
Machine-Learning-Based Prediction of Plant Cuticle–Air Partition Coefficients for Organic Pollutants: Revealing Mechanisms from a Molecular Structure Perspective

Abstract

1. Introduction
2. Results and Discussion
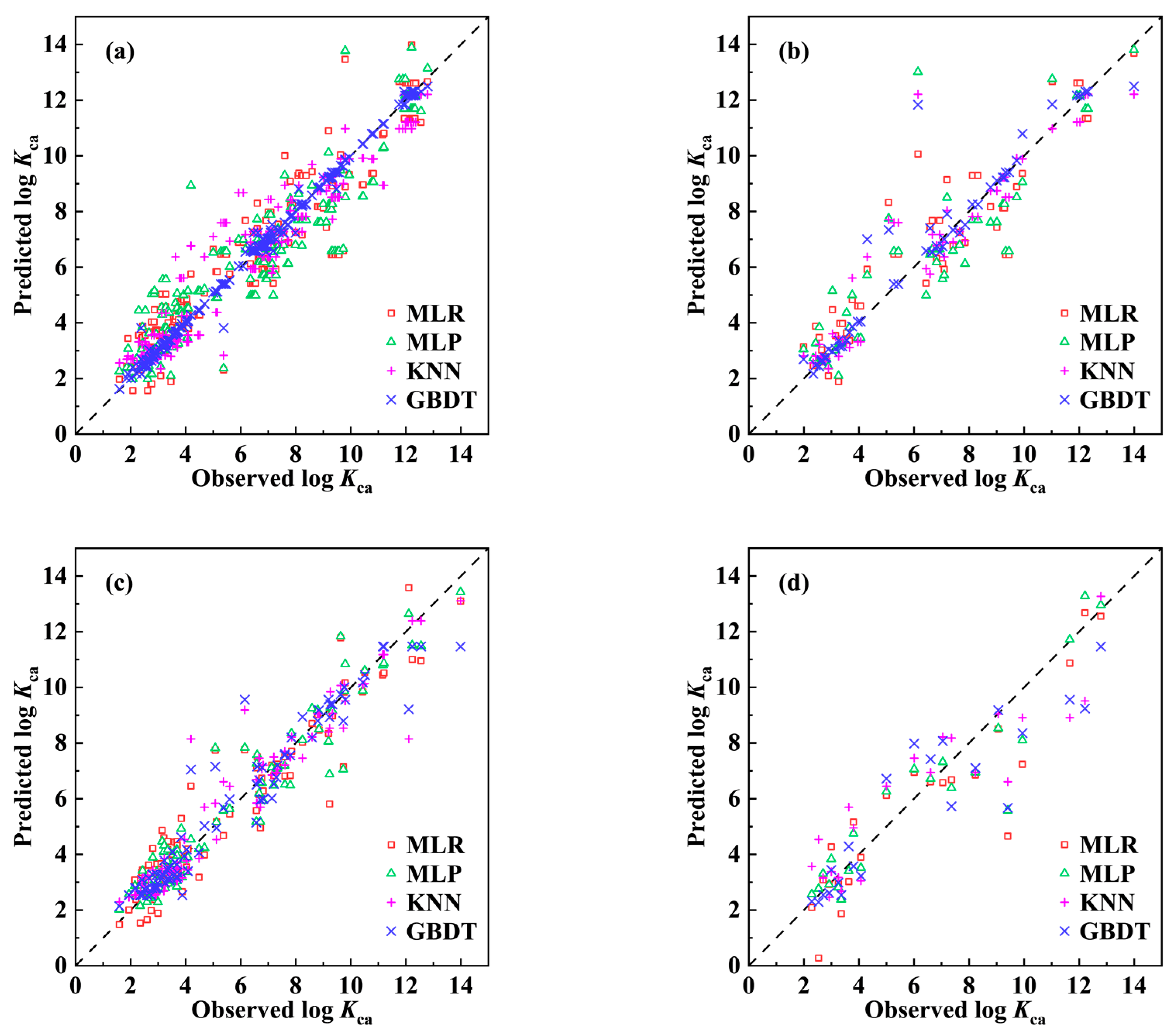
2.1. Development of the QSPR Model
- ntra = 204, = 0.873, = 0.869, = 0.872, RMSEtra = 1.101;
- next = 51, = 0.839, = 0.835, RMSEext = 1.296.
- ntra = 84, = 0.891, = 0.874, = 0.886, RMSEtra = 0.987;
- next = 22, = 0.833, = 0.807, RMSEext = 1.466.
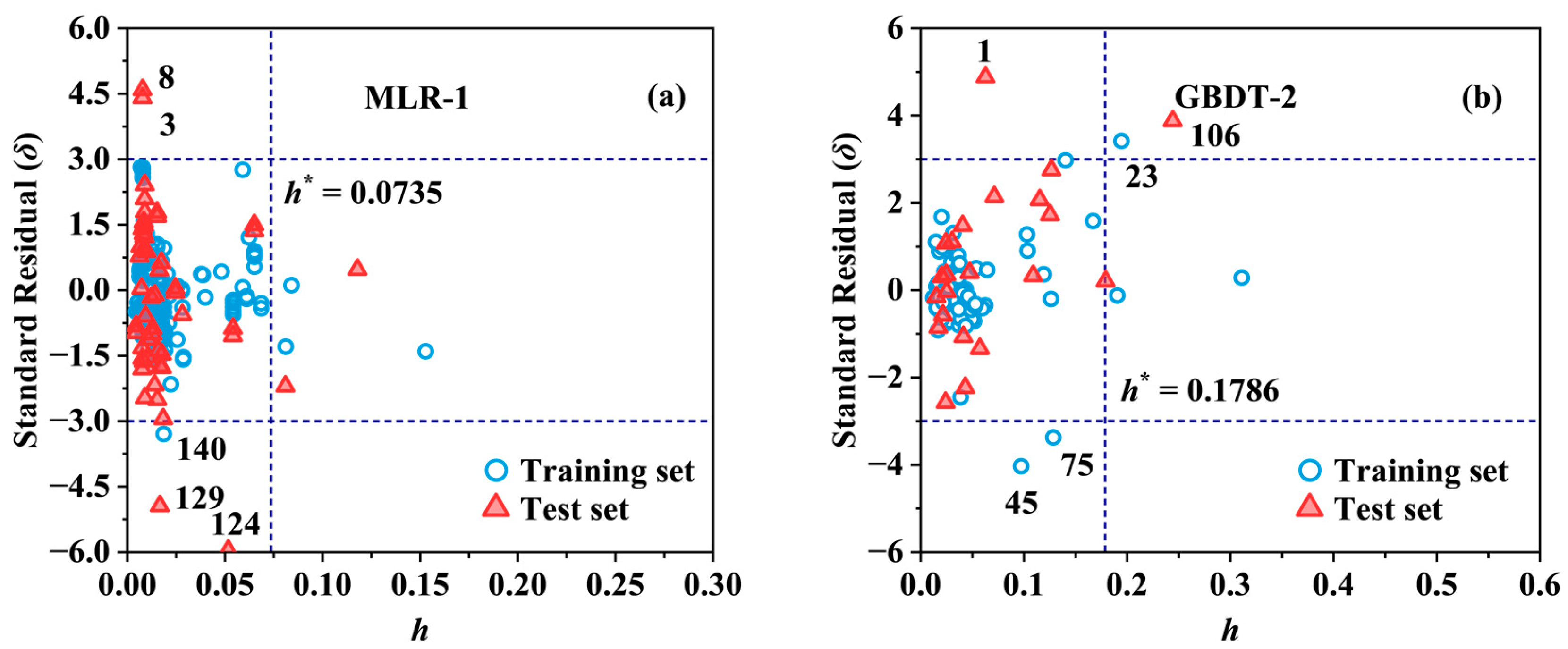
2.2. Applicability Domain
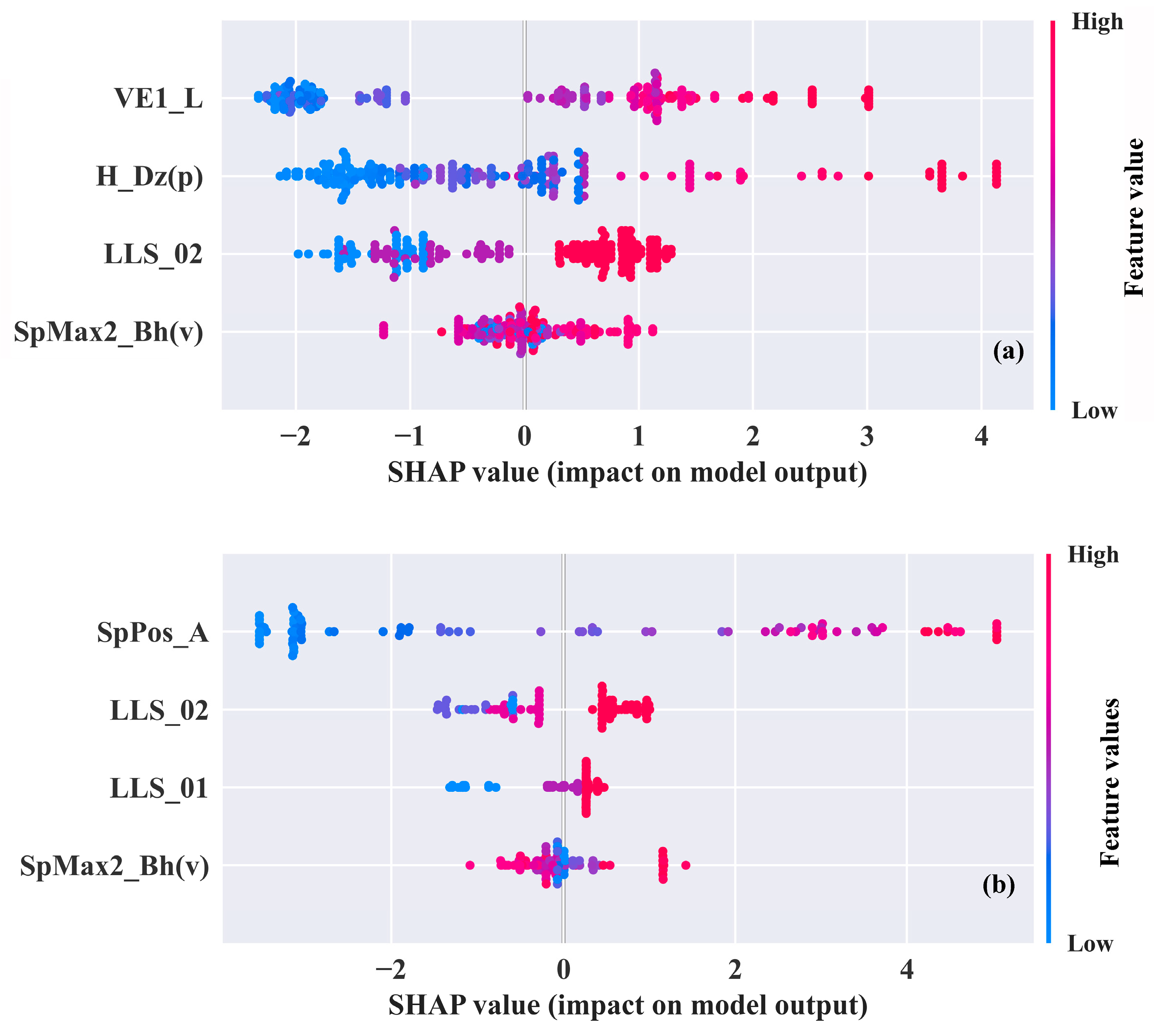
2.3. Mechanism Interpretation
2.4. Model Comparison
3. Materials and Methods
3.1. Dataset Preparation
3.2. Descriptor Generation and Filtering
3.3. Model Development and Validation
3.4. Model Interpretation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Talaiekhozani, A.; Rezania, S.; Kim, K.-H.; Sanaye, R.; Amani, A.M. Recent advances in photocatalytic removal of organic and inorganic pollutants in air. J. Clean. Prod. 2021, 278, 123895. [Google Scholar] [CrossRef]
- Welke, B.; Ettlinger, K.; Riederer, M. Sorption of Volatile Organic Chemicals in Plant Surfaces. Environ. Sci. Technol. 1998, 32, 1099–1104. [Google Scholar] [CrossRef]
- Li, Q.; Chen, B. Organic Pollutant Clustered in the Plant Cuticular Membranes: Visualizing the Distribution of Phenanthrene in Leaf Cuticle Using Two-Photon Confocal Scanning Laser Microscopy. Environ. Sci. Technol. 2014, 48, 4774–4781. [Google Scholar] [CrossRef]
- Collins, C.D.; Finnegan, E. Modeling the Plant Uptake of Organic Chemicals, Including the Soil−Air−Plant Pathway. Environ. Sci. Technol. 2010, 44, 998–1003. [Google Scholar] [CrossRef] [PubMed]
- Sabljic, A.; Guesten, H.; Schoenherr, J.; Riederer, M. Modeling plant uptake of airborne organic chemicals. 1. Plant cuticle/water partitioning and molecular connectivity. Environ. Sci. Technol. 1990, 24, 1321–1326. [Google Scholar] [CrossRef]
- Eddula, S.; Xu, A.; Jiang, C.; Huang, J.; Tirumala, P.; Liu, G.; Acree, W.E.; Abraham, M.H. Abraham solvation parameter model: Updated correlations for describing solute partitioning into plant cuticles from water and from air. Phys. Chem. Liq. 2021, 59, 716–732. [Google Scholar] [CrossRef]
- Chefetz, B.; Xing, B. Relative Role of Aliphatic and Aromatic Moieties as Sorption Domains for Organic Compounds: A Review. Environ. Sci. Technol. 2009, 43, 1680–1688. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, Z.; Tan, F.; Rodgers, T.F.M.; Hou, M.; Yang, Y.; Li, X. Ornamental houseplants as potential biosamplers for indoor pollution of organophosphorus flame retardants. Sci. Total Environ. 2021, 767, 144433. [Google Scholar] [CrossRef]
- Zhao, X.; He, M.; Shang, H.; Yu, H.; Wang, H.; Li, H.; Piao, J.; Quinto, M.; Li, D. Biomonitoring polycyclic aromatic hydrocarbons by Salix matsudana leaves: A comparison with the relevant air content and evaluation of environmental parameter effects. Atmos. Environ. 2018, 181, 47–53. [Google Scholar] [CrossRef]
- Platts, J.A.; Abraham, M.H. Partition of Volatile Organic Compounds from Air and from Water into Plant Cuticular Matrix: An LFER Analysis. Environ. Sci. Technol. 2000, 34, 318–323. [Google Scholar] [CrossRef]
- Keymeulen, R.; De Bruyn, G.; Van Langenhove, H. Headspace gas chromatographic determination of the plant cuticle–air partition coefficients for monocyclic aromatic hydrocarbons as environmental compartment. J. Chromatogr. 1997, 774, 213–221. [Google Scholar] [CrossRef]
- Barber, J.L.; Thomas, G.O.; Kerstiens, G.; Jones, K.C. Current issues and uncertainties in the measurement and modelling of air–vegetation exchange and within-plant processing of POPs. Environ. Pollut. 2004, 128, 99–138. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Dai, C.; Zhou, Y.; Peng, H.; Yi, K.; Qin, P.; Luo, S.; Zhang, X. Comparisons of three plant species in accumulating polycyclic aromatic hydrocarbons (PAHs) from the atmosphere: A review. Environ. Sci. Pollut. Res. 2018, 25, 16548–16566. [Google Scholar] [CrossRef] [PubMed]
- Fernández, V.; Bahamonde, H.A.; Javier Peguero-Pina, J.; Gil-Pelegrín, E.; Sancho-Knapik, D.; Gil, L.; Goldbach, H.E.; Eichert, T. Physico-chemical properties of plant cuticles and their functional and ecological significance. J. Exp. Bot. 2017, 68, 5293–5306. [Google Scholar] [CrossRef]
- Qi, X.; Li, X.; Yao, H.; Huang, Y.; Cai, X.; Chen, J.; Zhu, H. Predicting plant cuticle-water partition coefficients for organic pollutants using pp-LFER model. Sci. Total Environ. 2020, 725, 138455. [Google Scholar] [CrossRef] [PubMed]
- Nabi, D.; Arey, J.S. Predicting Partitioning and Diffusion Properties of Nonpolar Chemicals in Biotic Media and Passive Sampler Phases by GC × GC. Environ. Sci. Technol. 2017, 51, 3001–3011. [Google Scholar] [CrossRef] [PubMed]
- Gui, B.; Xu, X.; Zhang, S.; Wang, Y.; Li, C.; Zhang, D.; Su, L.; Zhao, Y. Prediction of organic compounds adsorbed by polyethylene and chlorinated polyethylene microplastics in freshwater using QSAR. Environ. Res. 2021, 197, 111001. [Google Scholar] [CrossRef]
- Qiu, Y.; Li, Z.; Zhang, T.; Zhang, P. Predicting aqueous sorption of organic pollutants on microplastics with machine learning. Water Res. 2023, 244, 120503. [Google Scholar] [CrossRef]
- Abouzari, M.; Pahlavani, P.; Izaditame, F.; Bigdeli, B. Estimating the chemical oxygen demand of petrochemical wastewater treatment plants using linear and nonlinear statistical models—A case study. Chemosphere 2021, 270, 129465. [Google Scholar] [CrossRef]
- Liu, X.; Lu, D.; Zhang, A.; Liu, Q.; Jiang, G. Data-Driven Machine Learning in Environmental Pollution: Gains and Problems. Environ. Sci. Technol. 2022, 56, 2124–2133. [Google Scholar] [CrossRef]
- Diéguez-Santana, K.; Nachimba-Mayanchi, M.M.; Puris, A.; Gutiérrez, R.T.; González-Díaz, H. Prediction of acute toxicity of pesticides for Americamysis bahia using linear and nonlinear QSTR modelling approaches. Environ. Res. 2022, 214, 113984. [Google Scholar] [CrossRef]
- Hamadache, M.; Benkortbi, O.; Hanini, S.; Amrane, A.; Khaouane, L.; Si Moussa, C. A Quantitative Structure Activity Relationship for acute oral toxicity of pesticides on rats: Validation, domain of application and prediction. J. Hazard. Mater. 2016, 303, 28–40. [Google Scholar] [CrossRef] [PubMed]
- Zhong, S.; Zhang, K.; Bagheri, M.; Burken, J.G.; Gu, A.; Li, B.; Ma, X.; Marrone, B.L.; Ren, Z.J.; Schrier, J.; et al. Machine Learning: New Ideas and Tools in Environmental Science and Engineering. Environ. Sci. Technol. 2021, 55, 12741–12754. [Google Scholar] [CrossRef]
- OECD. Guidance Document on the Validation of (Quantitative) Structure-Activity Relationship [(Q)SAR] Models; OECD: Paris, France, 2014. [Google Scholar] [CrossRef]
- Zhu, T.; Tao, C. Prediction models with multiple machine learning algorithms for POPs: The calculation of PDMS-air partition coefficient from molecular descriptor. J. Hazard. Mater. 2022, 423, 127037. [Google Scholar] [CrossRef]
- Chirico, N.; Gramatica, P. Real External Predictivity of QSAR Models. Part 2. New Intercomparable Thresholds for Different Validation Criteria and the Need for Scatter Plot Inspection. J. Chem. Inf. Model. 2012, 52, 2044–2058. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, R.K.; Kumar, V.; Roy, K. Ecotoxicological QSTR and QSTTR Modeling for the Prediction of Acute Oral Toxicity of Pesticides against Multiple Avian Species. Environ. Sci. Technol. 2022, 56, 335–348. [Google Scholar] [CrossRef] [PubMed]
- Shahi, A.; Vafaei Molamahmood, H.; Faraji, N.; Long, M. Quantitative structure-activity relationship for the oxidation of organic contaminants by peracetic acid using GA-MLR method. J. Environ. Manag. 2022, 310, 114747. [Google Scholar] [CrossRef]
- Wang, L.; Chen, B.; Zhang, T. Predicting hydrolysis kinetics for multiple types of halogenated disinfection byproducts via QSAR models. Chem. Eng. J. 2018, 342, 372–385. [Google Scholar] [CrossRef]
- Galimberti, F.; Moretto, A.; Papa, E. Application of chemometric methods and QSAR models to support pesticide risk assessment starting from ecotoxicological datasets. Water Res. 2020, 174, 115583. [Google Scholar] [CrossRef]
- Xiao, R.; Ye, T.; Wei, Z.; Luo, S.; Yang, Z.; Spinney, R. Quantitative Structure–Activity Relationship (QSAR) for the Oxidation of Trace Organic Contaminants by Sulfate Radical. Environ. Sci. Technol. 2015, 49, 13394–13402. [Google Scholar] [CrossRef]
- Lu, H.; Liu, W.; Yang, F.; Zhou, H.; Liu, F.; Yuan, H.; Chen, G.; Jiao, Y. Thermal Conductivity Estimation of Diverse Liquid Aliphatic Oxygen-Containing Organic Compounds Using the Quantitative Structure–Property Relationship Method. ACS Omega 2020, 5, 8534–8542. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.; Li, Y.; Yu, Y.; Wang, Z.; Xu, T.; Chen, J.; Lin, J.; Li, X. Development of models predicting biodegradation rate rating with multiple linear regression and support vector machine algorithms. Chemosphere 2020, 253, 126666. [Google Scholar] [CrossRef]
- Peng, D.; Picchioni, F. Prediction of toxicity of Ionic Liquids based on GC-COSMO method. J. Hazard. Mater. 2020, 398, 122964. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Jin, L.; Yu, H.; Lv, L.; Chen, C.-E.; Ying, G.-G. Understanding and predicting the diffusivity of organic chemicals for diffusive gradients in thin-films using a QSPR model. Sci. Total Environ. 2020, 706, 135691. [Google Scholar] [CrossRef] [PubMed]
- Gobas, F.A.P.C.; McNeil, E.J.; Lovett-Doust, L.; Haffner, G.D. Bioconcentration of chlorinated aromatic hydrocarbons in aquatic macrophytes. Environ. Sci. Technol. 1991, 25, 924–929. [Google Scholar] [CrossRef]
- Eichenlaub, J.; Rakowska, P.W.; Kloskowski, A. User-assisted methodology targeted for building structure interpretable QSPR models for boosting CO2 capture with ionic liquids. J. Mol. Liq. 2022, 350, 118511. [Google Scholar] [CrossRef]
- Sanches-Neto, F.O.; Dias-Silva, J.R.; de Oliveira, V.M.; Aquilanti, V.; Carvalho-Silva, V.H. Evaluating and elucidating the reactivity of OH radicals with atmospheric organic pollutants: Reaction kinetics and mechanisms by machine learning. Atmos. Environ. 2022, 275, 119019. [Google Scholar] [CrossRef]
- Monge, A.; Arrault, A.; Marot, C.; Morin-Allory, L. Managing, profiling and analyzing a library of 2.6 million compounds gathered from 32 chemical providers. Mol. Divers. 2006, 10, 389–403. [Google Scholar] [CrossRef]
- Heda, P.; Ravishankar, S.; Shankar, A.; Chaganti, S.; Rajan, D.; Parekh, R.; Renganathan, G. Identifying promising anticancer Sulforaphane derivatives using QSAR, Docking, and ADME studies. J. Stud. Res. 2021, 10. [Google Scholar] [CrossRef]
- Dobričić, V.; Bošković, J.; Vukadinović, D.; Vladimirov, S.; Čudina, O. Estimation of lipophilicity and design of new 17β-carboxamide glucocorticoids using RP-HPLC and quantitative structure-retention relationships analysis. Acta Chromatogr. 2021, 34, 130–137. [Google Scholar] [CrossRef]
- Cao, C.; Nian, B.; Li, Y.; Wu, S.; Liu, Y. Multiple Hydrogen-Bonding Interactions Enhance the Solubility of Starch in Natural Deep Eutectic Solvents: Molecule and Macroscopic Scale Insights. J. Agric. Food Chem. 2019, 67, 12366–12373. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Fan, T.; Sun, G.; Zhao, L.; Zhong, R.; Peng, Y. Systematic QSAR and iQCCR modelling of fused/non-fused aromatic hydrocarbons (FNFAHs) carcinogenicity to rodents: Reducing unnecessary chemical synthesis and animal testing. Green Chem. 2022, 24, 5304–5319. [Google Scholar] [CrossRef]
- Ibrahim, M.T.; Uzairu, A.; Uba, S.; Shallangwa, G.A. Computational modeling of novel quinazoline derivatives as potent epidermal growth factor receptor inhibitors. Heliyon 2020, 6, e03289. [Google Scholar] [CrossRef] [PubMed]
- Sikorska, C. Toward predicting vertical detachment energies for superhalogen anions exclusively from 2-D structures. Chem. Phys. Lett. 2015, 625, 157–163. [Google Scholar] [CrossRef]
- Khan, K.; Khan, P.M.; Lavado, G.; Valsecchi, C.; Pasqualini, J.; Baderna, D.; Marzo, M.; Lombardo, A.; Roy, K.; Benfenati, E. QSAR modeling of Daphnia magna and fish toxicities of biocides using 2D descriptors. Chemosphere 2019, 229, 8–17. [Google Scholar] [CrossRef]
- Congreve, M.; Carr, R.; Murray, C.; Jhoti, H. A ‘Rule of Three’ for fragment-based lead discovery? Drug Discov. Today 2003, 8, 876–877. [Google Scholar] [CrossRef] [PubMed]
- Vios, V.S.L.; Billones, J.B. Cluster and multi-linear regression analyses guided identification of molecular descriptors that account for cyclooxygenase activities. J. Chem. Pharm. Res. 2015, 7, 735–742. [Google Scholar]
- Wang, W.; Pan, Y.; Zhu, Y.; Xu, H.; Zhou, L.; Noh, H.M.; Jeong, J.H.; Liu, X.; Li, L. Bond energy, site preferential occupancy and Eu2+/3+ co-doping system induced by Eu3+ self-reduction in Ca10M(PO4)7 (M = Li, Na, K) crystals. Dalton Trans. 2018, 47, 6507–6518. [Google Scholar] [CrossRef]
- Abudour, A.M.; Mohammad, S.A.; Robinson, R.L., Jr.; Gasem, K.A.M. Generalized binary interaction parameters for the Peng–Robinson equation of state. Fluid Phase Equilib. 2014, 383, 156–173. [Google Scholar] [CrossRef]
- Soteras, I.; Curutchet, C.; Bidon-Chanal, A.; Dehez, F.; Ángyán, J.G.; Orozco, M.; Chipot, C.; Luque, F.J. Derivation of Distributed Models of Atomic Polarizability for Molecular Simulations. J. Chem. Theory Comput. 2007, 3, 1901–1913. [Google Scholar] [CrossRef]
- Yang, Z.; Luo, S.; Wei, Z.; Ye, T.; Spinney, R.; Chen, D.; Xiao, R. Rate constants of hydroxyl radical oxidation of polychlorinated biphenyls in the gas phase: A single−descriptor based QSAR and DFT study. Environ. Pollut. 2016, 211, 157–164. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Yang, X.; Zhang, S.; Guo, T.L.; Zhao, B.; Du, Q.; Chen, J. Polarizability and aromaticity index govern AhR-mediated potencies of PAHs: A QSAR with consideration of freely dissolved concentrations. Chemosphere 2021, 268, 129343. [Google Scholar] [CrossRef] [PubMed]
- Rojas Villa, C.X.; Duchowicz, P.R.; Tripaldi, P.; Pis Diez, R. Quantitative Structure-Property Relationships for Predicting the Retention Indices of Fragrances on Stationary Phases of Different Polarity. J. Argent. Chem. Soc. 2017, 104, 173–193. [Google Scholar]
- Mansouri, K.; Ringsted, T.; Ballabio, D.; Todeschini, R.; Consonni, V. Quantitative Structure–Activity Relationship Models for Ready Biodegradability of Chemicals. J. Chem. Inf. Model. 2013, 53, 867–878. [Google Scholar] [CrossRef] [PubMed]
- Consonni, V.; Todeschini, R. Multivariate Analysis of Molecular Descriptors. In Statistical Modelling of Molecular Descriptors in QSAR/QSPR; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2012; pp. 111–147. [Google Scholar]
- Wang, Z.; Su, Y.; Shen, W.; Jin, S.; Clark, J.H.; Ren, J.; Zhang, X. Predictive deep learning models for environmental properties: The direct calculation of octanol–water partition coefficients from molecular graphs. Green Chem. 2019, 21, 4555–4565. [Google Scholar] [CrossRef]
- Borhani, T.N.G.; Saniedanesh, M.; Bagheri, M.; Lim, J.S. QSPR prediction of the hydroxyl radical rate constant of water contaminants. Water Res. 2016, 98, 344–353. [Google Scholar] [CrossRef] [PubMed]
- Thandra, D.R.; Bojja, R.R.; Allikayala, R. Synthesis, spectral studies, molecular structure determination by single crystal X-ray diffraction of (E)-1-(((3-fluoro-4-morpholinophenyl)imino)methyl)napthalen-2-ol and computational studies by Austin model-1(AM1), MM2 and DFT/B3LYP. SN Appl. Sci. 2020, 2, 1765. [Google Scholar] [CrossRef]
- Mauri, A. alvaDesc: A Tool to Calculate and Analyze Molecular Descriptors and Fingerprints. In Ecotoxicological QSARs; Roy, K., Ed.; Springer: New York, NY, USA, 2020; pp. 801–820. [Google Scholar]
- Islam, M.N.; Huang, L.; Siciliano, S.D. Inclusion of molecular descriptors in predictive models improves pesticide soil-air partitioning estimates. Chemosphere 2020, 248, 126031. [Google Scholar] [CrossRef]
- Glienke, J.; Schillberg, W.; Stelter, M.; Braeutigam, P. Prediction of degradability of micropollutants by sonolysis in water with QSPR—A case study on phenol derivates. Ultrason. Sonochem. 2022, 82, 105867. [Google Scholar] [CrossRef]
- Shao, Y.; Liu, J.; Wang, M.; Shi, L.; Yao, X.; Gramatica, P. Integrated QSPR models to predict the soil sorption coefficient for a large diverse set of compounds by using different modeling methods. Atmos. Environ. 2014, 88, 212–218. [Google Scholar] [CrossRef]
- Cao, L.; Zhu, P.; Zhao, Y.; Zhao, J. Using machine learning and quantum chemistry descriptors to predict the toxicity of ionic liquids. J. Hazard. Mater. 2018, 352, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Xie, L.; Zhang, D.; Xu, X.; Xu, L. Application of Machine Learning Methods to Predict the Air Half-Lives of Persistent Organic Pollutants. Molecules 2023, 28, 7457. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Li, J.-J.; Wang, Q.; Jia, Q.; Yan, F.; Luo, Z.-H.; Zhou, Y.-N. Computer-aided estimation of kinetic rate constant for degradation of volatile organic compounds by hydroxyl radical: An improved model using quantum chemical and norm descriptors. Chem. Eng. Sci. 2022, 248, 117244. [Google Scholar] [CrossRef]
- IBM Corp. IBM SPSS Statistics for Windows; International Business Machines Corporation: Armonk, NY, USA, 2011; Available online: https://www.ibm.com/analytics/spss-statistics-software (accessed on 13 January 2020).
- Ling, Y.; Klemes, M.J.; Steinschneider, S.; Dichtel, W.R.; Helbling, D.E. QSARs to predict adsorption affinity of organic micropollutants for activated carbon and β-cyclodextrin polymer adsorbents. Water Res. 2019, 154, 217–226. [Google Scholar] [CrossRef]
- Saavedra, L.M.; Romanelli, G.P.; Duchowicz, P.R. A non-conformational QSAR study for plant-derived larvicides against Zika Aedes aegypti L. vector. Environ. Sci. Pollut. Res. 2020, 27, 6205–6214. [Google Scholar] [CrossRef]
- Python Software Foundation. Python Programming Language; Python Software Foundation: Beaverton, OR, USA, 2021; Available online: https://www.python.org/ (accessed on 26 January 2022).
- Parinet, J. Prediction of pesticide retention time in reversed-phase liquid chromatography using quantitative-structure retention relationship models: A comparative study of seven molecular descriptors datasets. Chemosphere 2021, 275, 130036. [Google Scholar] [CrossRef]
- De, P.; Kar, S.; Ambure, P.; Roy, K. Prediction reliability of QSAR models: An overview of various validation tools. Arch. Toxicol. 2022, 96, 1279–1295. [Google Scholar] [CrossRef]
- Gramatica, P.; Sangion, A. A Historical Excursus on the Statistical Validation Parameters for QSAR Models: A Clarification Concerning Metrics and Terminology. J. Chem. Inf. Model. 2016, 56, 1127–1131. [Google Scholar] [CrossRef]
- Samad, A.; Garuda, S.; Vogt, U.; Yang, B. Air pollution prediction using machine learning techniques—An approach to replace existing monitoring stations with virtual monitoring stations. Atmos. Environ. 2023, 310, 119987. [Google Scholar] [CrossRef]
- Yang, Y.-T.; Ni, H.-G. Predictive in silico models for aquatic toxicity of cosmetic and personal care additive mixtures. Water Res. 2023, 236, 119981. [Google Scholar] [CrossRef]
- Gramatica, P. Principles of QSAR models validation: Internal and external. QSAR Comb. Sci. 2007, 26, 694–701. [Google Scholar] [CrossRef]
- Djaković Sekulić, T.; Jović, B.; Ivančev-Tumbas, I.; Panglisch, S. In silico modelling of selected organic substances adsorption from water onto activated carbon. Chem. Eng. Sci. 2024, 287, 119765. [Google Scholar] [CrossRef]
- Lavado, G.J.; Baderna, D.; Gadaleta, D.; Ultre, M.; Roy, K.; Benfenati, E. Ecotoxicological QSAR modeling of the acute toxicity of organic compounds to the freshwater crustacean Thamnocephalus platyurus. Chemosphere 2021, 280, 130652. [Google Scholar] [CrossRef] [PubMed]
- Gély, C.A.; Picard-Hagen, N.; Chassan, M.; Garrigues, J.-C.; Gayrard, V.; Lacroix, M.Z. Contribution of Reliable Chromatographic Data in QSAR for Modelling Bisphenol Transport across the Human Placenta Barrier. Molecules 2023, 28, 500. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Sun, G.; Fan, T.; Li, F.; Xu, Y.; Zhang, N.; Zhao, L.; Zhong, R. Ecotoxicological QSAR study of fused/non-fused polycyclic aromatic hydrocarbons (FNFPAHs): Assessment and priority ranking of the acute toxicity to Pimephales promelas by QSAR and consensus modeling methods. Sci. Total Environ. 2023, 876, 162736. [Google Scholar] [CrossRef] [PubMed]
- Derki, N.-E.H.; Kerassa, A.; Belaidi, S.; Derki, M.; Yamari, I.; Samadi, A.; Chtita, S. Computer-Aided Strategy on 5-(Substituted Benzylidene) Thiazolidine-2,4-Diones to Develop New and Potent PTP1B Inhibitors: QSAR Modeling, Molecular Docking, Molecular Dynamics, PASS Predictions, and DFT Investigations. Molecules 2024, 29, 822. [Google Scholar] [CrossRef] [PubMed]
- Lundberg, S.M.; Erion, G.; Chen, H.; DeGrave, A.; Prutkin, J.M.; Nair, B.; Katz, R.; Himmelfarb, J.; Bansal, N.; Lee, S.-I. From local explanations to global understanding with explainable AI for trees. Nat. Mach. Intell. 2020, 2, 56–67. [Google Scholar] [CrossRef]
- Wojtuch, A.; Jankowski, R.; Podlewska, S. How can SHAP values help to shape metabolic stability of chemical compounds? J. Cheminf. 2021, 13, 74. [Google Scholar] [CrossRef]
- Zheng, S.; Guo, W.; Li, C.; Sun, Y.; Zhao, Q.; Lu, H.; Si, Q.; Wang, H. Application of machine learning and deep learning methods for hydrated electron rate constant prediction. Environ. Res. 2023, 231, 115996. [Google Scholar] [CrossRef]
- Abdollahi, A.; Pradhan, B. Explainable artificial intelligence (XAI) for interpreting the contributing factors feed into the wildfire susceptibility prediction model. Sci. Total Environ. 2023, 879, 163004. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Datasets | Descriptors | Parameters | p | VIF |
|---|---|---|---|---|
| (I) | VE1_L | coefficient sum of the last eigenvector (absolute values) from Laplace matrix | <0.001 | 7.527 |
| LLS_02 | modified lead-like score from Monge et al. (8 rules) | <0.001 | 2.816 | |
| H_Dz(p) | Harary-like index from Barysz matrix weighted by polarizability | <0.001 | 9.343 | |
| SpMax2_Bh(v) | largest eigenvalue n. 2 of Burden matrix weighted by van der Waals volume | <0.001 | 2.937 | |
| (II) | SpPos_A | spectral positive sum from adjacency matrix | <0.001 | 6.594 |
| LLS_02 | modified lead-like score from Monge et al. (8 rules) | <0.001 | 2.011 | |
| LLS_01 | modified lead-like score from Congreve et al. (6 rules) | <0.001 | 2.125 | |
| SpMax2_Bh(v) | largest eigenvalue n. 2 of Burden matrix weighted by van der Waals volume | <0.001 | 3.128 |
| Models | Training Set | Test Set | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ntra | MAEtra | RMSEtra | stra | next | MAEext | RMSEext | sext | CCC | ||||||
| Threshold | - | >0.7 | >0.6 | >0.6 | - | - | - | - | >0.7 | >0.6 | - | - | - | >0.85 |
| MLR-1 | 204 | 0.873 | 0.869 | 0.872 | 0.874 | 1.101 | 1.114 | 51 | 0.839 | 0.835 | 1.024 | 1.296 | 1.364 | 0.914 |
| MLP-1 | 0.850 | 0.678 | 0.676 | 0.917 | 1.194 | 1.209 | 0.790 | 0.784 | 1.009 | 1.485 | 1.564 | 0.889 | ||
| KNN-1 | 0.920 | 0.742 | 0.778 | 0.638 | 0.871 | 0.882 | 0.859 | 0.855 | 0.706 | 1.214 | 1.279 | 0.925 | ||
| GBDT-1 | 0.995 | 0.936 | 0.964 | 0.114 | 0.224 | 0.227 | 0.911 | 0.902 | 0.411 | 1.001 | 1.054 | 0.952 | ||
| MLR-2 | 84 | 0.891 | 0.874 | 0.886 | 0.725 | 0.987 | 1.018 | 22 | 0.833 | 0.807 | 1.006 | 1.466 | 1.668 | 0.902 |
| MLP-2 | 0.921 | 0.798 | 0.809 | 0.629 | 0.843 | 0.869 | 0.887 | 0.884 | 0.798 | 1.139 | 1.296 | 0.940 | ||
| KNN-2 | 0.919 | 0.661 | 0.802 | 0.536 | 0.855 | 0.881 | 0.821 | 0.815 | 1.176 | 1.435 | 1.632 | 0.891 | ||
| GBDT-2 | 0.925 | 0.756 | 0.864 | 0.504 | 0.821 | 0.847 | 0.837 | 0.811 | 1.097 | 1.451 | 1.651 | 0.891 | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tao, T.; Tao, C.; Zhu, T. Machine-Learning-Based Prediction of Plant Cuticle–Air Partition Coefficients for Organic Pollutants: Revealing Mechanisms from a Molecular Structure Perspective. Molecules 2024, 29, 1381. https://doi.org/10.3390/molecules29061381
Tao T, Tao C, Zhu T. Machine-Learning-Based Prediction of Plant Cuticle–Air Partition Coefficients for Organic Pollutants: Revealing Mechanisms from a Molecular Structure Perspective. Molecules. 2024; 29(6):1381. https://doi.org/10.3390/molecules29061381
Chicago/Turabian StyleTao, Tianyun, Cuicui Tao, and Tengyi Zhu. 2024. "Machine-Learning-Based Prediction of Plant Cuticle–Air Partition Coefficients for Organic Pollutants: Revealing Mechanisms from a Molecular Structure Perspective" Molecules 29, no. 6: 1381. https://doi.org/10.3390/molecules29061381
APA StyleTao, T., Tao, C., & Zhu, T. (2024). Machine-Learning-Based Prediction of Plant Cuticle–Air Partition Coefficients for Organic Pollutants: Revealing Mechanisms from a Molecular Structure Perspective. Molecules, 29(6), 1381. https://doi.org/10.3390/molecules29061381