
Enzymatic Synthesis of New Acetoacetate–Ursodeoxycholic Acid Hybrids as Potential Therapeutic Agents and Useful Synthetic Scaffolds as Well


 ,
,  , , ,
, , ,  ,
,  ,
, 
Abstract
:1. Introduction
2. Results and Discussion
2.1. State of the Art on Enzyme-Catalyzed Esterification of Bile Acids
2.2. Screening of the Biocatalysts
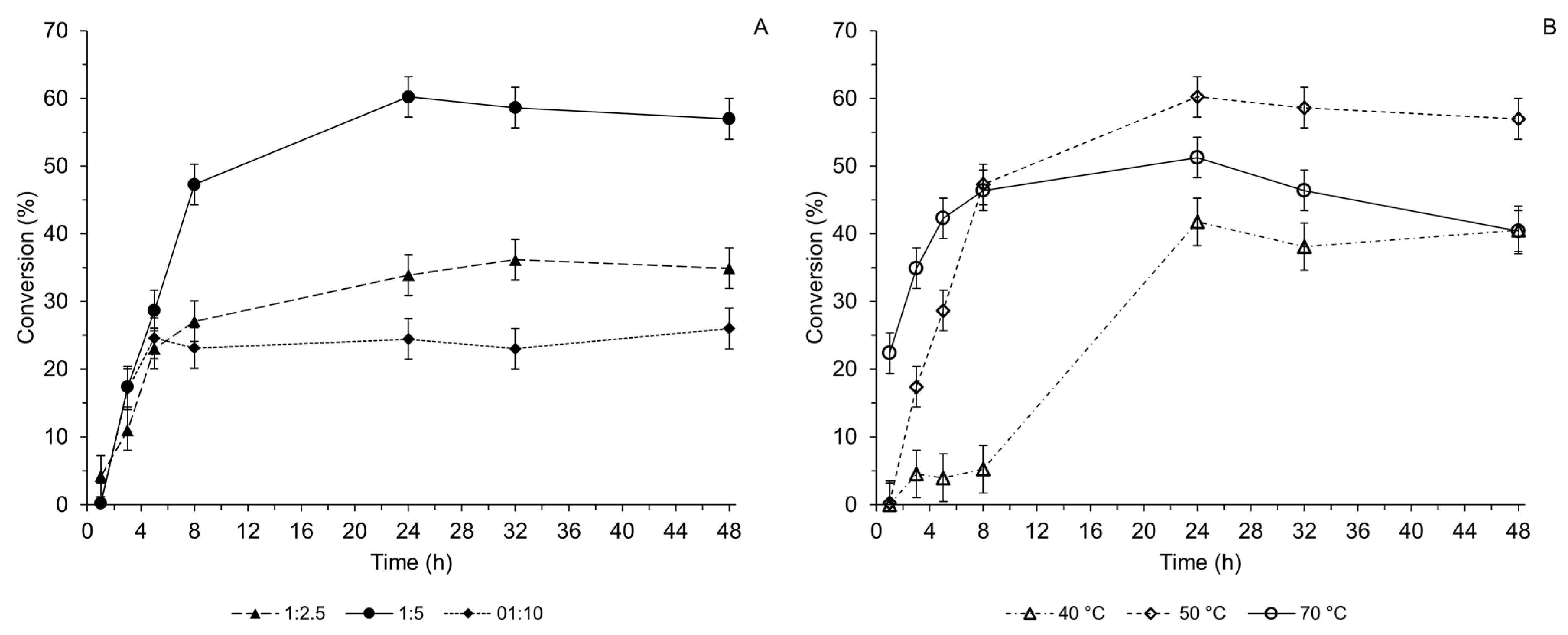
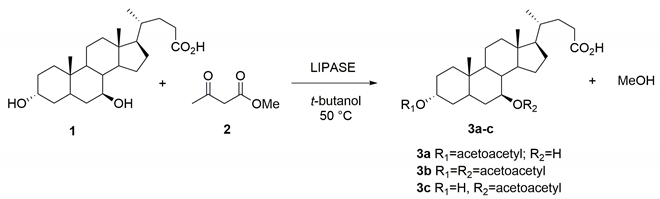
2.3. Optimized Synthesis of the 3α-Acetoacetoxy UDCA 3a
2.4. Chemo-Enzymatic Synthesis of the 3α,7β-bis-Acetoacetoxy UDCA 3b and 7β-Acetoacetoxy UDCA 3c
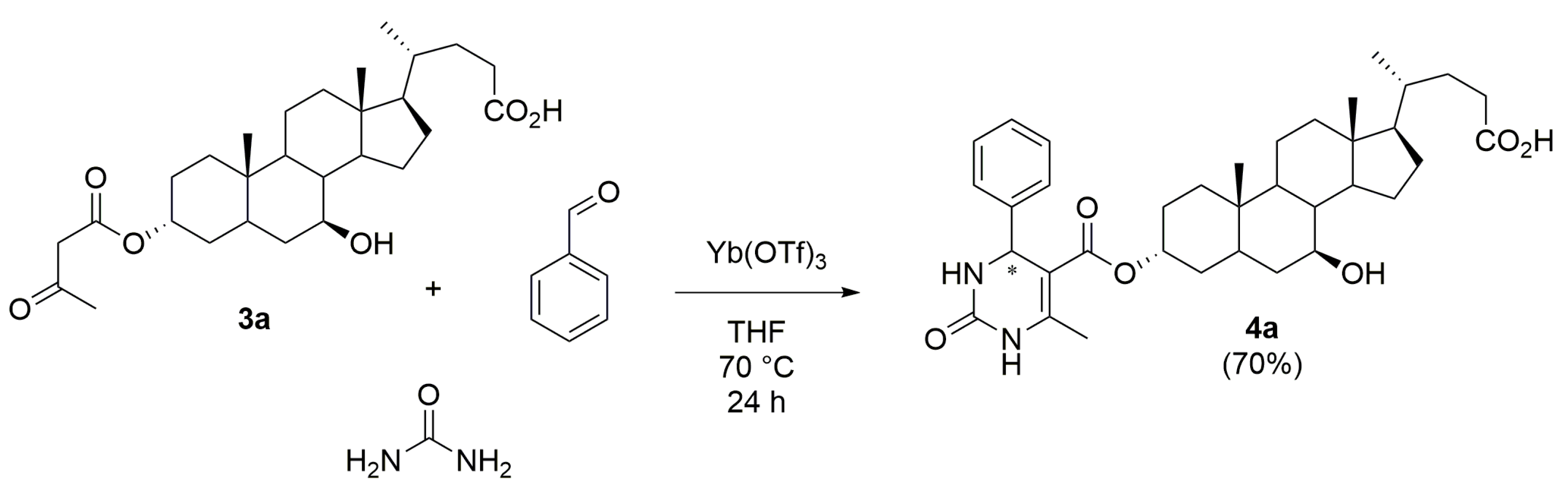
2.5. Synthesis of the 3α-(4-Phenyl-6-methyl-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxyl)-UDCA 4a
3. Materials and Methods
3.1. General Information
3.2. Optimized Procedure for the Synthesis of 3α-Acetoacetoxy Ursodeoxycholic Acid 3a
3.3. Procedure for the Synthesis of 3α,7β-bis-Acetoacetoxy Ursodeoxycholic Acid 3b
3.4. Procedure for the Synthesis of 7β-Acetoacetoxy Ursodeoxycholic Acid 3c and of the Corresponding Ethyl Ester 3d
3.5. Procedure for the Synthesis of 3α-(4-Phenyl-6-methyl-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxyl)-UDCA 4a
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Alkhzem, A.H.; Woodman, T.J.; Blagbrough, I.S. Design and Synthesis of Hybrid Compounds as Novel Drugs and Medicines. RSC Adv. 2022, 12, 19470–19484. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.K.; Kumar, A.; Singh, H.; Sonawane, P.; Paliwal, H.; Thareja, S.; Pathak, P.; Grishina, M.; Jaremko, M.; Emwas, A.-H.; et al. Concept of Hybrid Drugs and Recent Advancements in Anticancer Hybrids. Pharmaceuticals 2022, 15, 1071. [Google Scholar] [CrossRef] [PubMed]
- Faustino, C.; Serafim, C.; Rijo, P.; Reis, C.P. Bile Acids and Bile Acid Derivatives: Use in Drug Delivery Systems and as Therapeutic Agents. Expert Opin. Drug Deliv. 2016, 13, 1133–1148. [Google Scholar] [CrossRef] [PubMed]
- Hagey, L.; Crombie, D.; Espinosa, E.; Carey, M.; Igimi, H.; Hofmann, A. Ursodeoxycholic Acid in the Ursidae: Biliary Bile Acids of Bears, Pandas, and Related Carnivores. J. Lipid Res. 1993, 34, 1911–1917. [Google Scholar] [CrossRef]
- Keely, S.J.; Steer, C.J.; Lajczak-McGinley, N.K. Ursodeoxycholic Acid: A Promising Therapeutic Target for Inflammatory Bowel Diseases? Am. J. Physiol.-Gastrointest. Liver Physiol. 2019, 317, G872–G881. [Google Scholar] [CrossRef]
- Kumar, D.; Tandon, R.K. Use of Ursodeoxycholic Acid in Liver Diseases. J. Gastroenterol. Hepatol. 2001, 16, 3–14. [Google Scholar] [CrossRef]
- Shah, R.A.; Kowdley, K. V Current and Potential Treatments for Primary Biliary Cholangitis. Lancet Gastroenterol. Hepatol. 2020, 5, 306–315. [Google Scholar] [CrossRef]
- Huang, F. Ursodeoxycholic Acid as a Potential Alternative Therapeutic Approach for Neurodegenerative Disorders: Effects on Cell Apoptosis, Oxidative Stress and Inflammation in the Brain. Brain Behav. Immun. Health 2021, 18, 100348. [Google Scholar] [CrossRef]
- Daruich, A.; Jaworski, T.; Henry, H.; Zola, M.; Youale, J.; Parenti, L.; Naud, M.-C.; Delaunay, K.; Bertrand, M.; Berdugo, M.; et al. Oral Ursodeoxycholic Acid Crosses the Blood Retinal Barrier in Patients with Retinal Detachment and Protects Against Retinal Degeneration in an Ex Vivo Model. Neurotherapeutics 2021, 18, 1325–1338. [Google Scholar] [CrossRef] [PubMed]
- Song, P.; Zhang, X.; Feng, W.; Xu, W.; Wu, C.; Xie, S.; Yu, S.; Fu, R. Biological Synthesis of Ursodeoxycholic Acid. Front. Microbiol. 2023, 14, 1140662. [Google Scholar] [CrossRef] [PubMed]
- von Haehling, S.; Schefold, J.C.; Jankowska, E.A.; Springer, J.; Vazir, A.; Kalra, P.R.; Sandek, A.; Fauler, G.; Stojakovic, T.; Trauner, M.; et al. Ursodeoxycholic Acid in Patients with Chronic Heart Failure. J. Am. Coll. Cardiol. 2012, 59, 585–592. [Google Scholar] [CrossRef]
- Ferraro, E.; Pozhidaeva, L.; Pitcher, D.S.; Mansfield, C.; Koh, J.H.B.; Williamson, C.; Aslanidi, O.; Gorelik, J.; Ng, F.S. Prolonged Ursodeoxycholic Acid Administration Reduces Acute Ischaemia-Induced Arrhythmias in Adult Rat Hearts. Sci. Rep. 2020, 10, 15284. [Google Scholar] [CrossRef] [PubMed]
- Adeyemi, O.; Alvarez-Laviada, A.; Schultz, F.; Ibrahim, E.; Trauner, M.; Williamson, C.; Glukhov, A.V.; Gorelik, J. Ursodeoxycholic Acid Prevents Ventricular Conduction Slowing and Arrhythmia by Restoring T-Type Calcium Current in Fetuses during Cholestasis. PLoS ONE 2017, 12, e0183167. [Google Scholar] [CrossRef] [PubMed]
- Goossens, J.-F.; Bailly, C. Ursodeoxycholic Acid and Cancer: From Chemoprevention to Chemotherapy. Pharmacol. Ther. 2019, 203, 107396. [Google Scholar] [CrossRef] [PubMed]
- Brevini, T.; Maes, M.; Webb, G.J.; John, B.V.; Fuchs, C.D.; Buescher, G.; Wang, L.; Griffiths, C.; Brown, M.L.; Scott, W.E.; et al. FXR Inhibition May Protect from SARS-CoV-2 Infection by Reducing ACE2. Nature 2023, 615, 134–142. [Google Scholar] [CrossRef] [PubMed]
- Krebs, H.A.; Wallace, P.G.; Hems, R.; Freedland, R.A. Rates of Ketone-Body Formation in the Perfused Rat Liver. Biochem. J. 1969, 112, 595–600. [Google Scholar] [CrossRef] [PubMed]
- Dilliraj, L.N.; Schiuma, G.; Lara, D.; Strazzabosco, G.; Clement, J.; Giovannini, P.; Trapella, C.; Narducci, M.; Rizzo, R. The Evolution of Ketosis: Potential Impact on Clinical Conditions. Nutrients 2022, 14, 3613. [Google Scholar] [CrossRef]
- Saris, C.G.J.; Timmers, S. Ketogenic Diets and Ketone Suplementation: A Strategy for Therapeutic Intervention. Front. Nutr. 2022, 9, 947567. [Google Scholar] [CrossRef]
- Pollay, M.; Alan Stevens, F. Starvation-induced Changes in Transport of Ketone Bodies across the Blood-brain Barrier. J. Neurosci. Res. 1980, 5, 163–172. [Google Scholar] [CrossRef]
- Garruti, G.; Baj, J.; Cignarelli, A.; Perrini, S.; Giorgino, F. Hepatokines, Bile Acids and Ketone Bodies Are Novel Hormones Regulating Energy Homeostasis. Front. Endocrinol. 2023, 14, 1154561. [Google Scholar] [CrossRef]
- Marchesi, E.; Gentili, V.; Bortolotti, D.; Preti, L.; Marchetti, P.; Cristofori, V.; Fantinati, A.; Rizzo, R.; Trapella, C.; Perrone, D.; et al. Dihydroartemisinin-Ursodeoxycholic Bile Acid Hybrids in the Fight against SARS-CoV-2. ACS Omega 2023, 8, 45078–45087. [Google Scholar] [CrossRef]
- Melloni, E.; Marchesi, E.; Preti, L.; Casciano, F.; Rimondi, E.; Romani, A.; Secchiero, P.; Navacchia, M.L.; Perrone, D. Synthesis and Biological Investigation of Bile Acid-Paclitaxel Hybrids. Molecules 2022, 27, 471. [Google Scholar] [CrossRef]
- Mishra, R.; Mishra, S. Updates in Bile Acid-Bioactive Molecule Conjugates and Their Applications. Steroids 2020, 159, 108639. [Google Scholar] [CrossRef]
- Navacchia, M.L.; Marchesi, E.; Perrone, D. Bile Acid Conjugates with Anticancer Activity: Most Recent Research. Molecules 2020, 26, 25. [Google Scholar] [CrossRef]
- Lei, K.; Yuan, M.; Zhou, T.; Ye, Q.; Zeng, B.; Zhou, Q.; Wei, A.; Guo, L. Research Progress in the Application of Bile Acid-Drug Conjugates: A “Trojan Horse” Strategy. Steroids 2021, 173, 108879. [Google Scholar] [CrossRef]
- Zappaterra, F.; Costa, S.; Summa, D.; Semeraro, B.; Cristofori, V.; Trapella, C.; Tamburini, E. Glyceric Prodrug of Ursodeoxycholic Acid (UDCA): Novozym 435-Catalyzed Synthesis of UDCA-Monoglyceride. Molecules 2021, 26, 5966. [Google Scholar] [CrossRef]
- García Liñares, G.; Antonela Zígolo, M.; Simonetti, L.; Longhi, S.A.; Baldessari, A. Enzymatic Synthesis of Bile Acid Derivatives and Biological Evaluation against Trypanosoma Cruzi. Bioorg. Med. Chem. 2015, 23, 4804–4814. [Google Scholar] [CrossRef] [PubMed]
- Chanquia, S.N.; Ripani, E.; Baldessari, A.; García Liñares, G. Bile Acids: Lipase-Catalyzed Synthesis of New Hyodeoxycholic Acid Derivatives. Steroids 2018, 140, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Zígolo, M.A.; García Liñares, G.; Baldessari, A. New Cholic Acid Derivatives: Biocatalytic Synthesis and Molecular Docking Study. Steroids 2016, 107, 10–19. [Google Scholar] [CrossRef] [PubMed]
- Sugai, T.; Takizawa, M.; Bakke, M.; Ohtsuka, Y.; Ohta, H. Efficient Lipase-Catalyzed Preparation of Long-Chain Fatty Acid Esters of Bile Acids: Biological Activity and Synthetic Application of the Products. Biosci. Biotechnol. Biochem. 1996, 60, 2059–2063. [Google Scholar] [CrossRef] [PubMed]
- Riva, S.; Bovara, R.; Ottolina, G.; Secundo, F.; Carrea, G. Regioselective Acylation of Bile Acid Derivatives with Candida Cylindracea Lipase in Anhydrous Benzene. J. Org. Chem. 1989, 54, 3161–3164. [Google Scholar] [CrossRef]
- Córdova, A.; Janda, K.D. A Highly Chemo- and Stereoselective Synthesis of Beta-Keto Esters via a Polymer-Supported Lipase Catalyzed Transesterfication. J. Org. Chem. 2001, 66, 1906–1909. [Google Scholar] [CrossRef] [PubMed]
- Sugiyama, H.; Ehata, K.; Nate, K.; Mizushima, A. Alpha-Diazoacetoacetic Acid Ester and Pro-Duction Thereof. JPH01265066A 23 October 1989. [Google Scholar]
- Cummings, L.O.; Vogel, H.A.; Bader, A.R. Beta-Carbonyl Carboxylic Acid Esters of Steroids. USPTO 2693476A 2 November 1954. [Google Scholar]
- Brandes, B.; Hoenke, S.; Schultz, C.; Deigner, H.-P.; Csuk, R. Converting Bile Acids into Mitocans. Steroids 2023, 189, 109148. [Google Scholar] [CrossRef] [PubMed]
- Benetti, S.; Romagnoli, R.; De Risi, C.; Spalluto, G.; Zanirato, V. Mastering.Beta.-Keto Esters. Chem. Rev. 1995, 95, 1065–1114. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
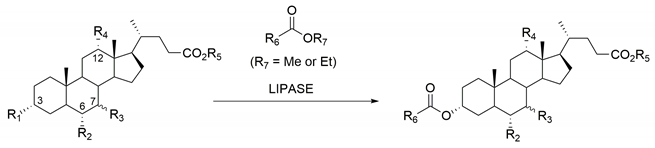
| Substrate | R1 | R2 | R3 | R4 | R5 | R6 | Enzyme | Ref. |
|---|---|---|---|---|---|---|---|---|
| Litocholic ac. | OH | H | H | H | H | CH3 | CAL-B | [27] |
| Chenodeoxycholic ac. | OH | H | 7α-OH | H | H | CH3 | CAL-B | [27] |
| Deoxycholic ac. | OH | H | H | OH | H | CH3 | CAL-B | [27] |
| Hyodeoxycholic ac. | OH | OH | H | H | H | CH3 | CAL-B | [28] |
| Ethyl cholate | OH | H | 7α-OH | OH | C2H5 | CH3 | CAL-B | [29] |
| Methyl cholate | OH | H | 7α-OH | OH | CH3 | CnH(2n+1) 1 | CAL-B | [30] |
| Methyl cholate | OH | H | 7α-OH | OH | CH3 | C3H7 | CCL Type VII | [31] |
| Methyl chenodeoxycholate | OH | H | 7α-OH | H | CH3 | C3H7 | CCL Type VII | [31] |
| Methyl deoxycholate | OH | H | H | OH | CH3 | C3H7 | CCL Type VII | [31] |
| Methyl ursodeoxycholate | OH | H | 7β-OH | H | CH3 | C3H7 | CCL Type VII | [31] |
| Biocatalyst Name | Lipase Type | Physical Form | Product | Conversion (%) 1 |
|---|---|---|---|---|
| Lipozyme 435 | lipase B from C. antarctica | Immobilized 2 | 3a | 57 |
| Novocor AD L | lipase A from C. antarctica | Solution 3 | no | - |
| Lipura Flex | lipase B from C. antarctica | Immobilized 2 | 3a | 61 |
| Lipozyme RM IM | 1,3-specific lipase from R. miehi | Immobilized 4 | no | - |
| Lipozyme TL IM | lipase from T. lanuginosus | Immobilized 5 | no | - |
| Palatase 20000 L | lipase from R. miehei | Solution 6 | no | - |

| Molar Ratio of 3b to Ethanol | Reaction Time (h) | 3c (%) 1 | 3d (%) 1 |
|---|---|---|---|
| 1:5 | 12 | 50 | 33 |
| 1:2 | 8 | 82 | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Venturi, V.; Marchesi, E.; Perrone, D.; Costa, V.; Catani, M.; Aprile, S.; Lerin, L.A.; Zappaterra, F.; Giovannini, P.P.; Preti, L. Enzymatic Synthesis of New Acetoacetate–Ursodeoxycholic Acid Hybrids as Potential Therapeutic Agents and Useful Synthetic Scaffolds as Well. Molecules 2024, 29, 1305. https://doi.org/10.3390/molecules29061305
Venturi V, Marchesi E, Perrone D, Costa V, Catani M, Aprile S, Lerin LA, Zappaterra F, Giovannini PP, Preti L. Enzymatic Synthesis of New Acetoacetate–Ursodeoxycholic Acid Hybrids as Potential Therapeutic Agents and Useful Synthetic Scaffolds as Well. Molecules. 2024; 29(6):1305. https://doi.org/10.3390/molecules29061305
Chicago/Turabian StyleVenturi, Valentina, Elena Marchesi, Daniela Perrone, Valentina Costa, Martina Catani, Simona Aprile, Lindomar Alberto Lerin, Federico Zappaterra, Pier Paolo Giovannini, and Lorenzo Preti. 2024. "Enzymatic Synthesis of New Acetoacetate–Ursodeoxycholic Acid Hybrids as Potential Therapeutic Agents and Useful Synthetic Scaffolds as Well" Molecules 29, no. 6: 1305. https://doi.org/10.3390/molecules29061305