
Design, Synthesis, and Biological Evaluation of Novel Coumarin Analogs Targeted against SARS-CoV-2
 , , ,
, , ,
Abstract
:1. Introduction
2. Results and Discussion
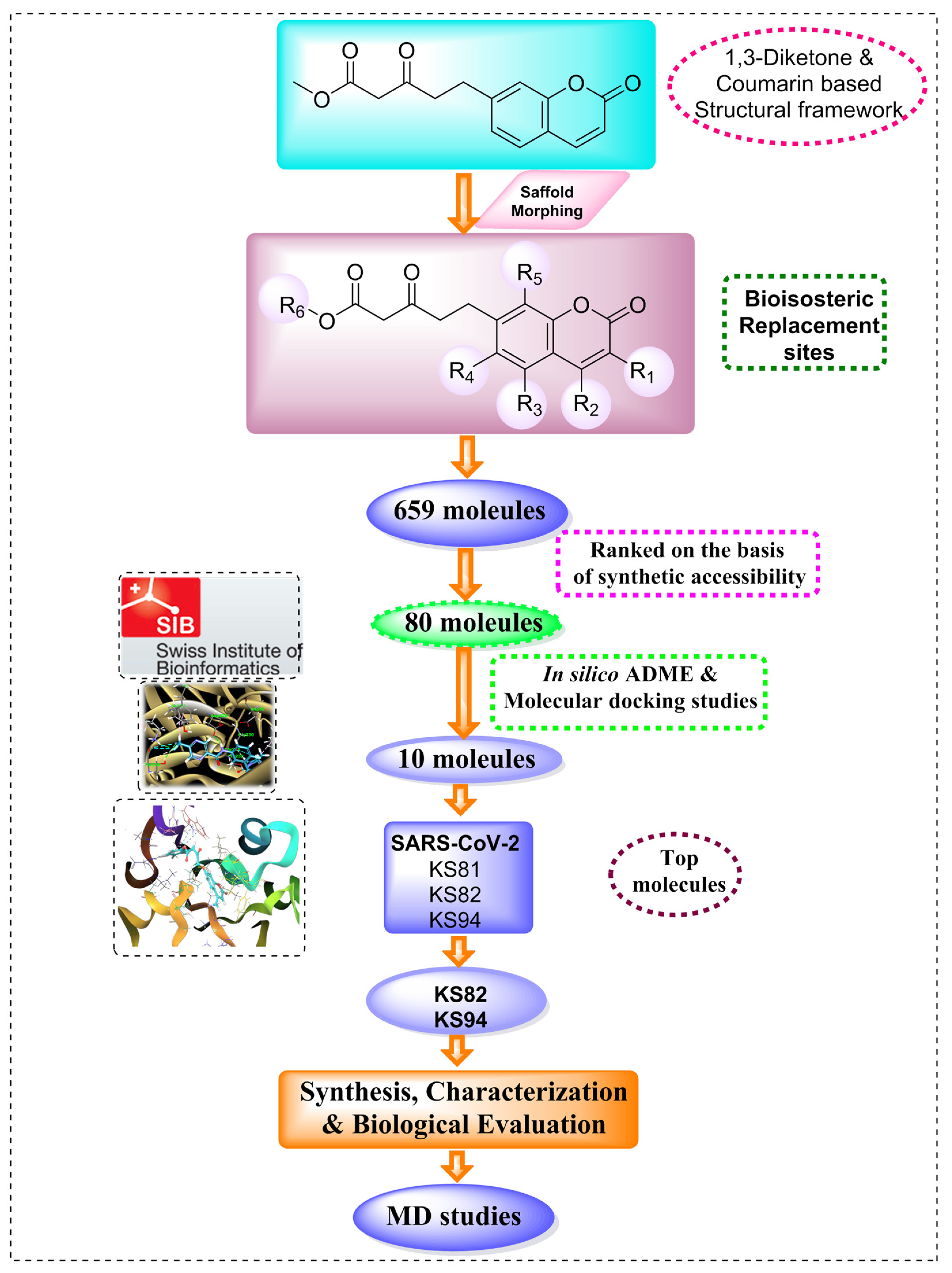
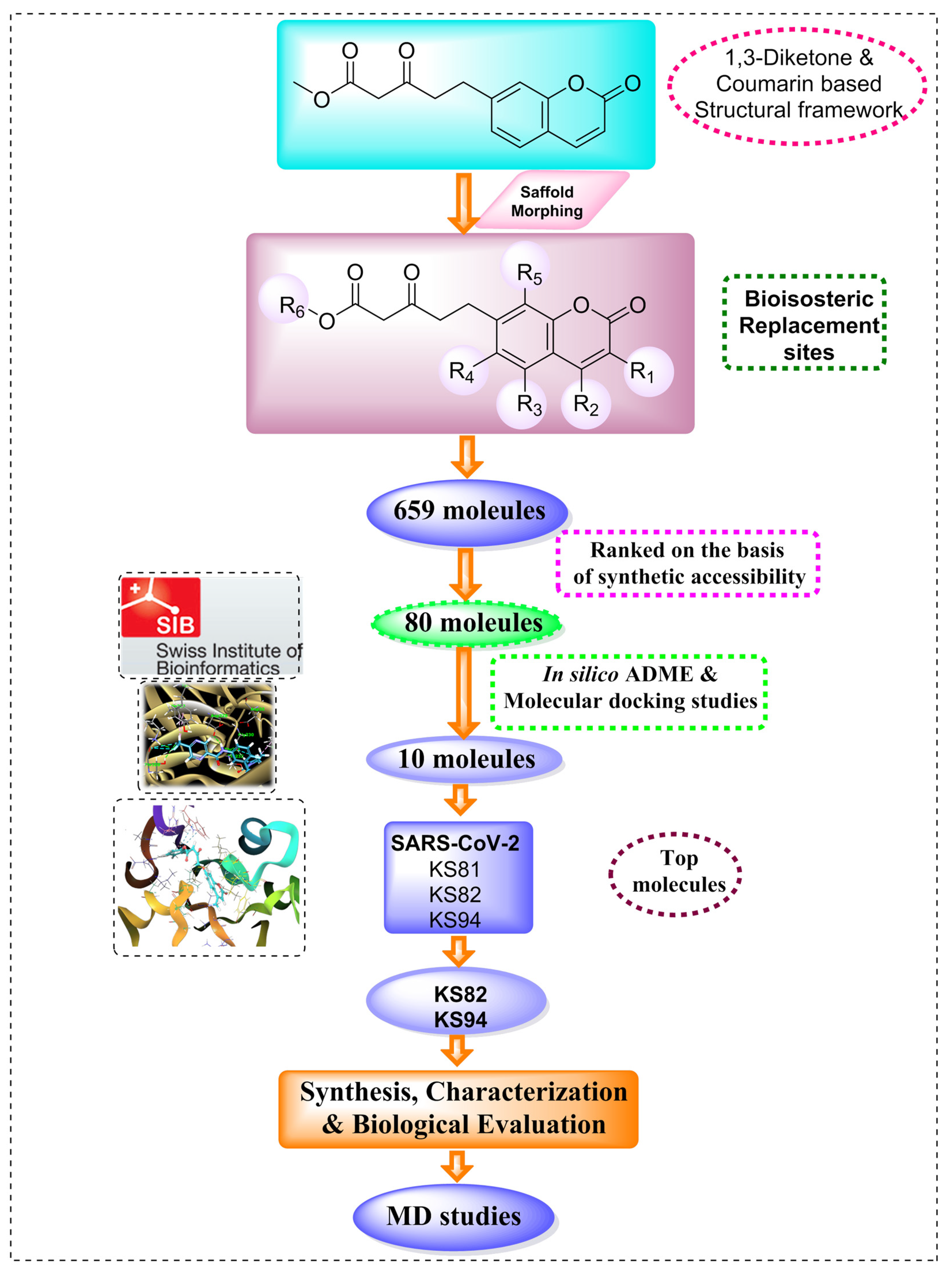
2.1. Scaffold Morphing
2.2. Pharmacokinetic Predictions
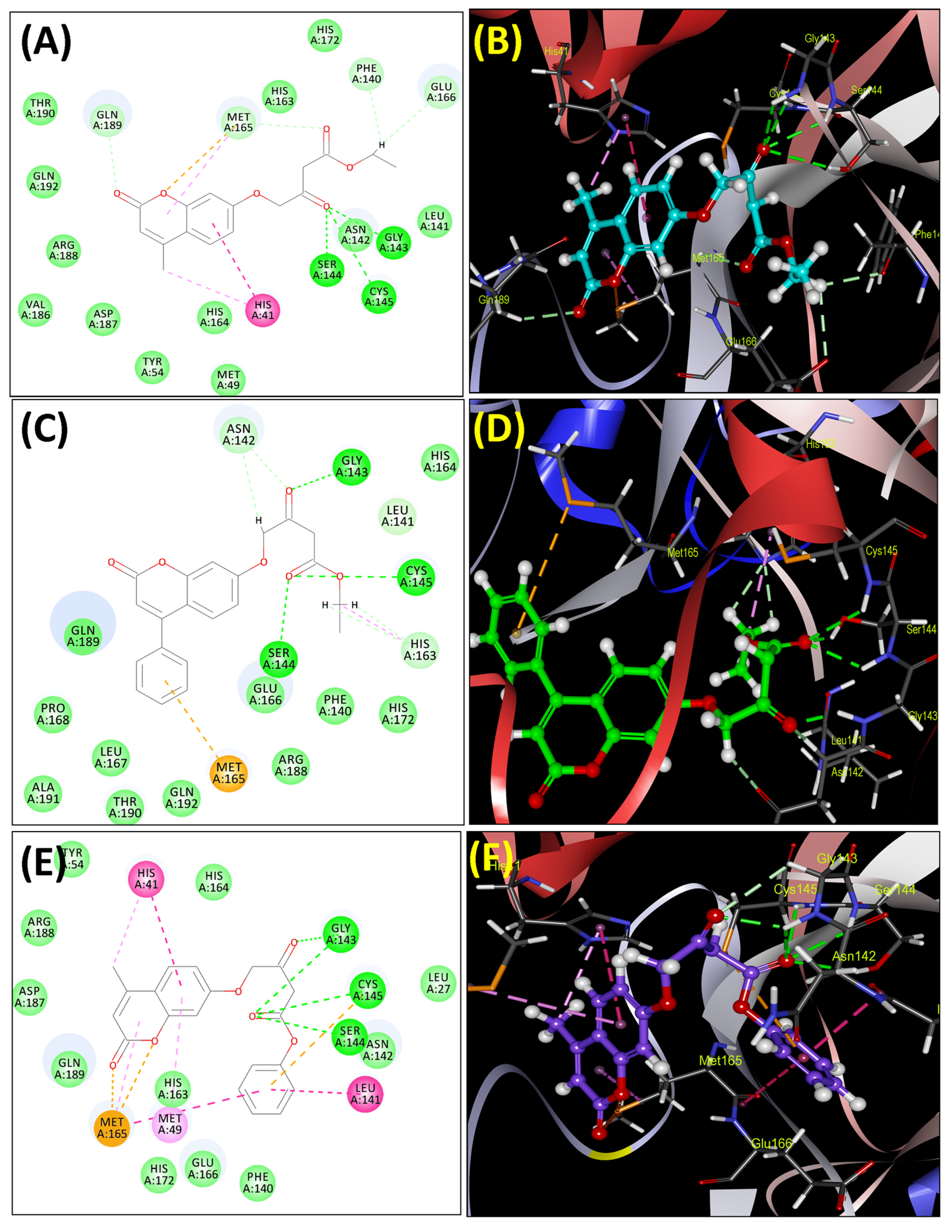
2.3. Molecular Docking Analysis
Molecular Docking Analysis with Mpro
2.4. Chemistry
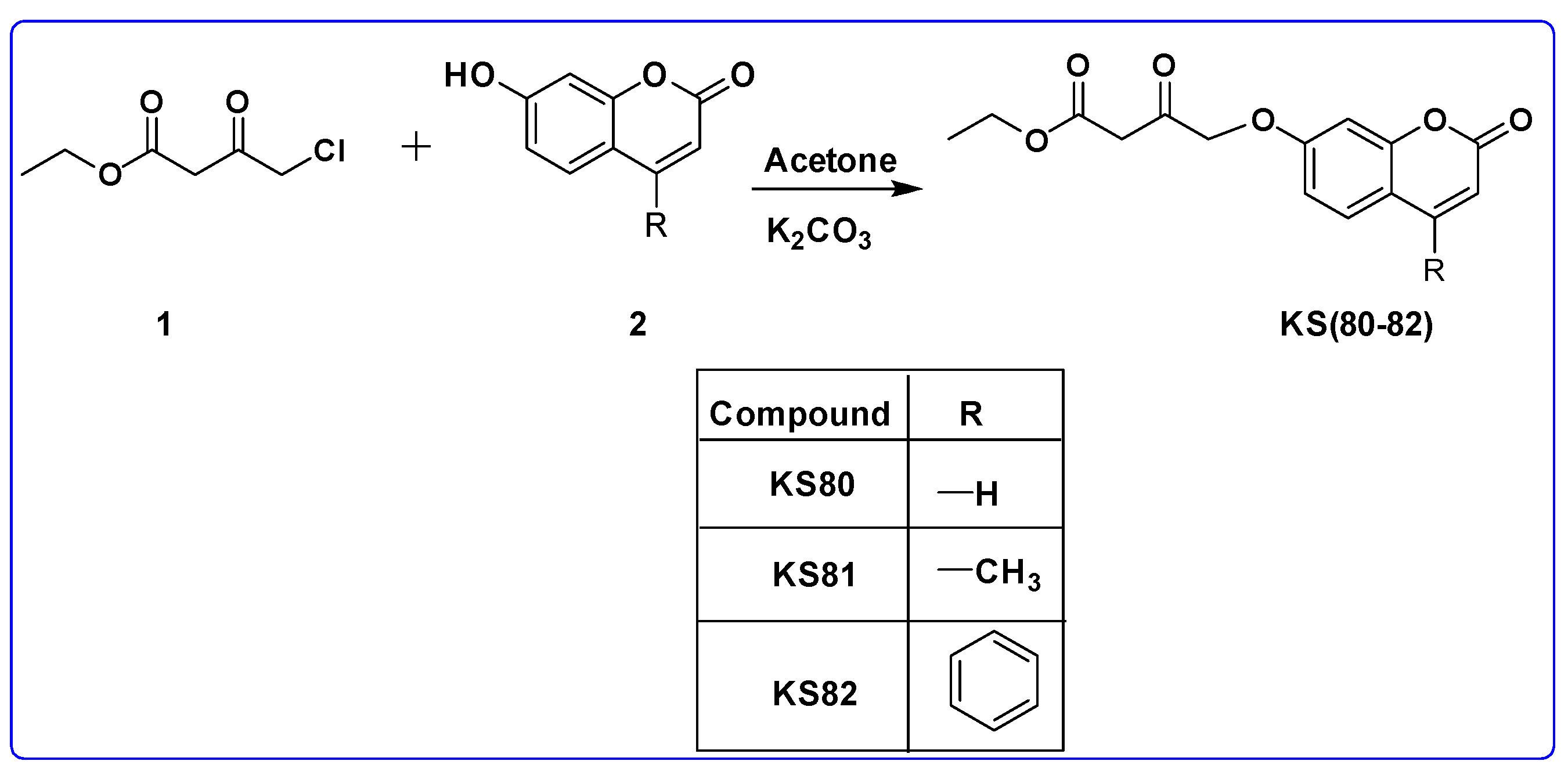
2.4.1. General Procedure for the Synthesis of KS(80–82)
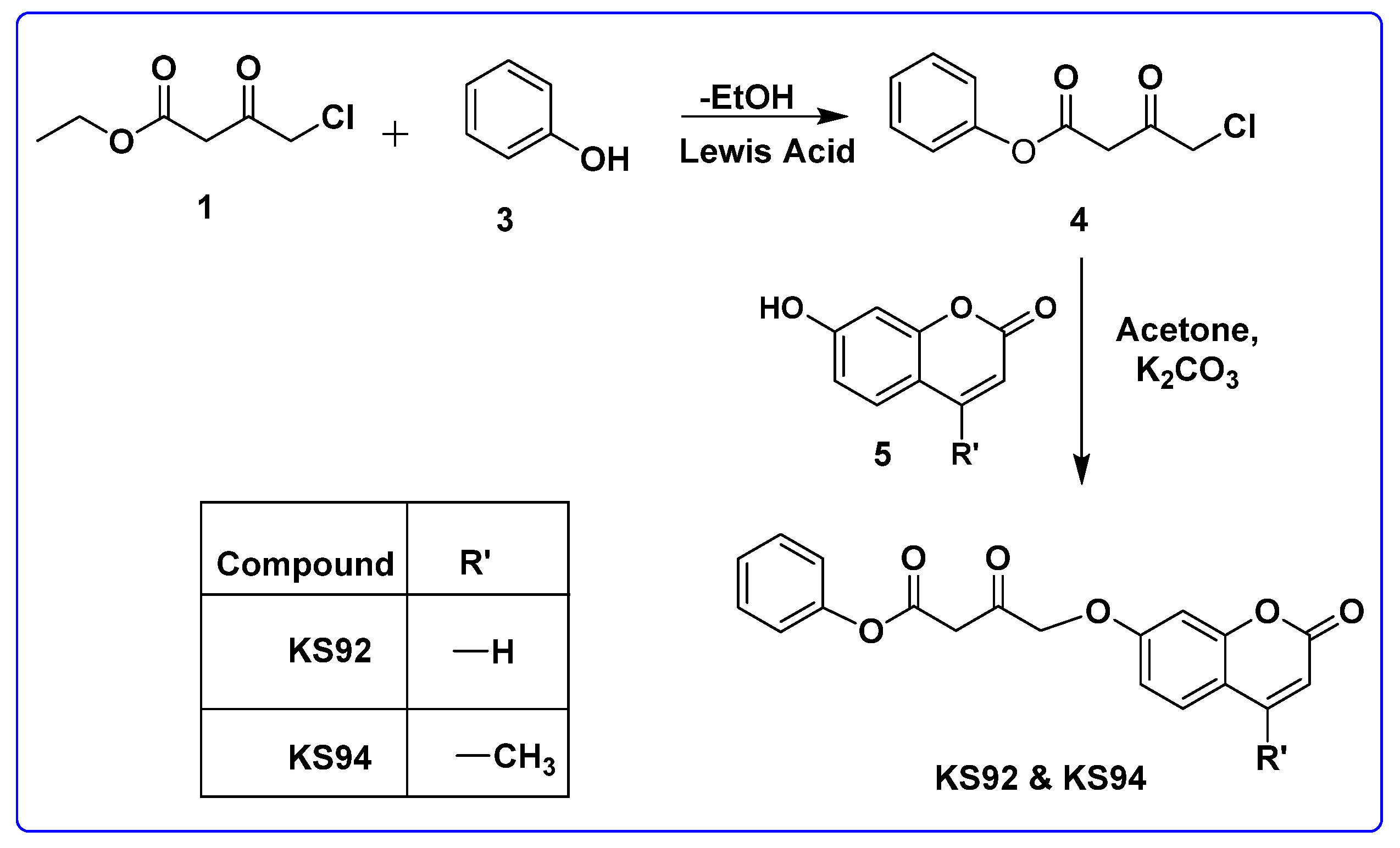
2.4.2. General Procedure for the Synthesis of KS92 and KS94
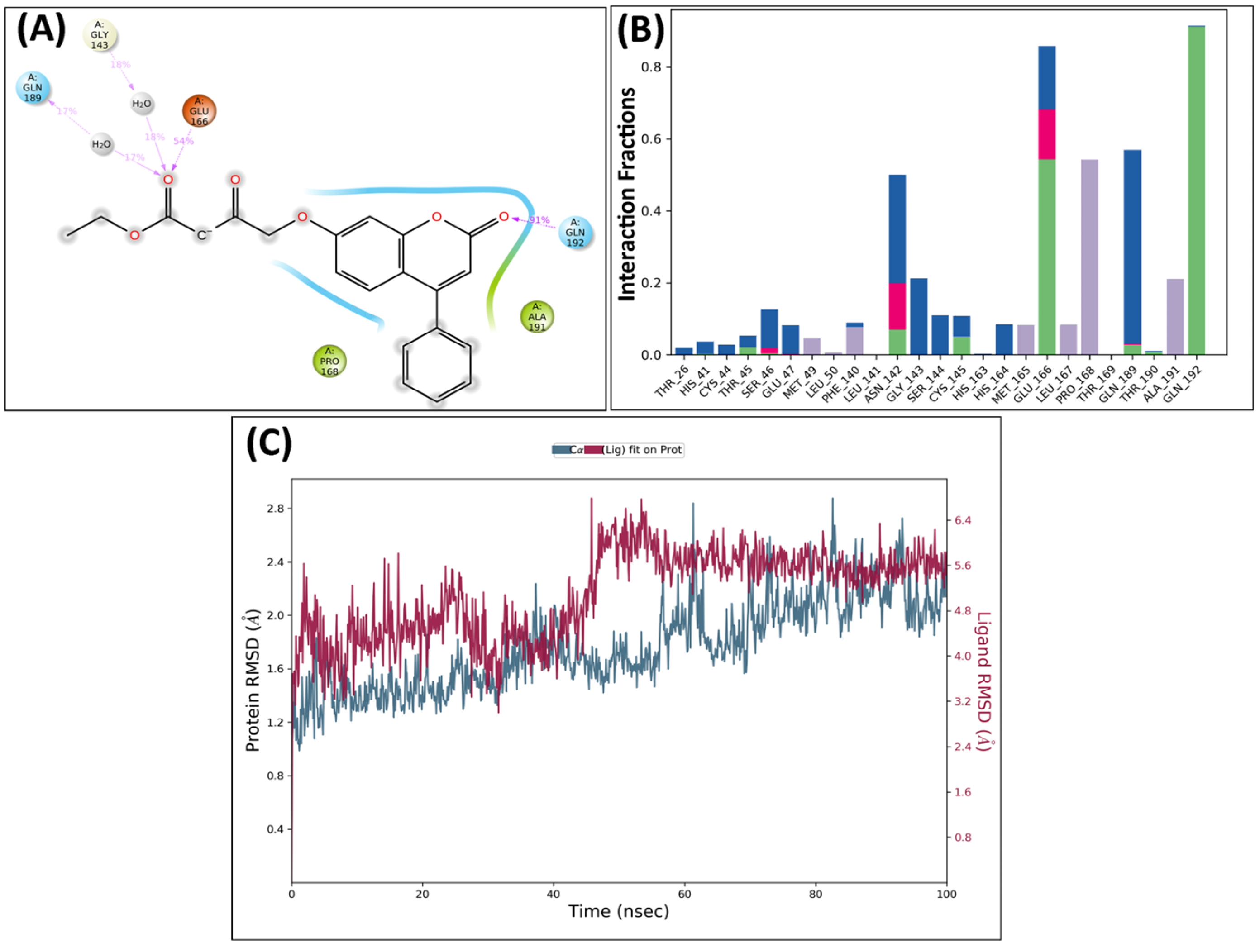
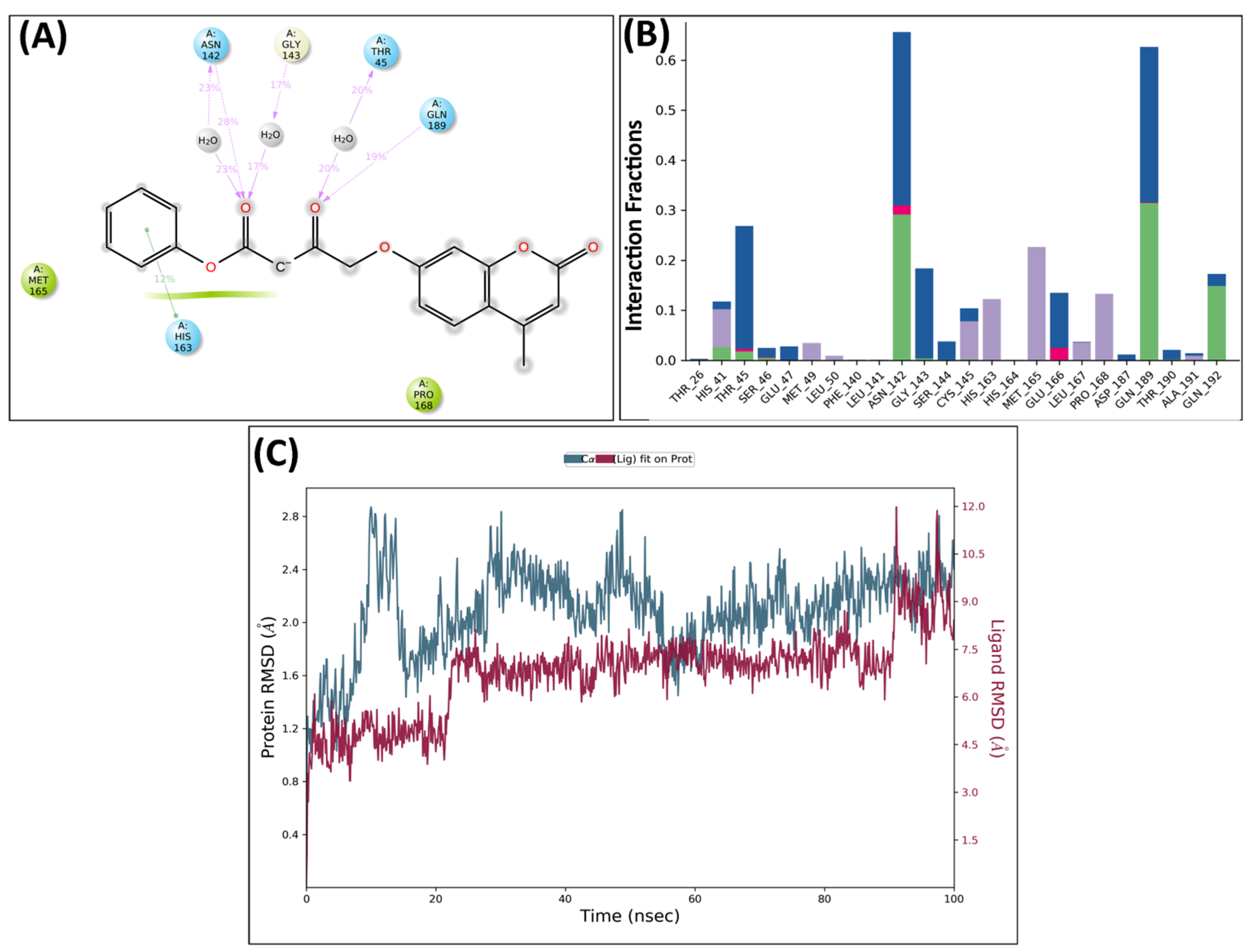
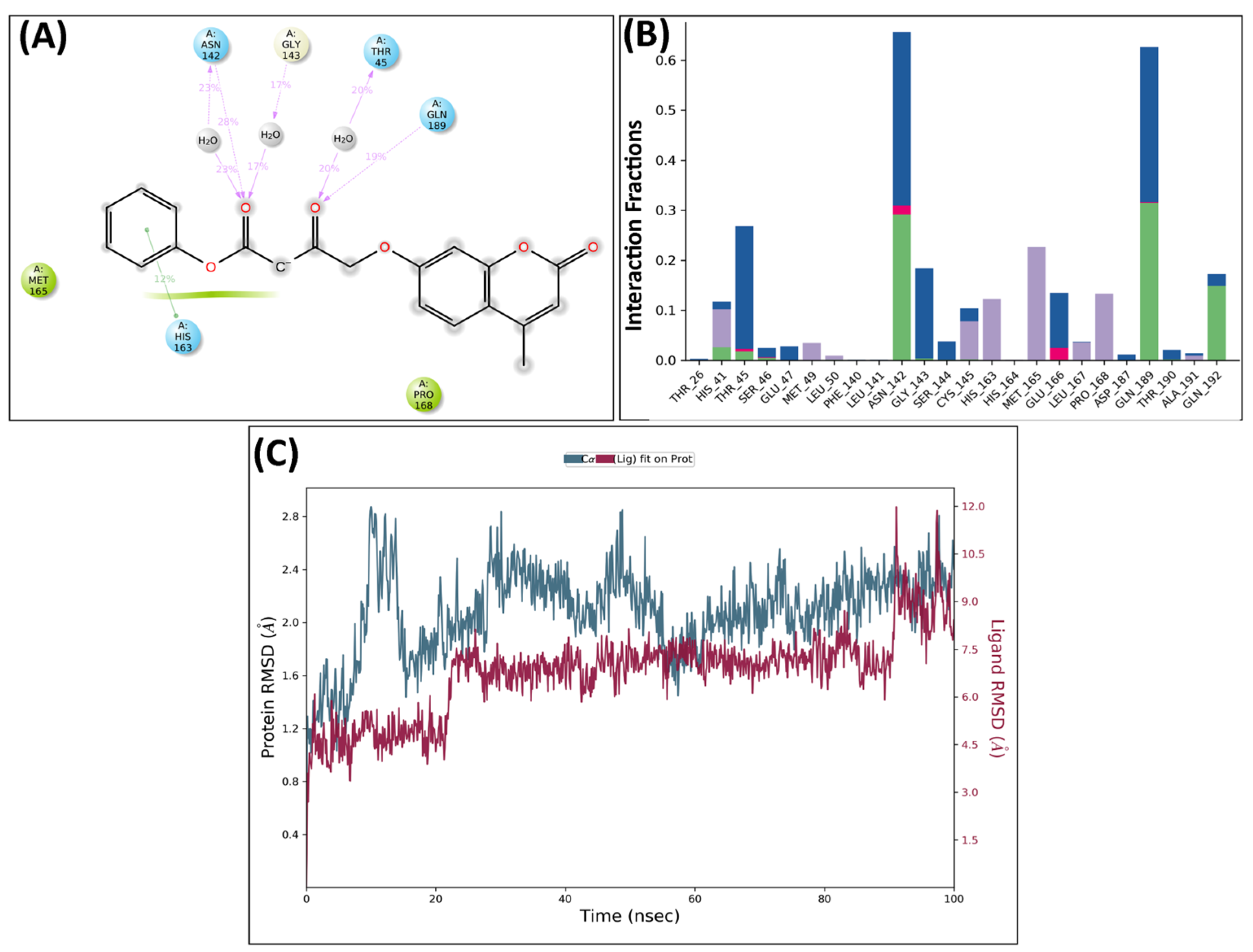
2.5. Molecular Dynamics Analysis
MD Analysis of KS82 and KS94 Complexed with Mpro
2.6. Anti-SARS-CoV2 Activity
2.6.1. Cell Viability Study
2.6.2. Anti-SARS-CoV2 Activity
3. Materials and Methods
3.1. Scaffold Morphing
3.2. ADME Prediction
3.3. Molecular Docking
3.4. Chemistry
3.5. Molecular Dynamics Studies
3.6. Toxicity Testing in the Cell Culture
3.7. Anti-SARS-CoV2 Activity
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Kang, C. Progress in developing inhibitors of SARS-CoV-2 3C-like protease. Microorganisms 2020, 8, 1250. [Google Scholar] [CrossRef] [PubMed]
- Taherizadeh, M.; Tabibzadeh, A.; Panahi, M.; Tameshkel, F.S.; Golahdooz, M.; Niya, M.H.K. An Introduction to SARS Coronavirus 2; Comparative Analysis with MERS and SARS Coronaviruses: A Brief Review. Iran. J. Public Health 2020, 49 (Suppl. 1), 30–37. [Google Scholar] [CrossRef]
- Sharun, K.; Tiwari, R.; Patel, S.K.; Karthik, K.; Yatoo, M.I.; Malik, Y.S.; Singh, K.P.; Panwar, P.K.; Harapan, H.; Singh, R.K.; et al. Coronavirus disease 2019 (COVID-19) in domestic animals and wildlife: Advances and prospects in the development of animal models for vaccine and therapeutic research. Hum. Vaccines Immunother. 2020, 16, 3043–3054. [Google Scholar] [CrossRef] [PubMed]
- Ye, Z.W.; Yuan, S.; Yuen, K.S.; Fung, S.Y.; Chan, C.P.; Jin, D.Y. Zoonotic origins of human coronaviruses. Int. J. Biol. Sci. 2020, 16, 1686. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Grunewald, M.; Perlman, S. Coronaviruses: An updated overview of their replication and pathogenesis. In Coronaviruses. Methods in Molecular Biology; Humana: New York, NY, USA, 2020; pp. 1–29. [Google Scholar] [CrossRef]
- Abu-Hashem, A.A.; Al-Hussain, S.A. The Synthesis, Antimicrobial Activity, and Molecular Docking of New 1, 2, 4-Triazole, 1, 2, 4-Triazepine, Quinoline, and Pyrimidine Scaffolds Condensed to Naturally Occurring Furochromones. Pharmaceuticals 2022, 15, 1232. [Google Scholar] [CrossRef] [PubMed]
- Jin, Z.; Du, X.; Xu, Y.; Deng, Y.; Liu, M.; Zhao, Y.; Zhang, B.; Li, X.; Zhang, L.; Peng, C.; et al. Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature 2020, 582, 289–293. [Google Scholar] [CrossRef] [PubMed]
- Ullrich, S.; Nitsche, C. The SARS-CoV-2 main protease as drug target. Bioorganic Med. Chem. Lett. 2020, 30, 127377. [Google Scholar] [CrossRef]
- Hu, Q.; Xiong, Y.; Zhu, G.H.; Zhang, Y.N.; Zhang, Y.W.; Huang, P.; Ge, G.B. The SARS-CoV-2 main protease (Mpro): Structure, function, and emerging therapies for COVID-19. MedComm 2022, 3, e151. [Google Scholar] [CrossRef]
- Abu-Hashem, A.A.; A Al-Hussain, S.; Zaki, M.E.A. Synthesis of novel benzodifuranyl; 1,3,5-triazines; 1,3,5-oxadiazepines; and thiazolopyrimidines derived from visnaginone and khellinone as anti-inflammatory and analgesic agents. Molecules 2020, 25, 220. [Google Scholar] [CrossRef]
- Shabir, G.; Shafique, I.; Saeed, A. Ultrasound assisted synthesis of 5–7 membered heterocyclic rings in organic molecules. J. Heterocycl. Chem. 2022, 59, 1669–1702. [Google Scholar] [CrossRef]
- Pivarcsik, T.; Tóth, G.; Szemerédi, N.; Bogdanov, A.; Spengler, G.; Kljun, J.; Kladnik, J.; Turel, I.; Enyedy, É.A. Comparison of solution chemical properties and biological activity of ruthenium complexes of selected β-diketone, 8-hydroxyquinoline and pyrithione ligands. Pharmaceuticals 2021, 14, 518. [Google Scholar] [CrossRef]
- Kaur, N.; Grewal, P.; Poonia, K. Dicarbonyl compounds in O-heterocycle synthesis. Synth. Commun. 2021, 51, 2423–2444. [Google Scholar] [CrossRef]
- Talbi, S.; Dib, M.; Bouissane, L.; Abderrafia, H.; Rabi, S.; Khouili, M. Recent Progress in the Synthesis of Heterocycles based on 1,3-diketones. Curr. Org. Synth. 2022, 19, 220–245. [Google Scholar] [CrossRef] [PubMed]
- Seifi, M.; Sheibani, H. Studies on condensation of 1,3-dicarbonyls with malononitrile: Synthesis of 2-pyridinones. Arab. J. Chem. 2017, 10, S2453–S2456. [Google Scholar] [CrossRef]
- de Gonzalo, G.; Alcántara, A.R. Recent Developments in the Synthesis of β-Diketones. Pharmaceuticals 2021, 14, 1043. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 1997, 23, 3–25. [Google Scholar] [CrossRef]
- Ferreira, J.C.; Fadl, S.; Villanueva, A.J.; Rabeh, W.M. Catalytic dyad residues His41 and Cys145 impact the catalytic activity and overall conformational fold of the main SARS-CoV-2 protease 3-chymotrypsin-like protease. Front. Chem. 2021, 9, 692168. [Google Scholar] [CrossRef]
- Khalifa, I.; Zhu, W.; Mohammed, H.H.H.; Dutta, K.; Li, C. Tannins inhibit SARS-CoV-2 through binding with cat-alytic dyad residues of 3CLpro: An in silico approach with 19 structural different hydrolysable tannins. J. Food Bio-Chem. 2020, 44, e13432. [Google Scholar]
- Mahajan, P.; Kaushal, J. Epidemic Trend of COVID-19 transmission in India during lockdown-1 phase. J. Community Health 2020, 45, 1291–1300. [Google Scholar] [CrossRef]
- Nagu, P.; Parashar, A.; Behl, T.; Mehta, V. CNS implications of COVID-19: A comprehensive review. Prog. Neurobiol. 2020, 32, 219–234. [Google Scholar] [CrossRef]
- Singh, M.; Nagpal, M.; Singh, V.; Sharma, A.; Dhingra, G.A.; Maman, P.; Puri, V. COVID-19: Epidemiology, patho-genicity and global updates. Int. J. Appl. Pharm. 2020, 12, 16–28. [Google Scholar] [CrossRef]
- Kaur, M.; Devi, G.; Nagpal, M.; Singh, M.; Dhingra, G.A.; Aggarwal, G. Antiviral Essential oils incorporated in nanocarriers: Strategy for prevention from COVID-19 and future infectious pandemics. Pharm. Nanotechnol. 2020, 8, 437–451. [Google Scholar] [CrossRef] [PubMed]
- Choudhary, S.; Silakari, O. Scaffold morphing of arbidol (umifenovir) in search of multi-targeting therapy halting the interaction of SARS-CoV-2 with ACE2 and other proteases involved in COVID-19. Virus Res. 2020, 289, 198146. [Google Scholar] [CrossRef]
- Vincetti, P.; Kaptein, S.J.F.; Costantino, G.; Neyts, J.; Radi, M. Scaffold morphing approach to expand the toolbox of broad-spectrum antivirals blocking dengue/Zika replication. ACS Med. Chem. Lett. 2019, 10, 558–563. [Google Scholar] [CrossRef] [PubMed]
- Shan, J.; Ji, C. MolOpt: A web server for drug design using bioisosteric transformation. Curr. Comput. Aided-Drug Des. 2020, 16, 460–466. [Google Scholar] [CrossRef] [PubMed]
- Doogue, M.P.; Polasek, T.M. The ABCD of clinical pharmacokinetics. Ther. Adv. Drug Saf. 2013, 4, 5–7. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Zhang, J.; Hu, C.Q.; Zhang, X.; Ma, B.; Zhang, P. In silico ADME and toxicity prediction of ceftazidime and its impurities. Front. Pharmacol. 2019, 10, 434. [Google Scholar] [CrossRef] [PubMed]
- Singh, M.; Kaur, M.; Singh, N.; Silakari, O. Exploration of multi-target potential of chromen-4-one based compounds in Alzheimer’s disease: Design, synthesis and biological evaluations. Bioorganic Med. Chem. 2017, 25, 6273–6285. [Google Scholar] [CrossRef]
- Kryger, G.; Silman, I.; Sussman, J.L. Structure of acetylcholinesterase complexed with E2020 (Aricept®): Implications for the design of new anti-Alzheimer drugs. Structure 1999, 7, 297–307. [Google Scholar] [CrossRef]
- Macalino, S.J.Y.; Gosu, V.; Hong, S.; Choi, S. Role of computer-aided drug design in modern drug discovery. Arch. Pharmacal Res. 2015, 38, 1686–1701. [Google Scholar] [CrossRef] [PubMed]
- Garg, S.; Anand, A.; Lamba, Y.; Roy, A. Molecular docking analysis of selected phytochemicals against SARS-CoV-2 M pro receptor. Vegetos 2020, 33, 766–781. [Google Scholar] [CrossRef]
- Wu, G.; Robertson, D.H.; Brooks, C.L., III; Vieth, M. Detailed analysis of grid-based molecular docking: A case study of CDOCKER—A CHARMm-based MD docking algorithm. J. Comput. Chem. 2003, 24, 1549–1562. [Google Scholar] [CrossRef]
- Shafiq, M.; Sherwani, Z.A.; Mushtaq, M.; Nur-E-Alam, M.; Ahmad, A.; Ul-Haq, Z. A deep learning-based theoretical protocol to identify potentially isoform-selective PI3Kα inhibitors. Mol. Divers. 2024, 1–18. [Google Scholar] [CrossRef]
- Kumar, D.; Kumari, K.; Jayaraj, A.; Singh, P. Development of a theoretical model for the inhibition of nsP3 of Chikungunya virus using pyranooxazoles. J. Biomol. Struct. Dyn. 2020, 38, 3018–3034. [Google Scholar] [CrossRef] [PubMed]
- Bowers, K.J.; Chow, E.; Xu, H.; Dror, R.O.; Eastwood, M.P.; Gregersen, B.A.; Klepeis, J.L.; Kolossvary, I.; Moraes, M.A.; Sac-erdoti, F.D.; et al. Scalable algorithms for molecular dynamics simulations on commodity clusters. In Proceedings of the 2006 ACM/IEEE Conference on Supercomputing, New York, NY, USA, 11 November 2006; p. 84. [Google Scholar]
- Li, Q.; Zhang, H.; Guan, S.; Du, J.; Zhang, Y.; Wang, S. Molecular dynamics simulation of the inhibition mechanism of factor XIa by Milvexian-like macrocyclic inhibitors. Comput. Theor. Chem. 2023, 1225, 114131. [Google Scholar] [CrossRef]
- Kumar, V.; Parate, S.; Danishuddin; Zeb, A.; Singh, P.; Lee, G.; Jung, T.S.; Lee, K.W.; Ha, M.W. 3D-QSAR-based pharmacophore modeling, virtual screening, and molecular dynamics simulations for the identification of spleen tyrosine kinase inhibitors. Front. Cell. Infect. Microbiol. 2022, 12, 909111. [Google Scholar] [CrossRef]
- Caly, L.; Druce, J.D.; Catton, M.G.; Jans, D.A.; Wagstaff, K.M. The FDA-approved drug ivermectin inhibits the replication of SARS-CoV-2 in vitro. Antivir. Res. 2020, 178, 104787. [Google Scholar] [CrossRef]
- Prajapat, S.K.; Mishra, L.; Khera, S.; Owusu, S.D.; Ahuja, K.; Sharma, P.; Choudhary, E.; Chhabra, S.; Kumar, N.; Singh, R.; et al. Methotrimeprazine is a neuroprotective antiviral in JEV infection via adaptive ER stress and autophagy. EMBO Mol. Med. 2024, 16, 185–217. [Google Scholar] [CrossRef]
- Kumar, S.; Singh, B.; Kumari, P.; Kumar, P.V.; Agnihotri, G.; Khan, S.; Beuria, T.K.; Syed, G.H.; Dixit, A. Identifi-cation of multipotent drugs for COVID-19 therapeutics with the evaluation of their SARS-CoV2 inhibitory activity. Comput. Struct. Biotechnol. J. 2021, 19, 1998–2017. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| S. No. | Molecular Structure | Cdocker Energy | Interactions Observed with the Binding Site of Mpro |
|---|---|---|---|
| 1 | 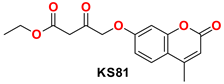 | −53.57 | Ser144, Gly143, Cys145, His41, Glu166, Leu141, Met49, Met165, and Phe140 |
| 2 | 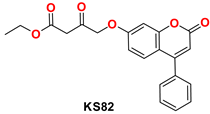 | −50.49 | Ser144, Pro168, Met49, Arg188, Gly143, His164, Cys145, and Glu166 |
| 3 | 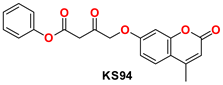 | −50.02 | Ser144, Cys145, Gly143, Glu166, Leu141, Met49, Arg188, Glu166, Phe140, and His41 |
| 4 | 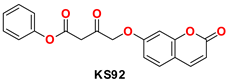 | −48.53 | Ser144, Cys145, Met49, His164, His41, Gly143, Glu166, Arg188, and Leu141 |
| 5 | 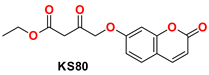 | −48.16 | Ser144, Gly143, Cys145, Tyr54, Met49, His41, His172, Arg188, Phe140, Leu141, and His163 |
| 6 | 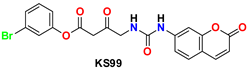 | −47.70 | Glu166, Ser144, Cys145, Gly143, Gln189, His41, Leu27, Pro168, and Met49 |
| 7 | 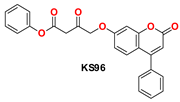 | −47.14 | Ser144, Gly143, Cys145, Met49, Met165, Met165, His41, Leu141, and Glu166 |
| 8 |  | −46.85 | Ser144, Gly143, Cys145, His164, Met49, Met49, Pro168, Leu141, His41, and His163 |
| 9 | 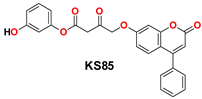 | −46.31 | Gly143, His164, Pro168, Met165, Cys145, Ser144, His41, Gln189, Glu166, and Met49 |
| 10 | 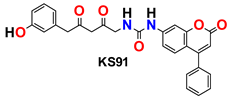 | −45.91 | Ser144, Gly143, Glu166, His164, Phe140, His172, Gln189, Leu141, Met49, Met165, and His41 |
| Compound Name | Concentration | % Cell Viability (30 h) |
|---|---|---|
| Remdesivir | 10 µM | 99.23 |
| KS82 | 1 μL added from the stock solution into 200 μL reaction | 98.34 |
| 1 μL added from 10-times-diluted solution into 200 μL reaction | 99.61 | |
| 1 μL added from 20-times-diluted solution into 200 μL reaction | 107.28 | |
| KS94 | 1 μL added from the stock solution into 200 μL reaction | 112.15 |
| 1 μL added from 10-times-diluted solution into 200 μL reaction | 113.57 | |
| 1 μL added from 20-times-diluted solution into 200 μL reaction | 115.55 |
| Compound Name | Concentration | % Cell Viability | % Inhibition |
|---|---|---|---|
| 30 h | |||
| Remdesivir | 10 µM | 99.23 | 96.85 |
| KS82 | 1 μL added from the stock solution | 98.34 | 76.23 |
| KS94 | 1 μL added from the stock solution | 112.15 | 69.48 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sharma, K.; Singh, M.; Sharma, P.; Sharma, S.C.; Mujwar, S.; Kapoor, M.; Mishra, K.K.; Wani, T.A. Design, Synthesis, and Biological Evaluation of Novel Coumarin Analogs Targeted against SARS-CoV-2. Molecules 2024, 29, 1406. https://doi.org/10.3390/molecules29061406
Sharma K, Singh M, Sharma P, Sharma SC, Mujwar S, Kapoor M, Mishra KK, Wani TA. Design, Synthesis, and Biological Evaluation of Novel Coumarin Analogs Targeted against SARS-CoV-2. Molecules. 2024; 29(6):1406. https://doi.org/10.3390/molecules29061406
Chicago/Turabian StyleSharma, Kirti, Manjinder Singh, Pratibha Sharma, Sumesh C. Sharma, Somdutt Mujwar, Mohit Kapoor, Krishna Kumar Mishra, and Tanveer A. Wani. 2024. "Design, Synthesis, and Biological Evaluation of Novel Coumarin Analogs Targeted against SARS-CoV-2" Molecules 29, no. 6: 1406. https://doi.org/10.3390/molecules29061406