM simulations using the CHARMM36 additive force field. J. Chem. Theory Comput. 2016, 12, 405–413. [Google Scholar] [CrossRef]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J.C. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef]
- Takeuchi, M.; Teshima, M.; Okubo, S.; Aoki, S. In silico and in vitro Identification of Compounds with Dual Pharmacological Activity against Metionyl-tRNA Synthetase and Isoleucyl-tRNA Synthetase of Staphylococcus aureus. ChemistrySelect 2023, 8, e202300460. [Google Scholar] [CrossRef]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A Linear Constraint Solver for Molecular Simulations. J. Comput. Chem. 1997, 18, 14631472. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N⋅log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ito, H.; Monobe, K.; Okubo, S.; Aoki, S. Identification of Novel Antimicrobial Compounds Targeting Mycobacterium tuberculosis S-Adenosyl-L-Homocysteine Hydrolase Using Dual Hierarchical In Silico Structure-Based Drug Screening. Molecules 2024, 29, 1303. https://doi.org/10.3390/molecules29061303
Ito H, Monobe K, Okubo S, Aoki S. Identification of Novel Antimicrobial Compounds Targeting Mycobacterium tuberculosis S-Adenosyl-L-Homocysteine Hydrolase Using Dual Hierarchical In Silico Structure-Based Drug Screening. Molecules. 2024; 29(6):1303. https://doi.org/10.3390/molecules29061303
Chicago/Turabian StyleIto, Hazuki, Kohei Monobe, Saya Okubo, and Shunsuke Aoki. 2024. "Identification of Novel Antimicrobial Compounds Targeting Mycobacterium tuberculosis S-Adenosyl-L-Homocysteine Hydrolase Using Dual Hierarchical In Silico Structure-Based Drug Screening" Molecules 29, no. 6: 1303. https://doi.org/10.3390/molecules29061303