
Organotransition Metal Chemistry of Terpenes: Syntheses, Structures, Reactivity and Molecular Rearrangements


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
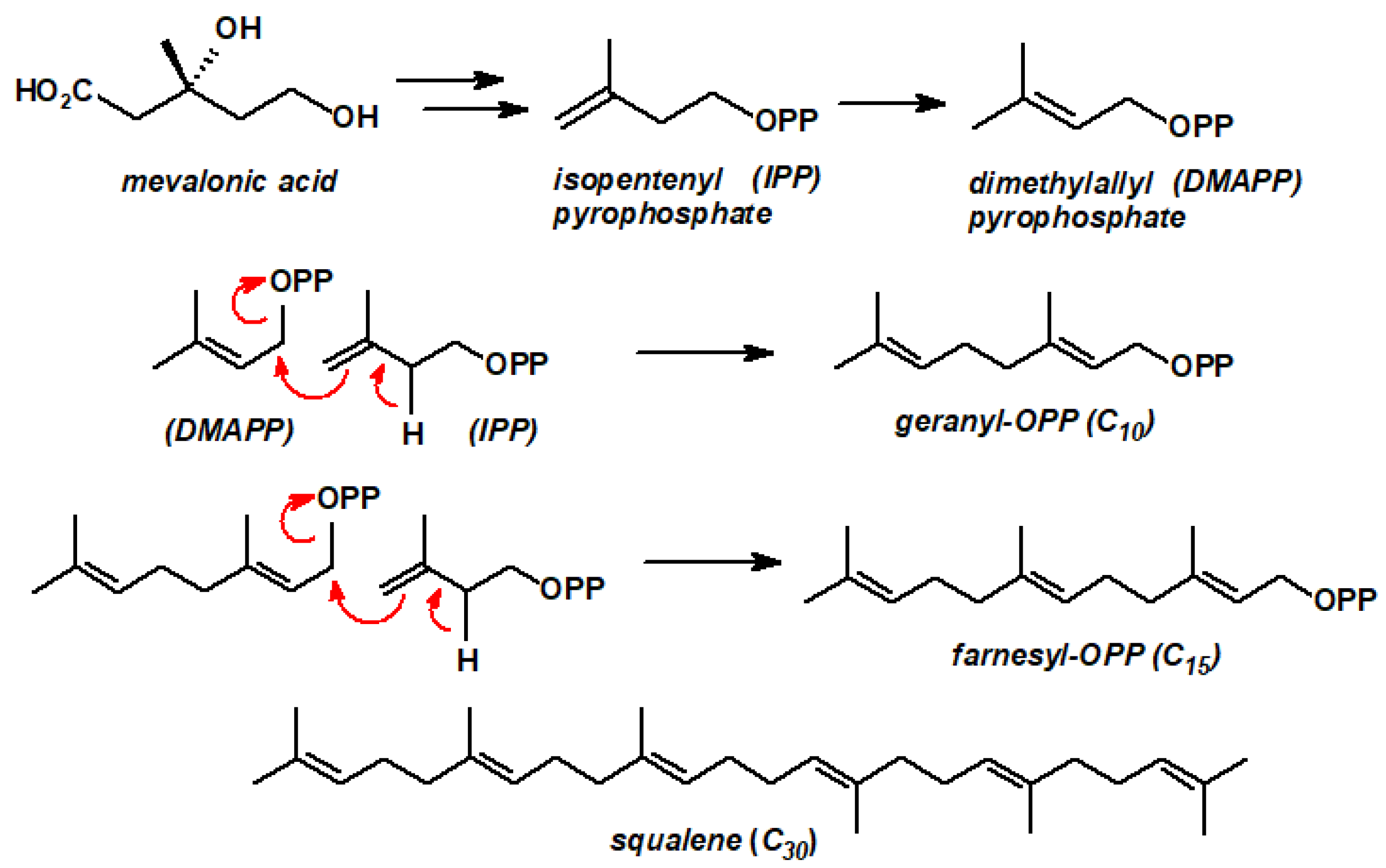{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
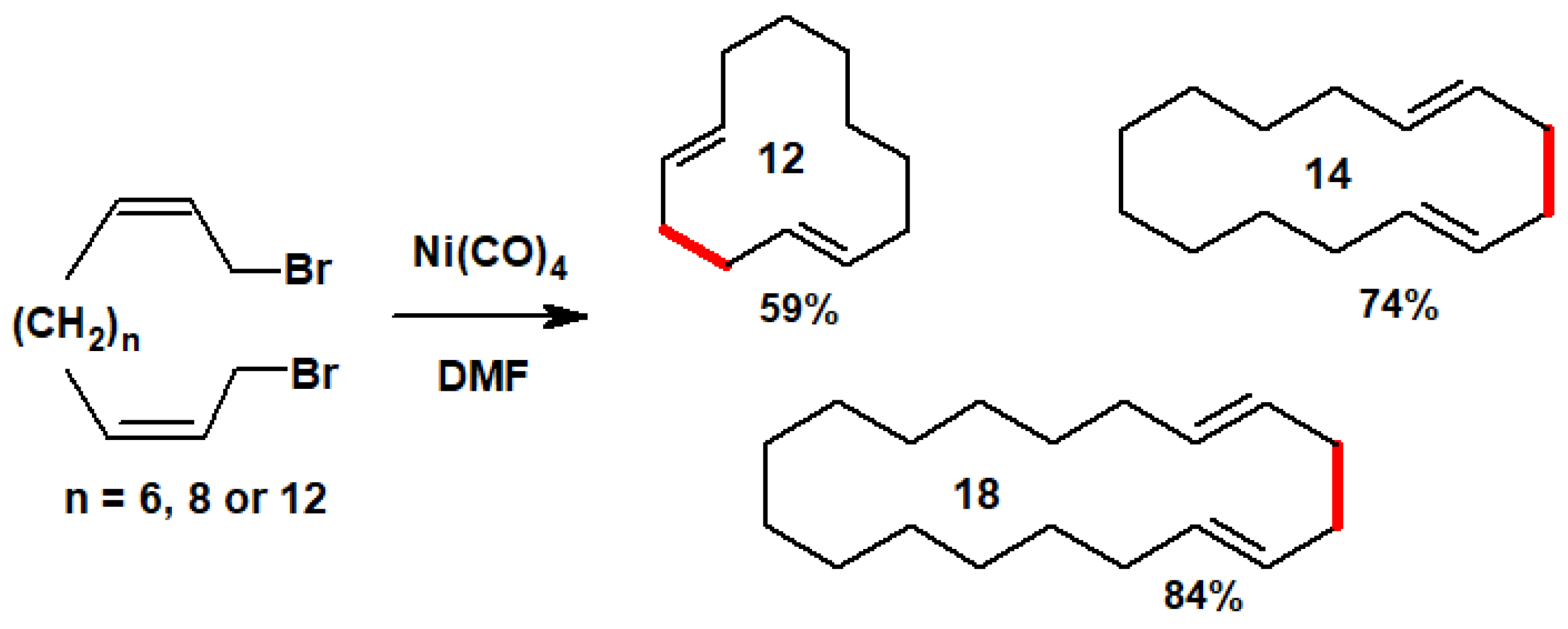{kind=link}
{kind=link}
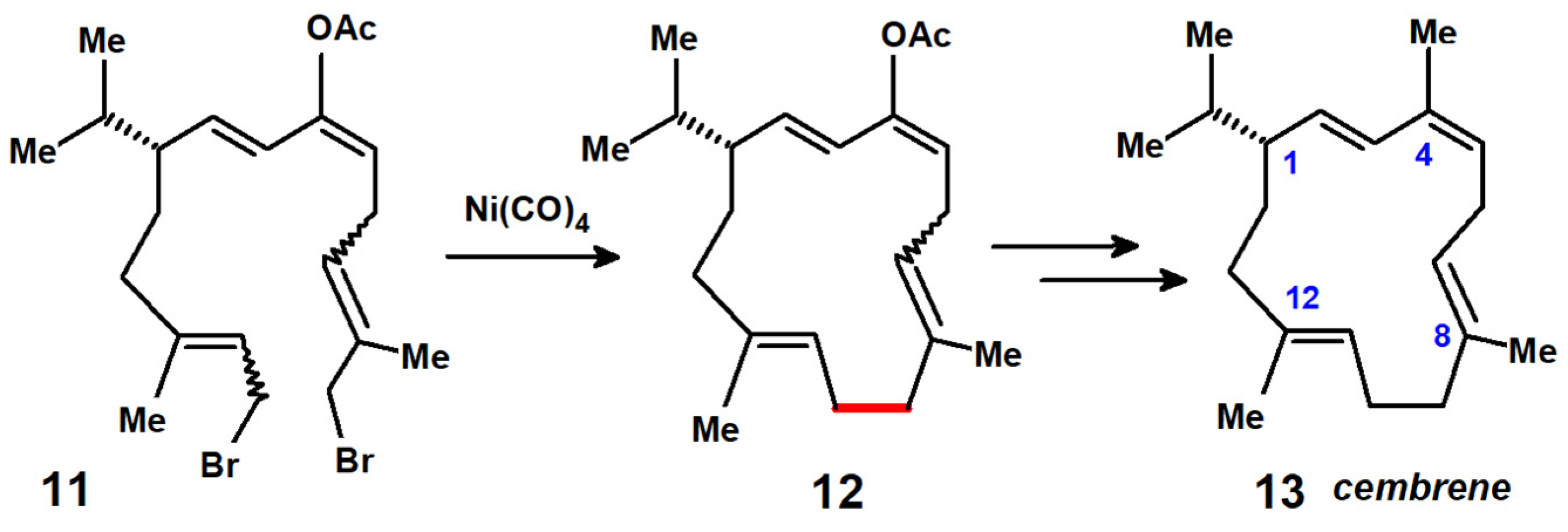{kind=link}
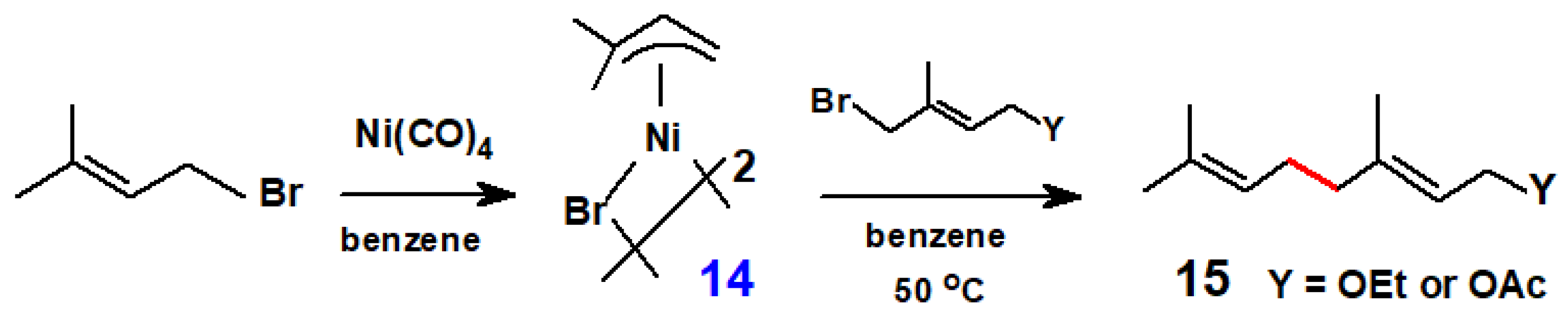{kind=link}
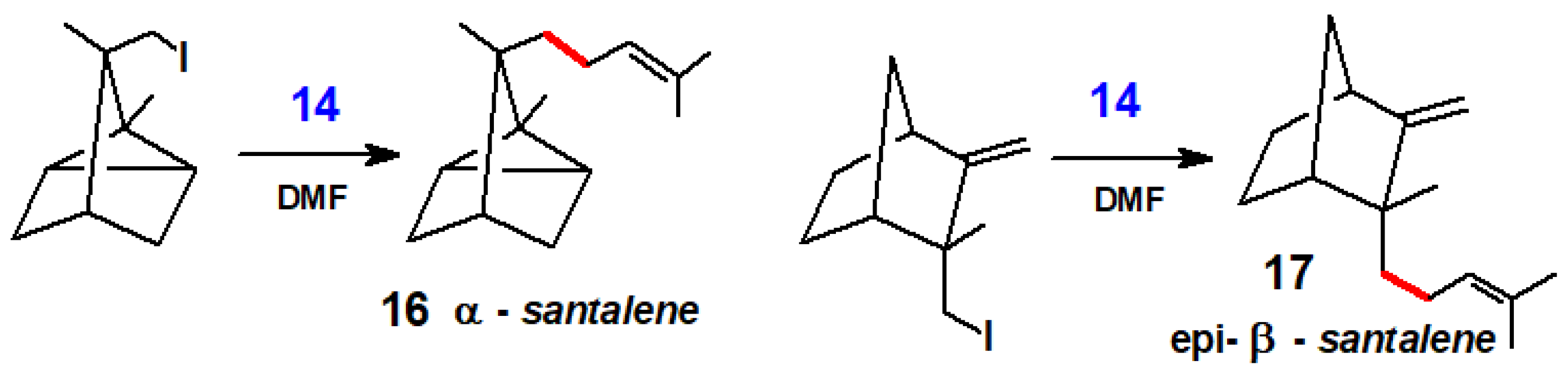{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
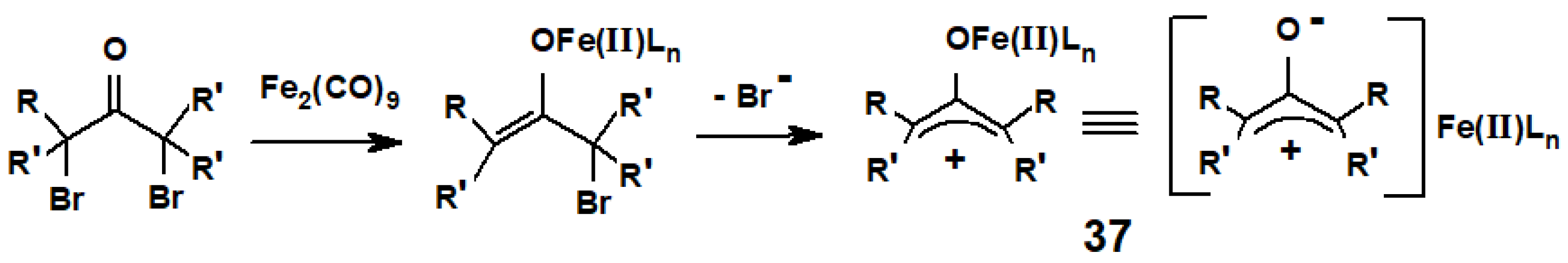{kind=link}
{kind=link}
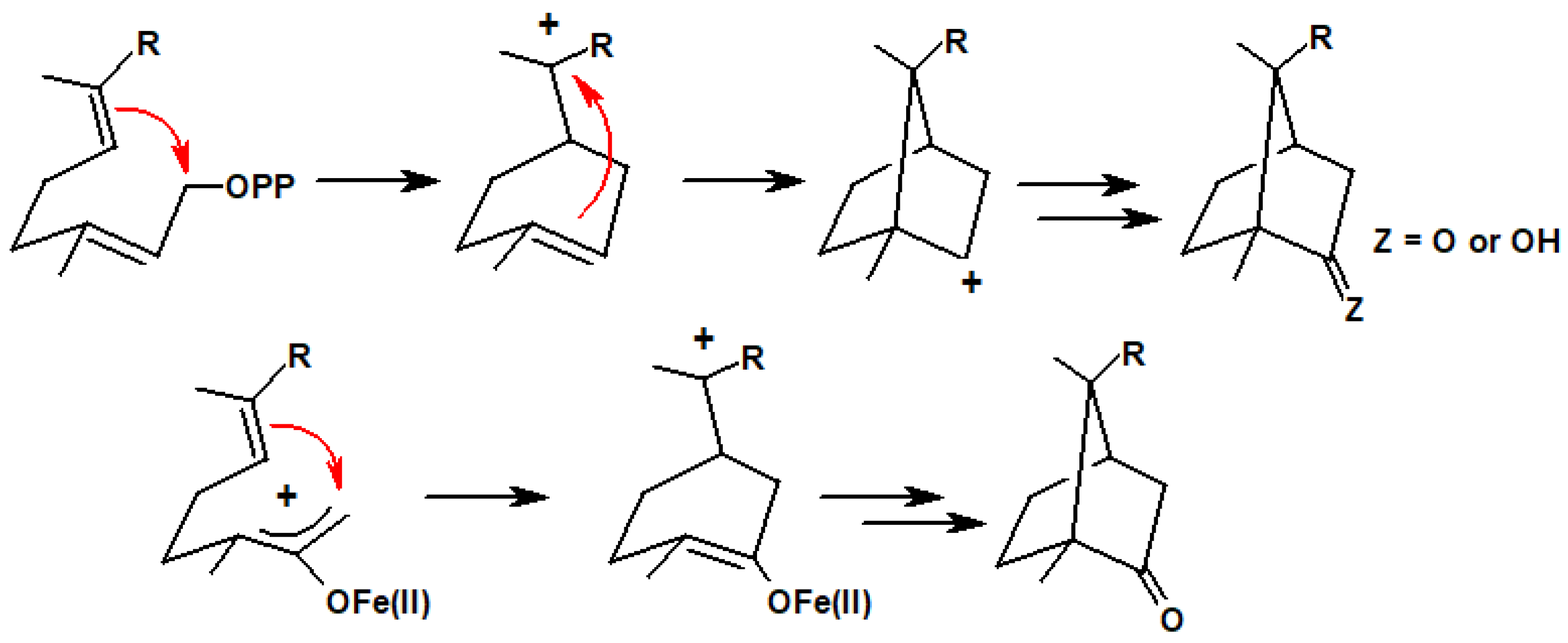{kind=link}
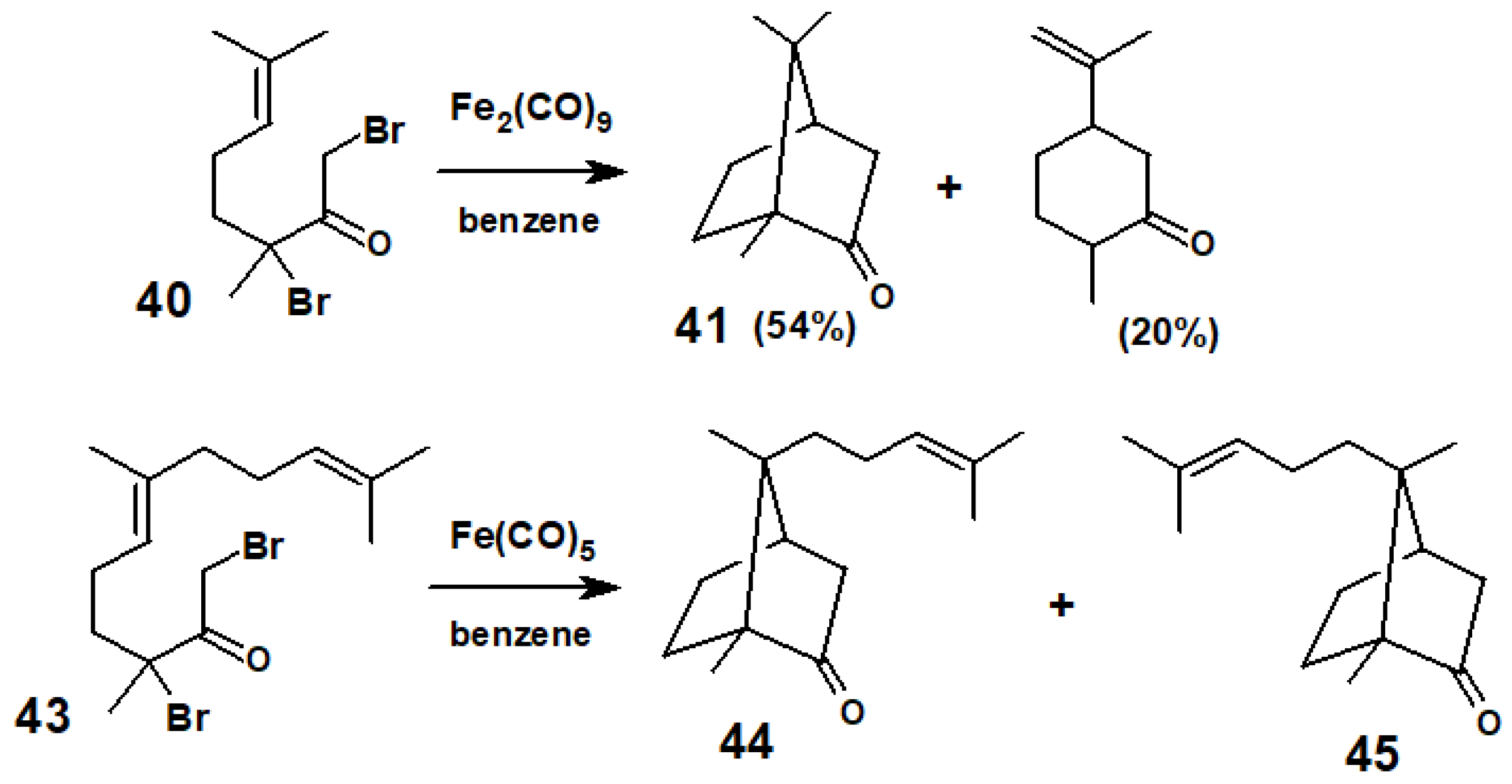{kind=link}
{kind=link}
{kind=link}
{kind=link}
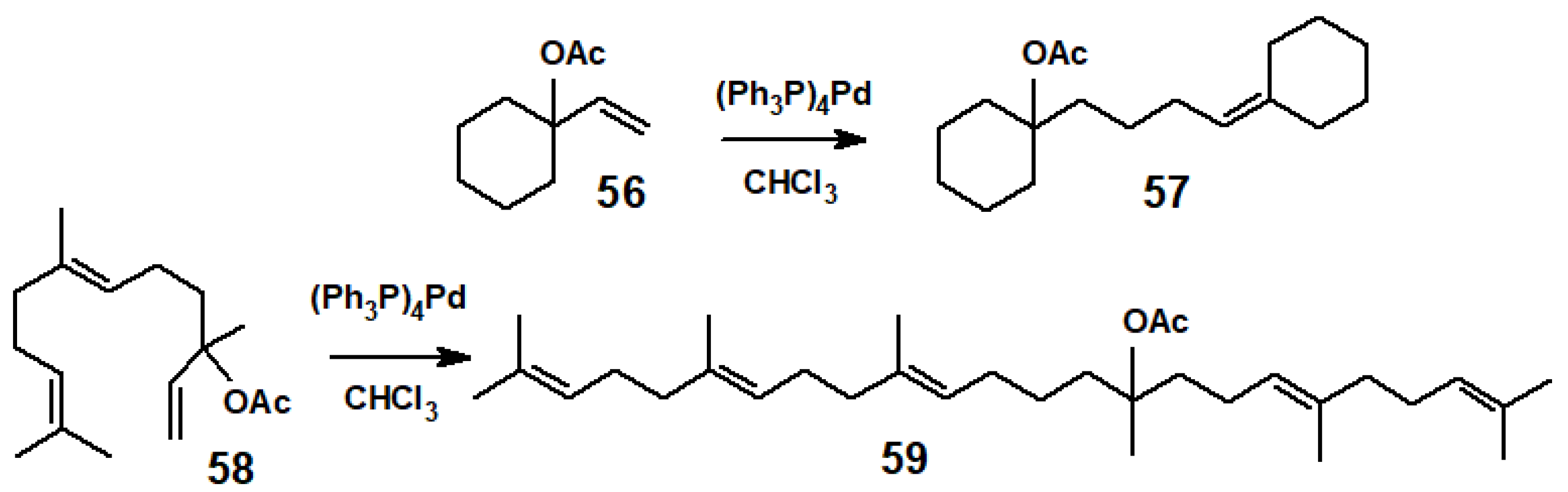{kind=link}
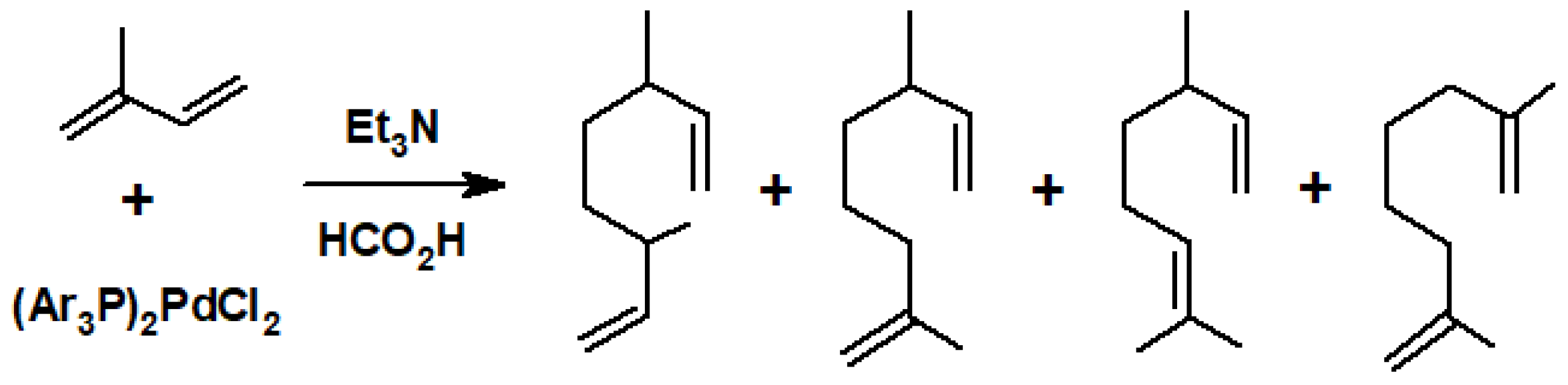{kind=link}
{kind=link}
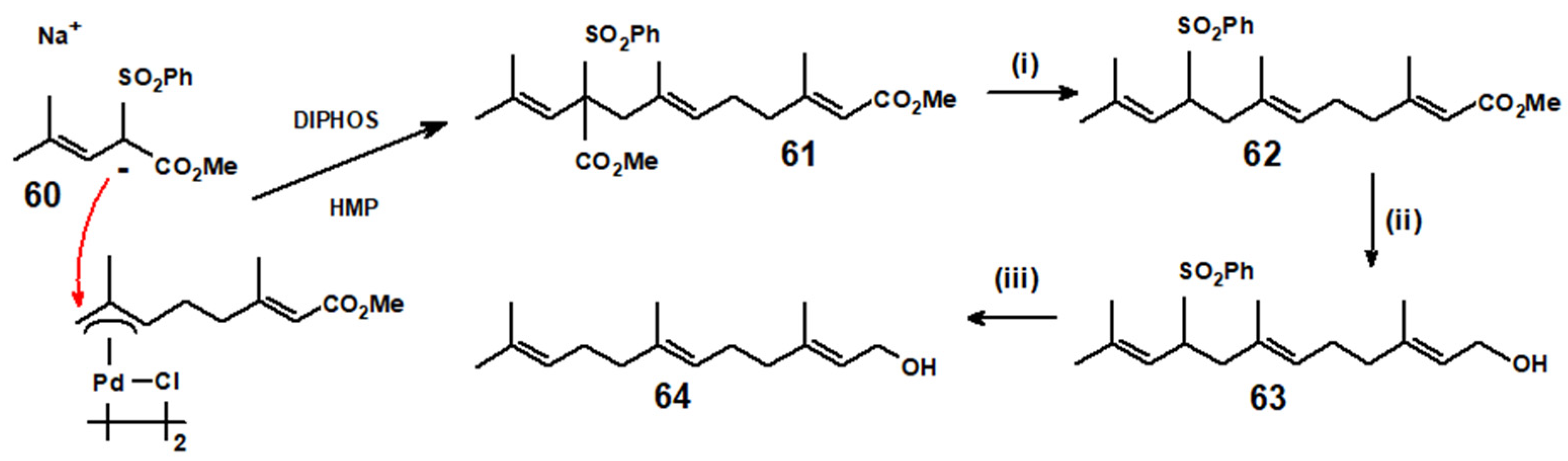{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
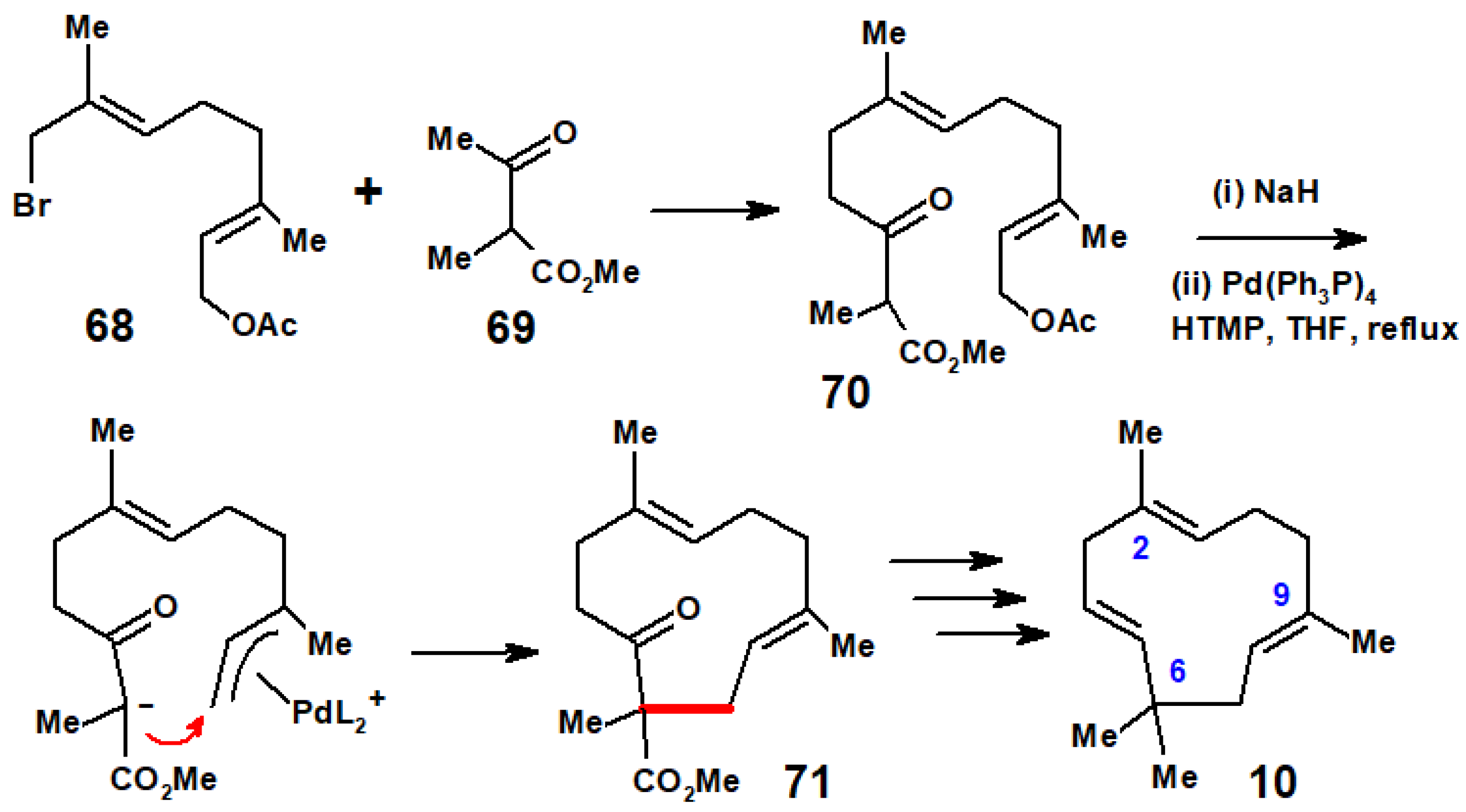{kind=link}
{kind=link}
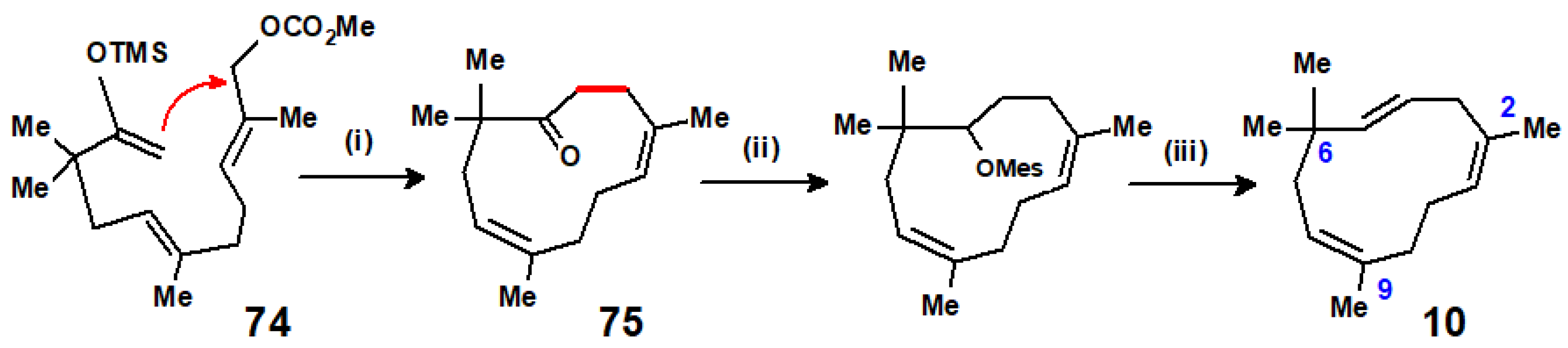{kind=link}
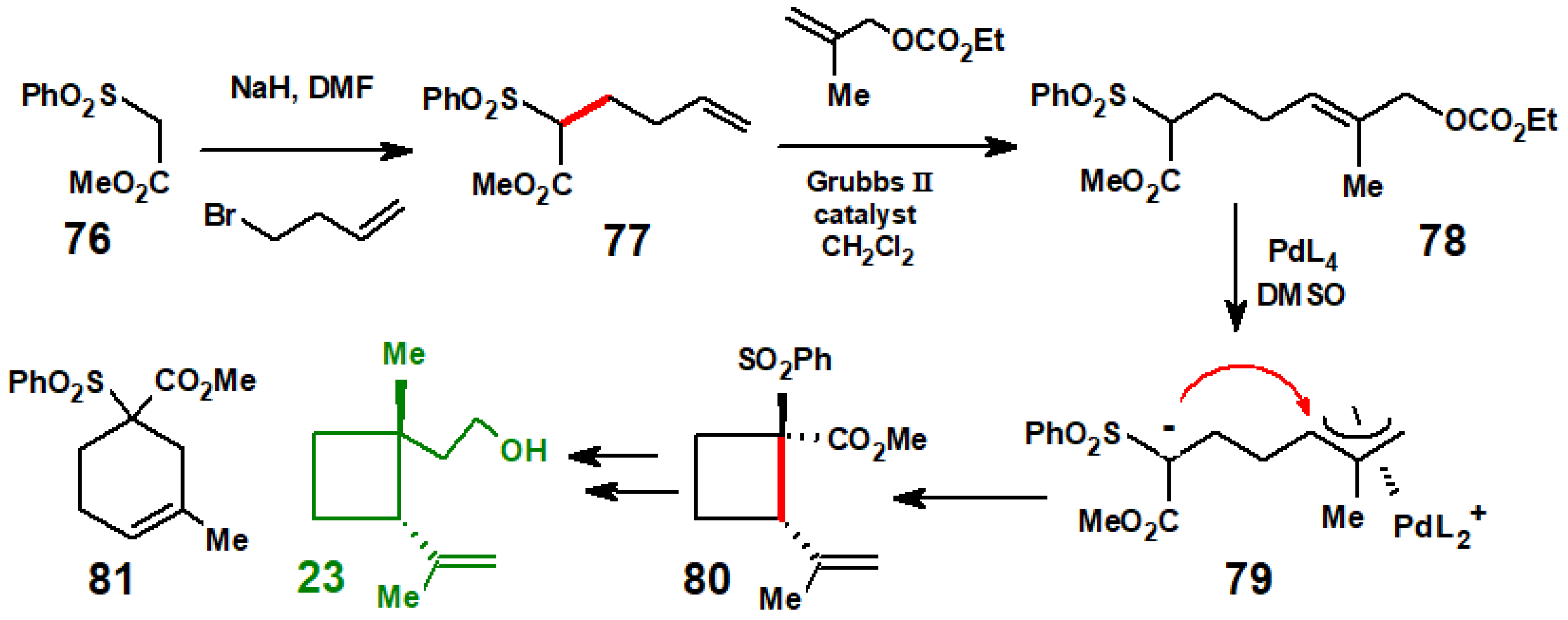{kind=link}
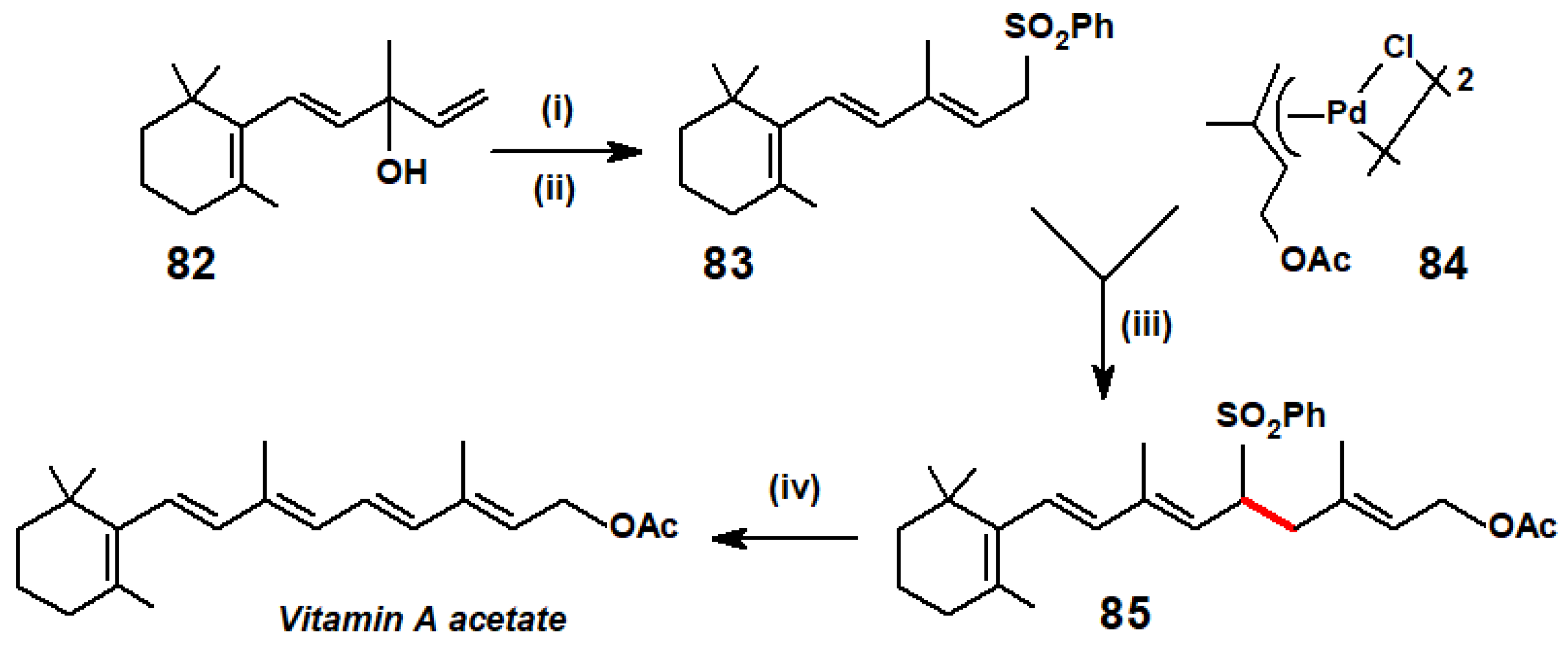{kind=link}
{kind=link}
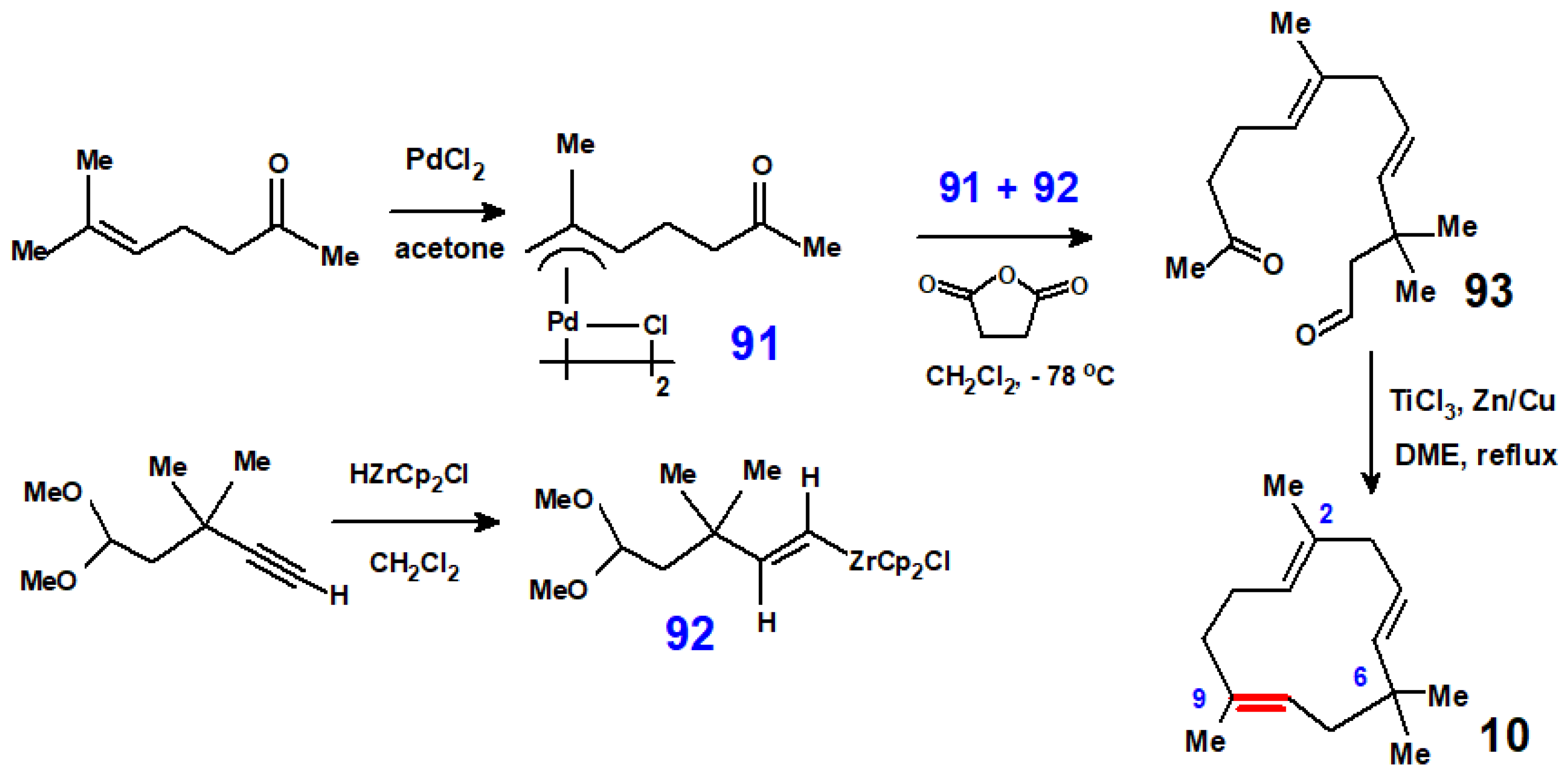{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
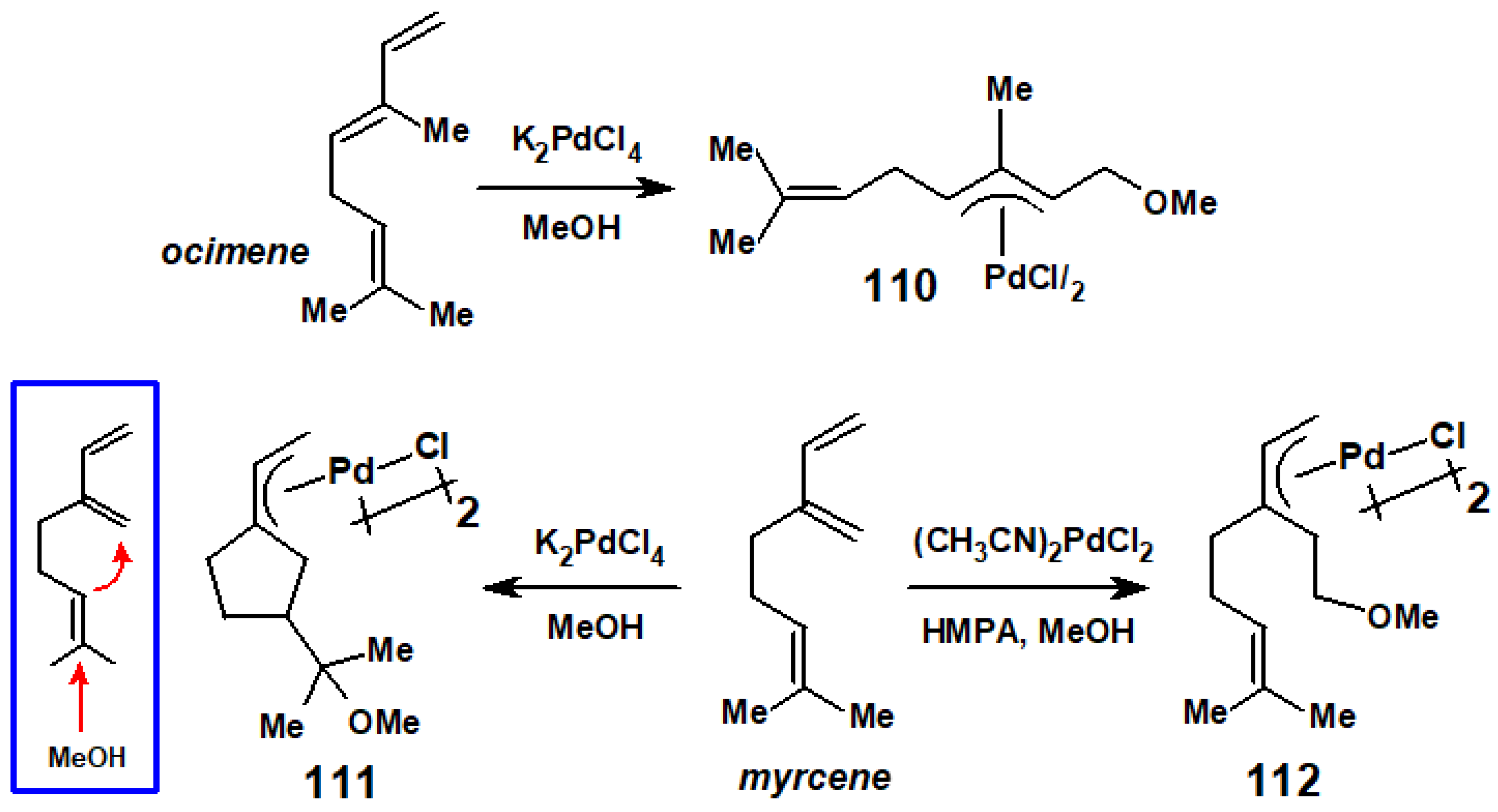{kind=link}
{kind=link}
{kind=link}
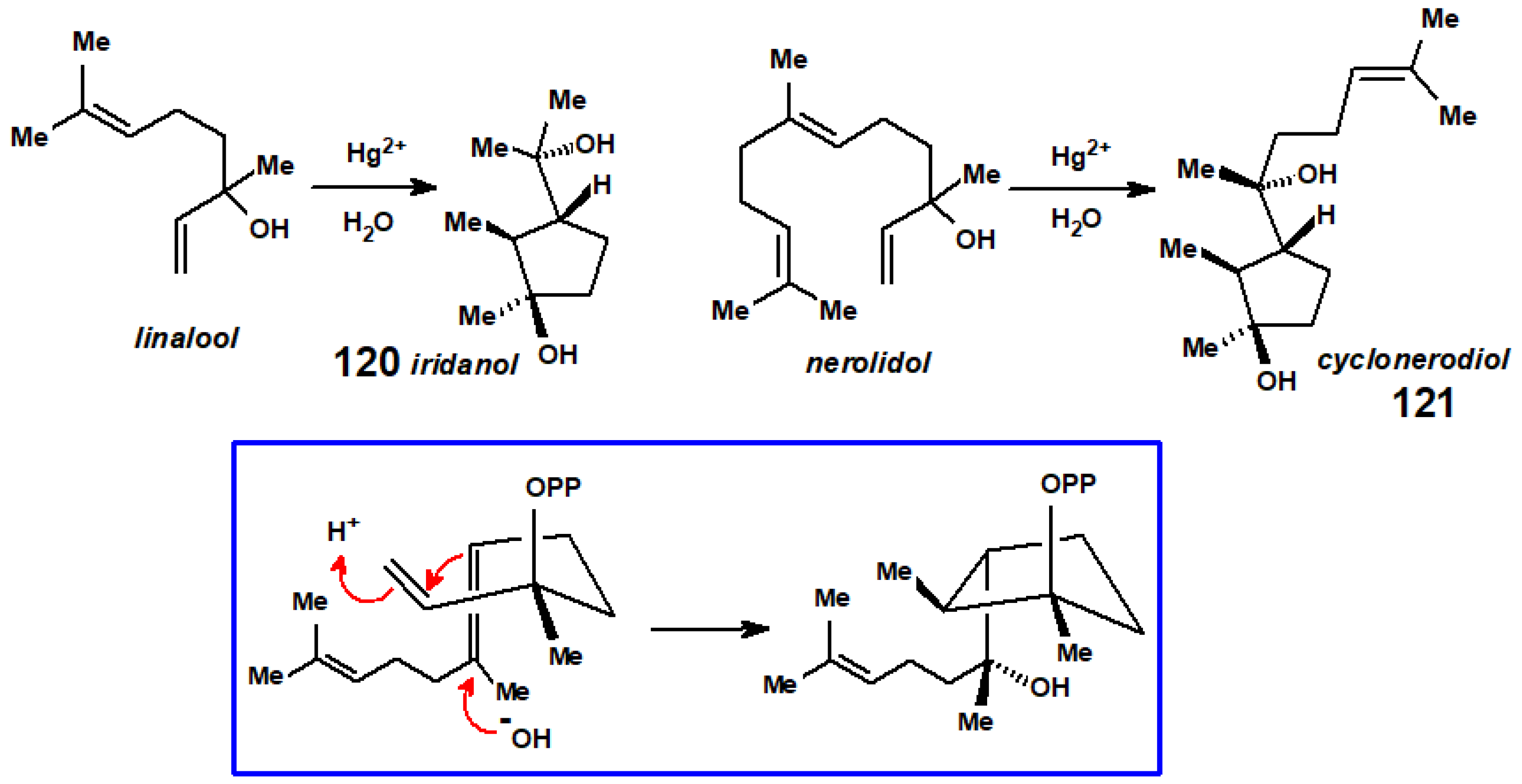{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
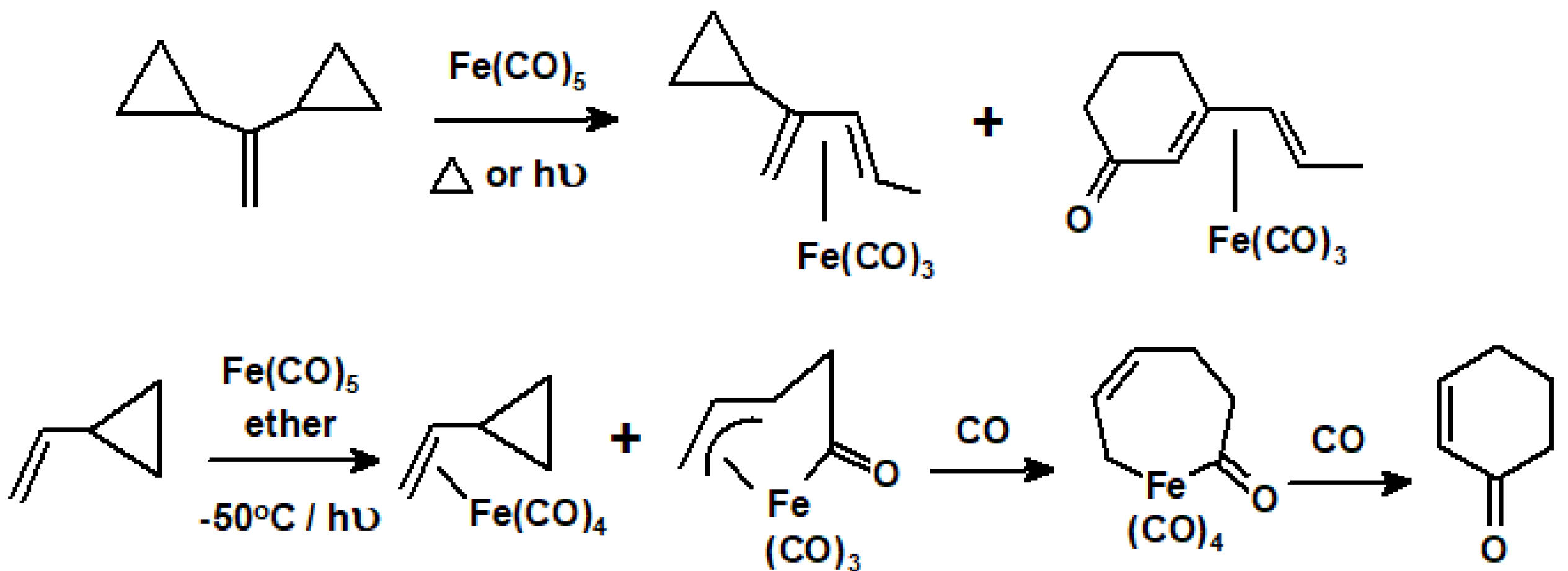{kind=link}
{kind=link}
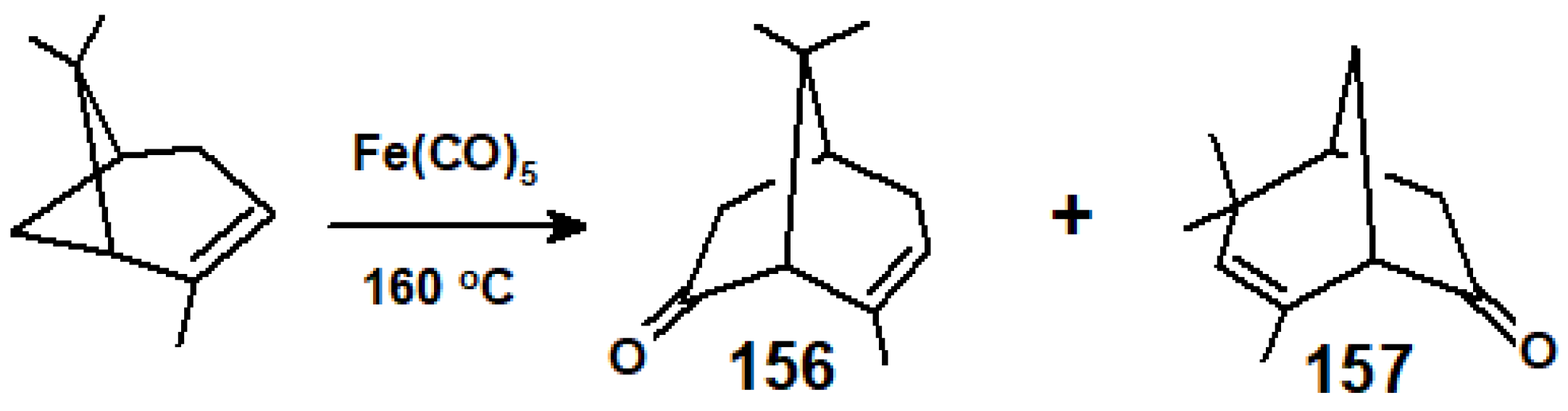{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
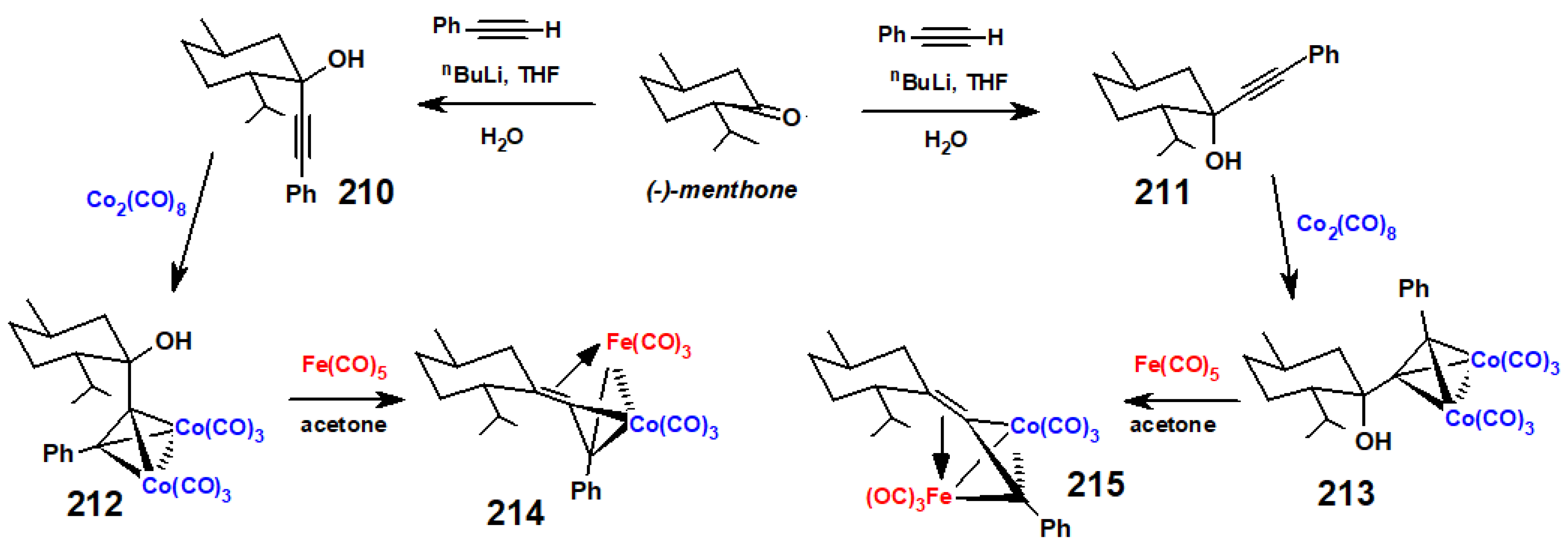{kind=link}
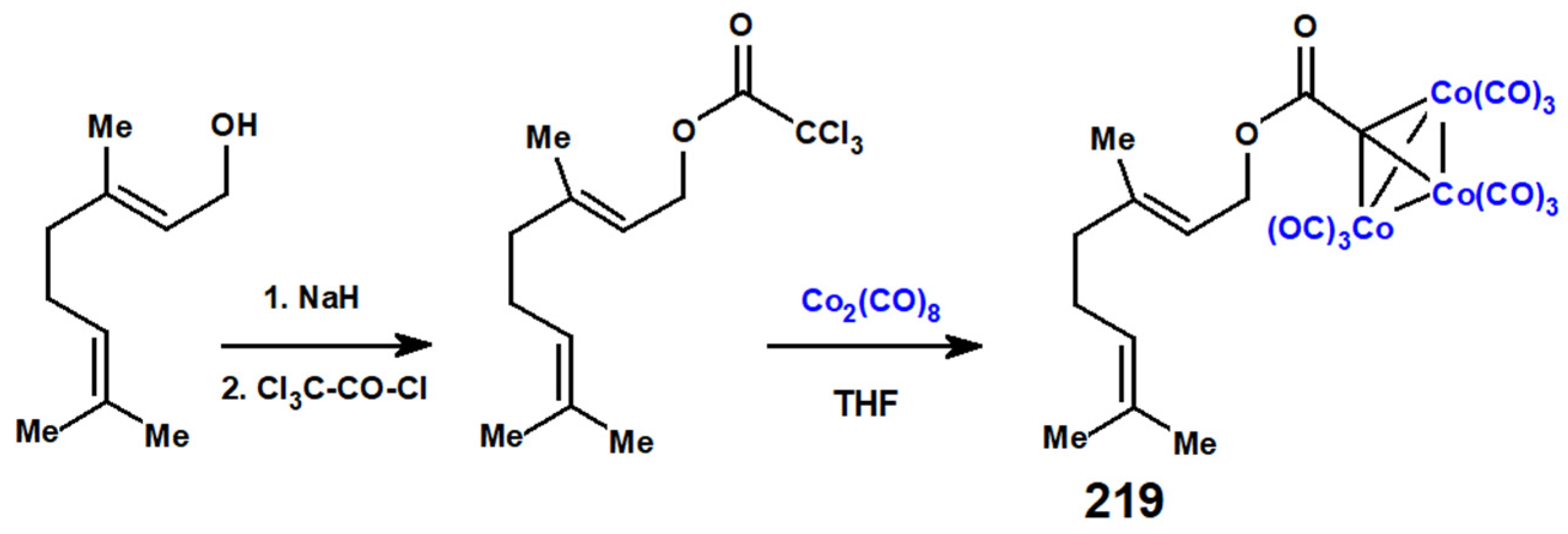
{kind=link}
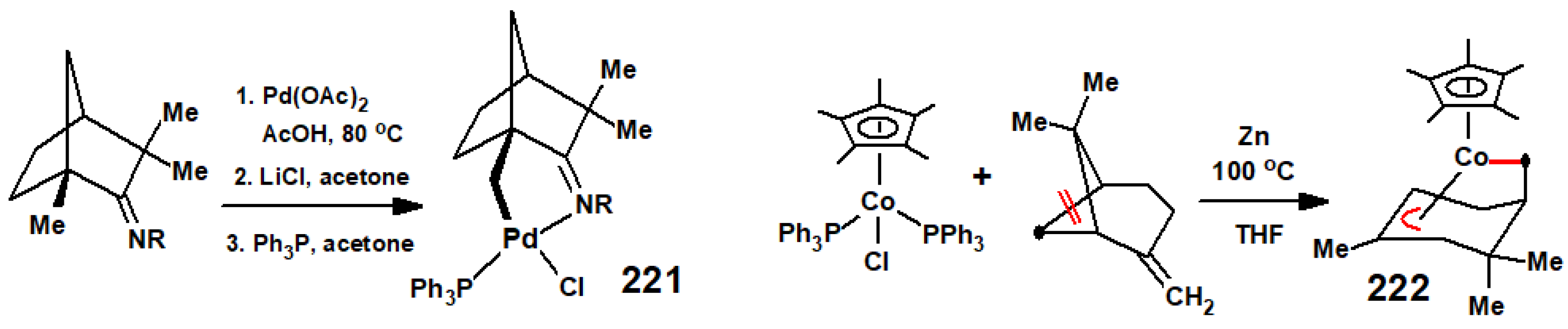
{kind=link}
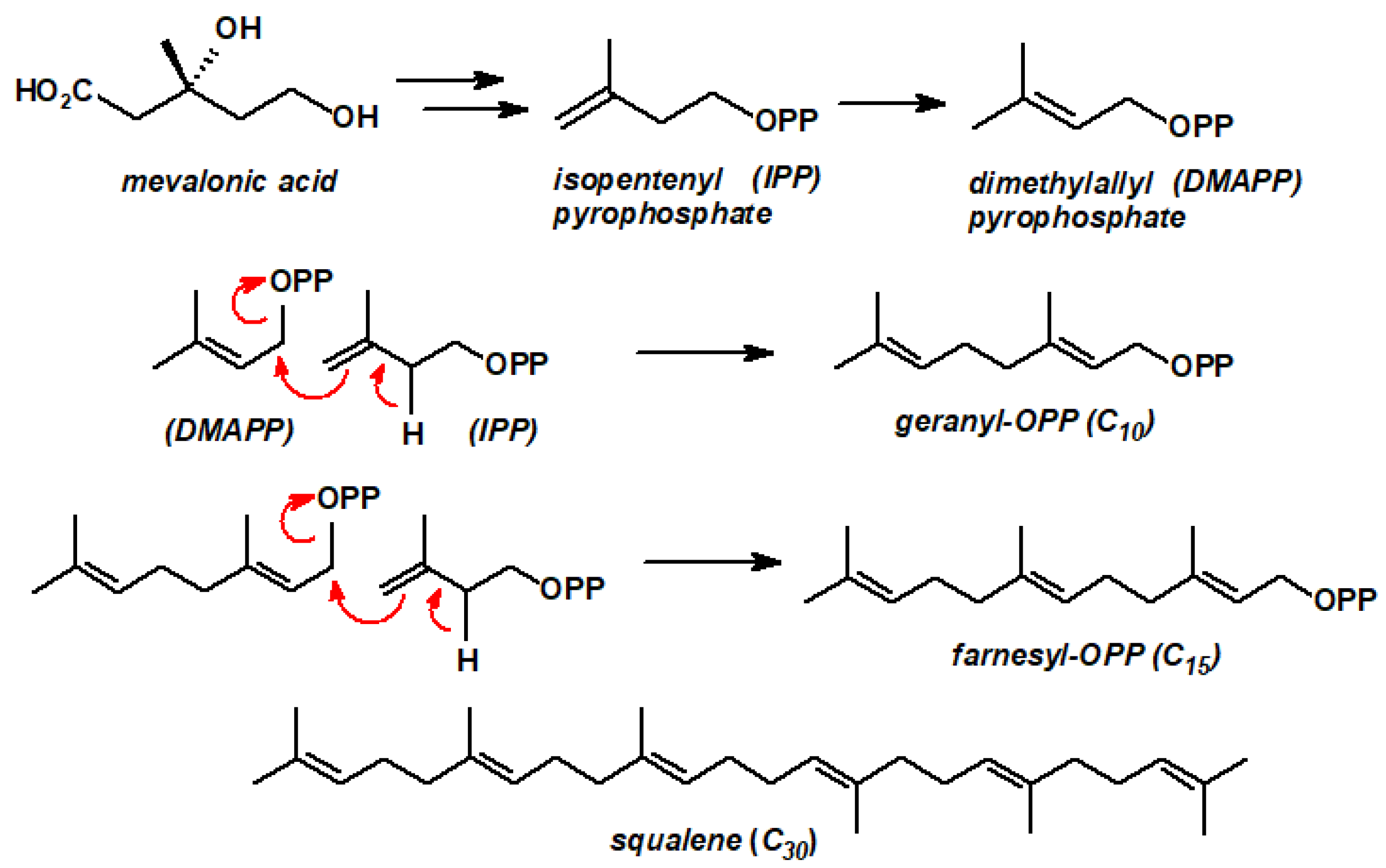
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Synthetic Overview
3. Organonickel Complexes as Coupling Reagents
3.1. Dehalogenations Using Ni(CO)4
3.2. π-Allyl-Nickel Complexes
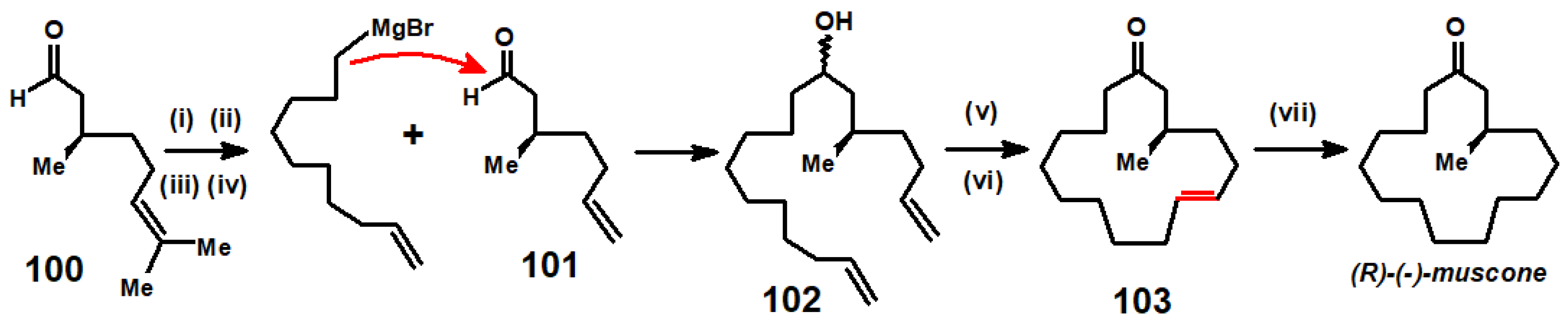
3.3. Syntheses of Grandisol and of Muscone
3.4. Organocobalt Complexes as Substitutes for Ni(CO)4
4. Organoiron Complexes as Coupling Reagents
4.1. Oxaallyl-Fe(II) Intermediates
4.2. Allyl-Iron(Dicarbonyl)(η5-Cyclopentadienyl) Complexes
5. Palladium Complexes as Coupling Reagents
5.1. The Heck Reaction
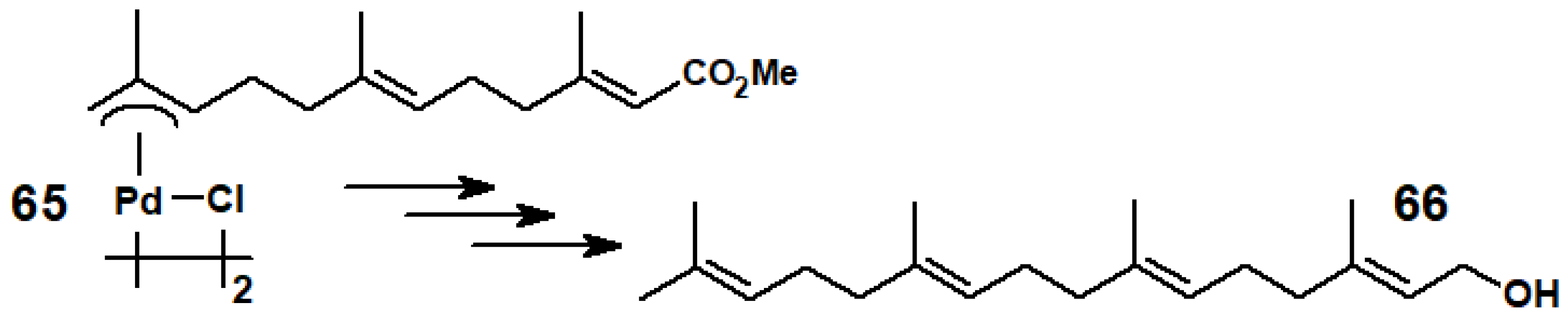
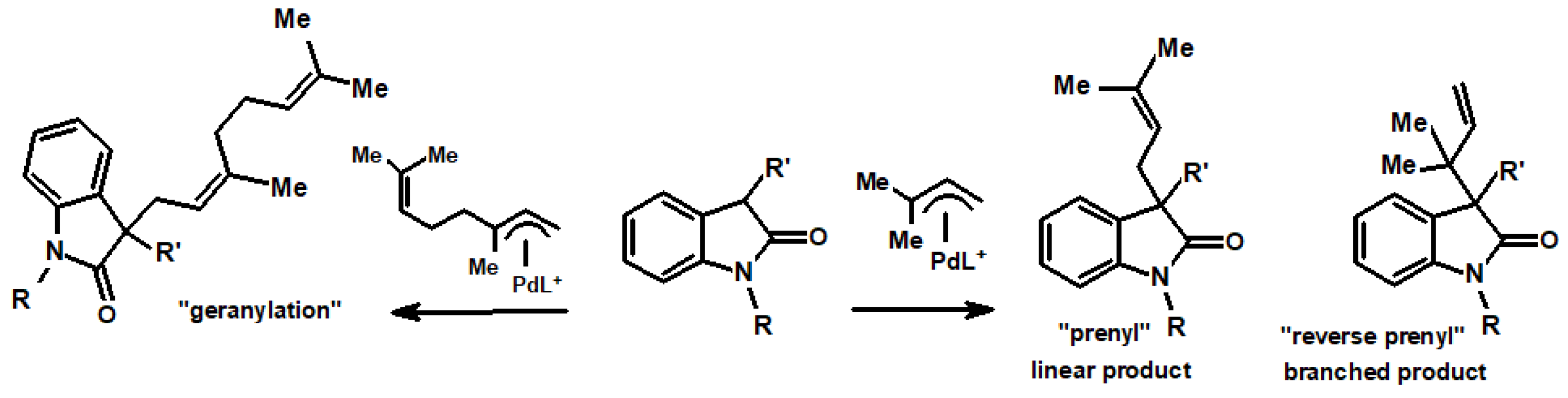
5.2. Prenylations and Geranylations Using π-Allyl-Palladium Complexes
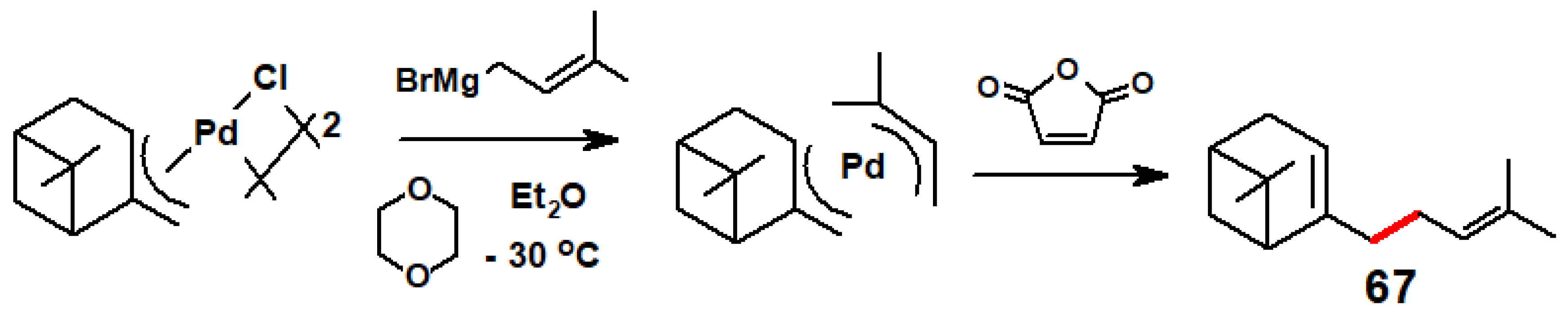
5.3. Palladium-Mediated Routes to Humulene, Grandisol, Vitamin A and Mokupalide
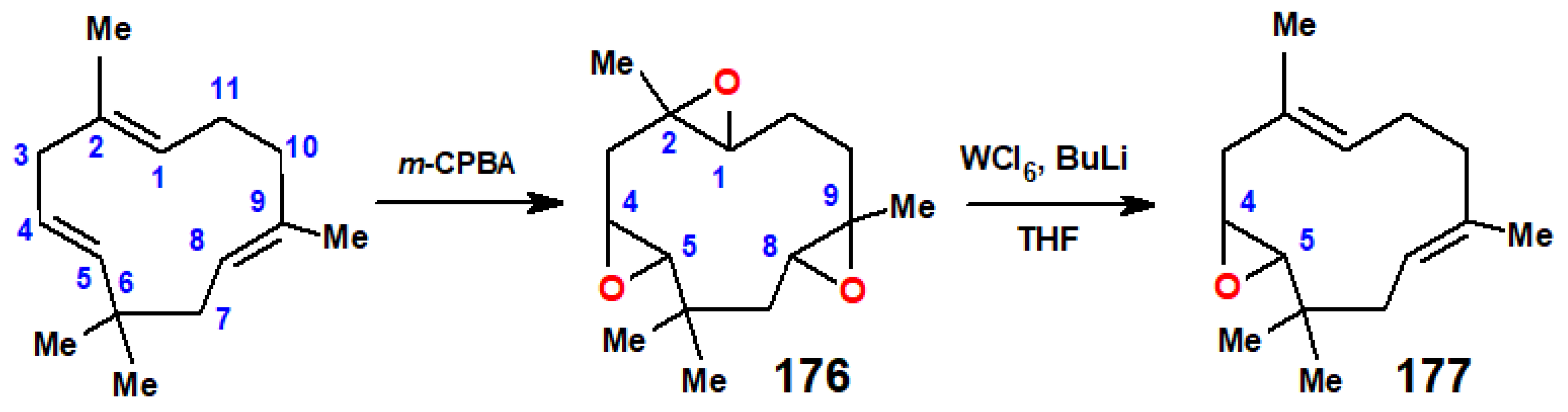
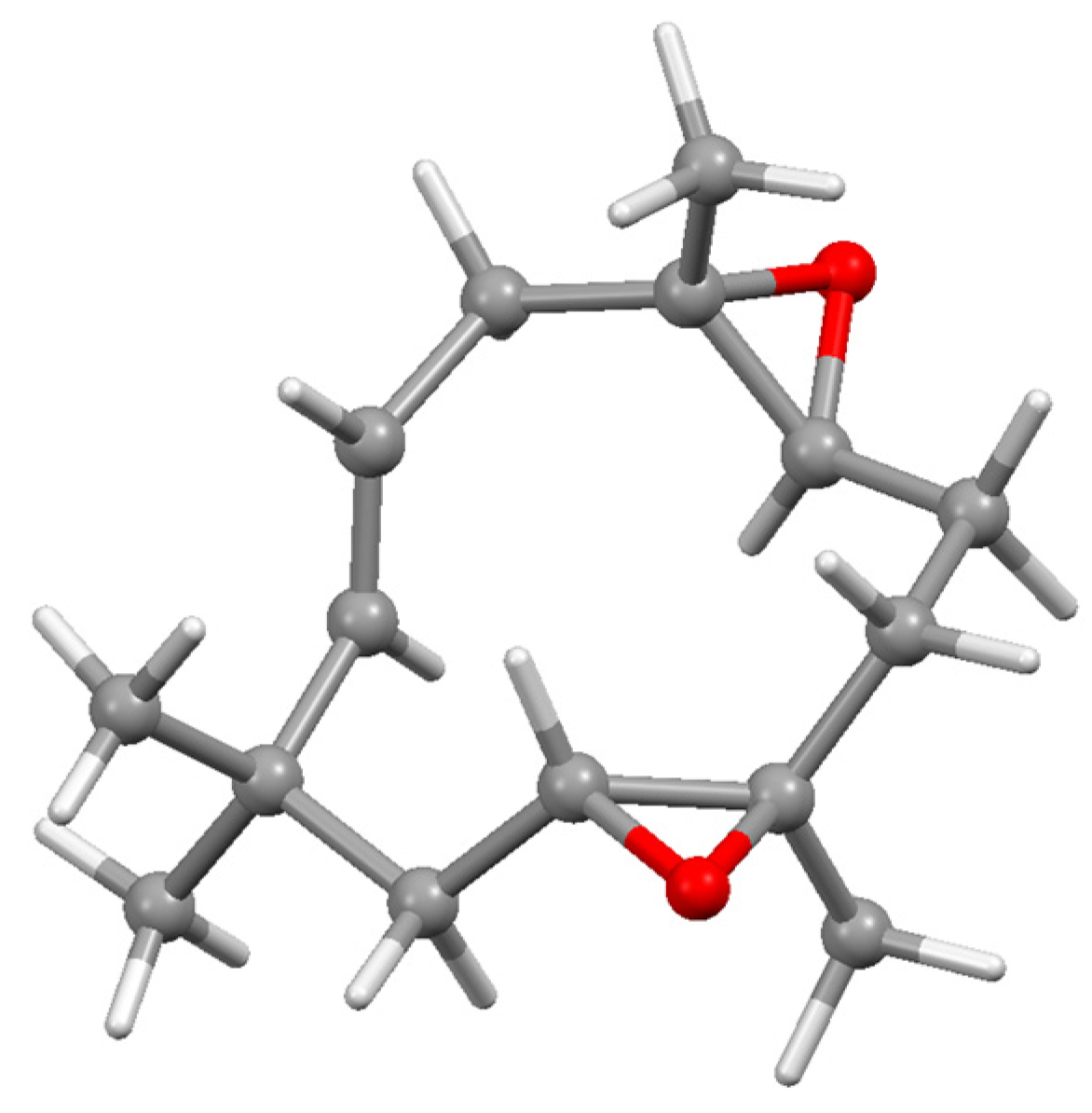
6. Terpenoid Synthesis Using Titanium and Zirconium Reagents
6.1. Schwartz’s Hydrozirconation Reaction
6.2. McMurry Dicarbonyl Coupling
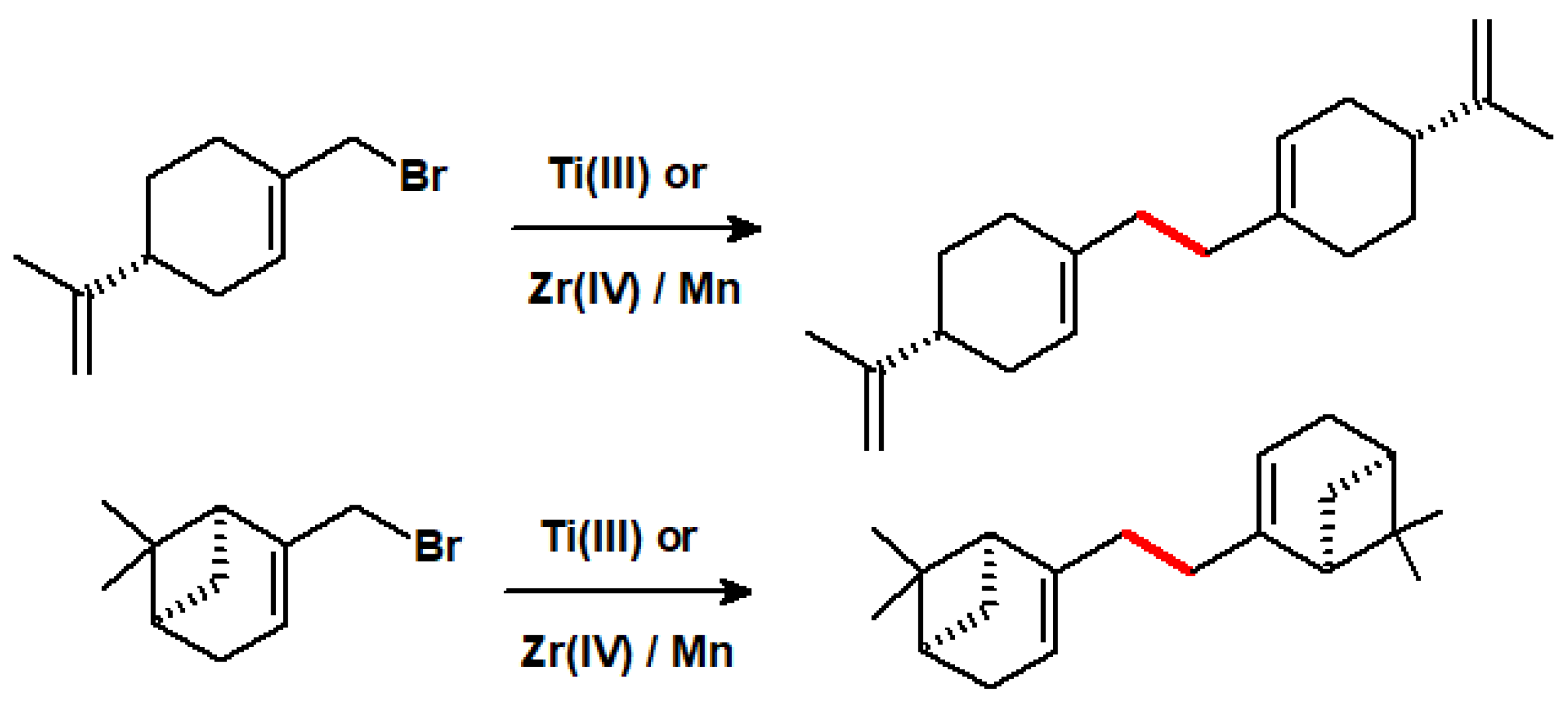
6.3. Ti(III) and Zr(III) as Dimerisation Catalysts
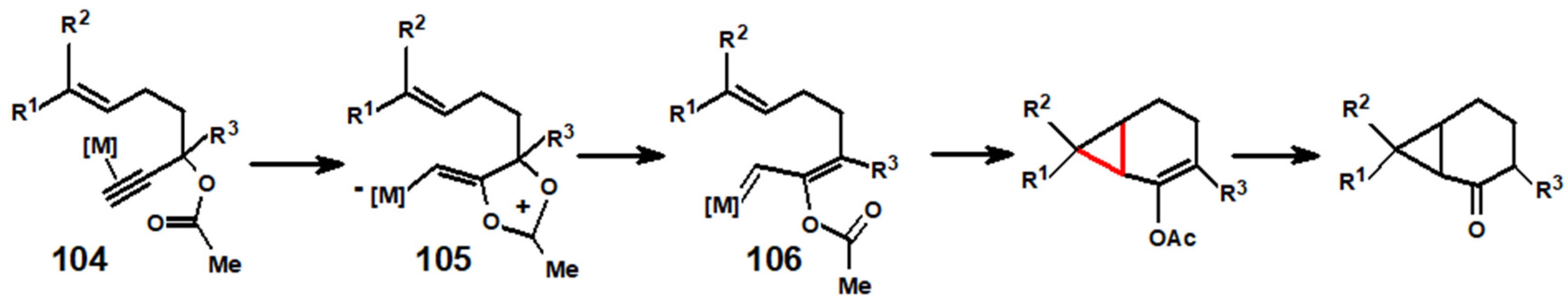
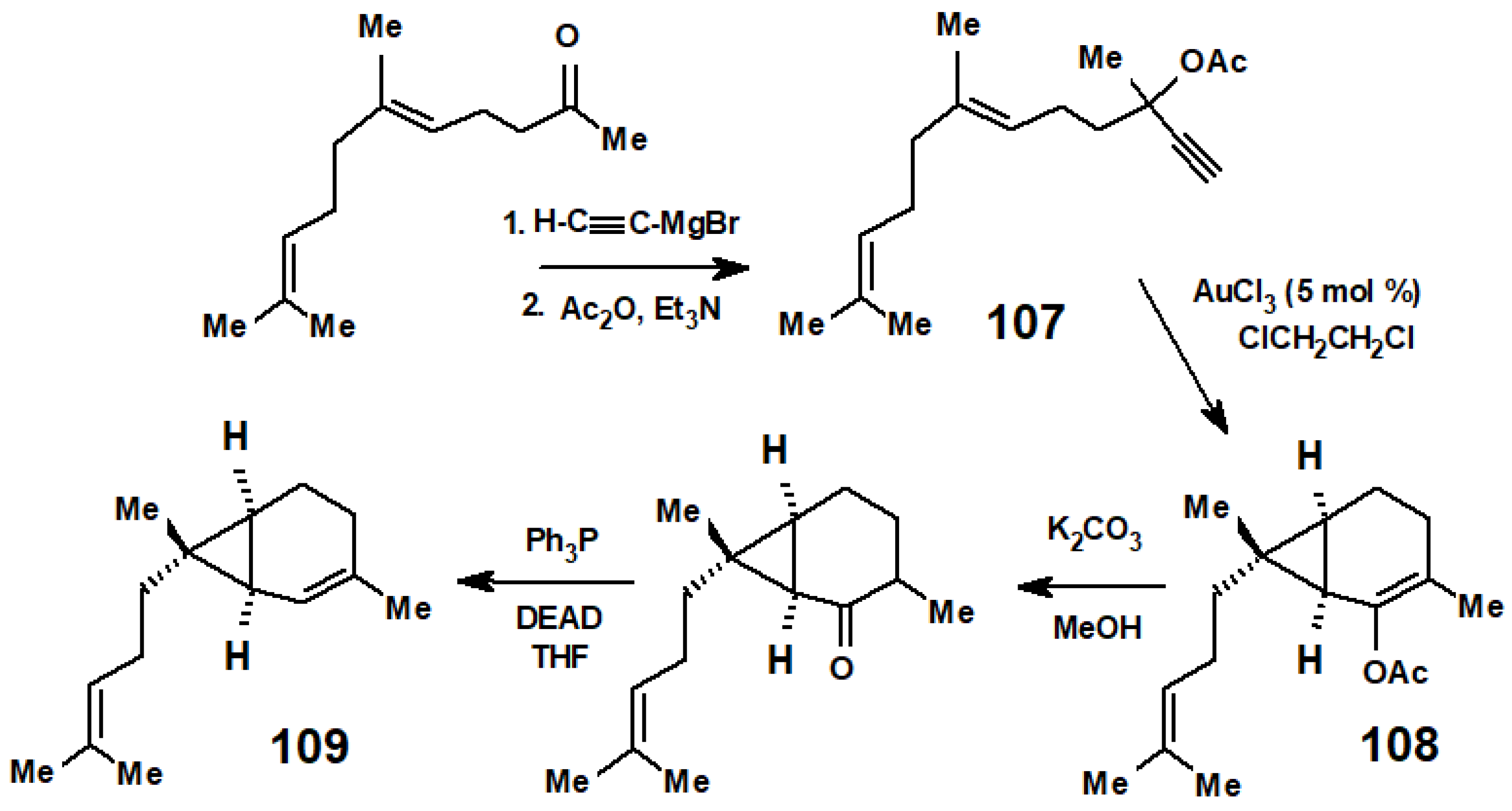
7. The Golden Route to Carenes
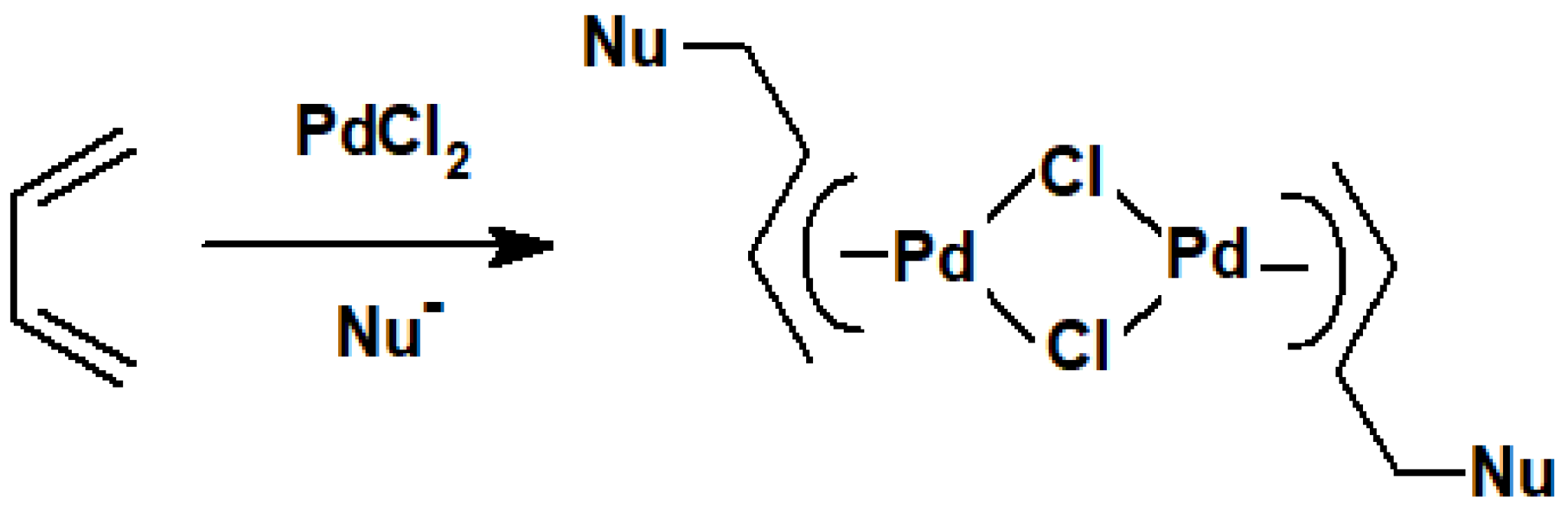
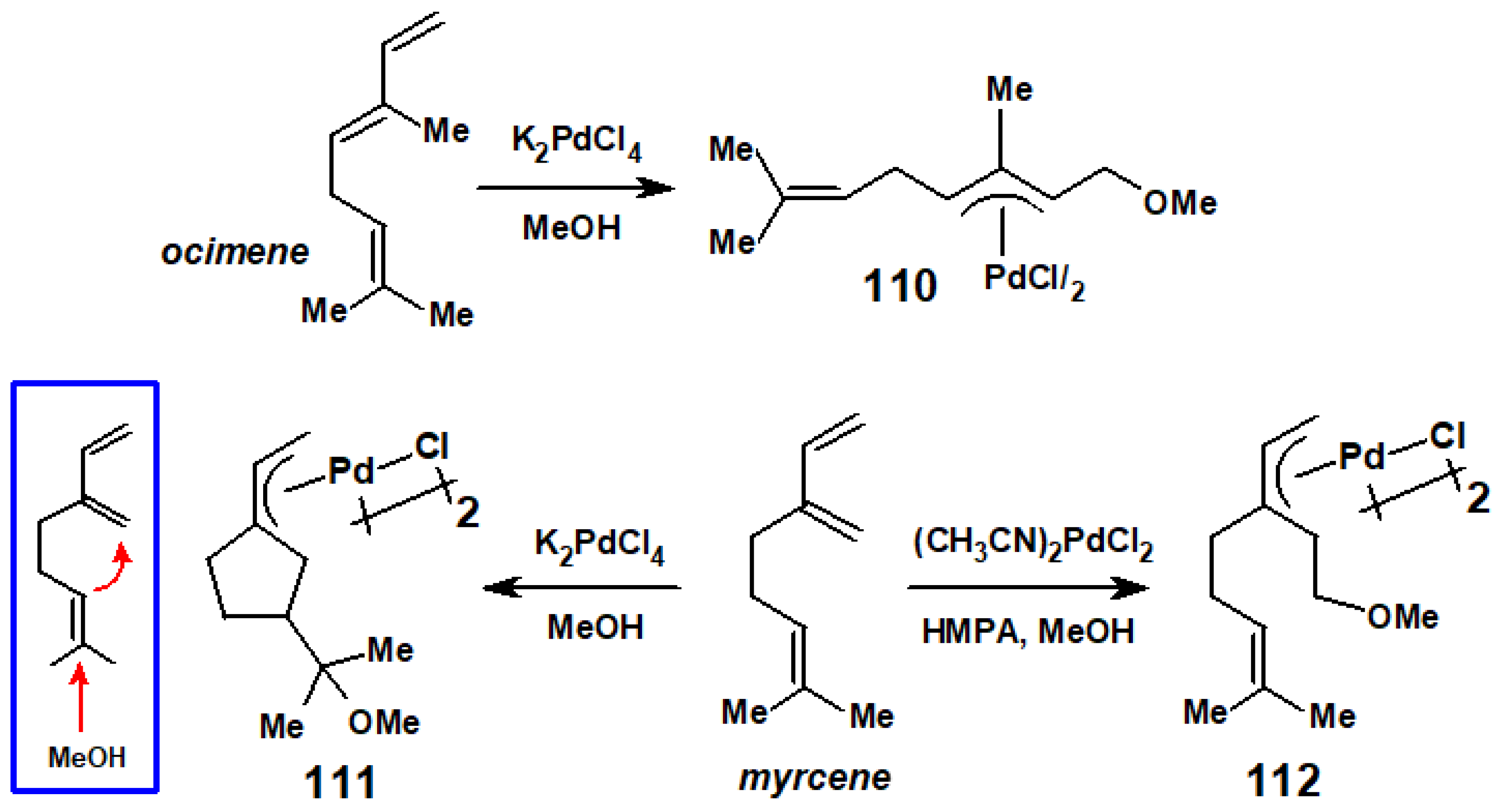
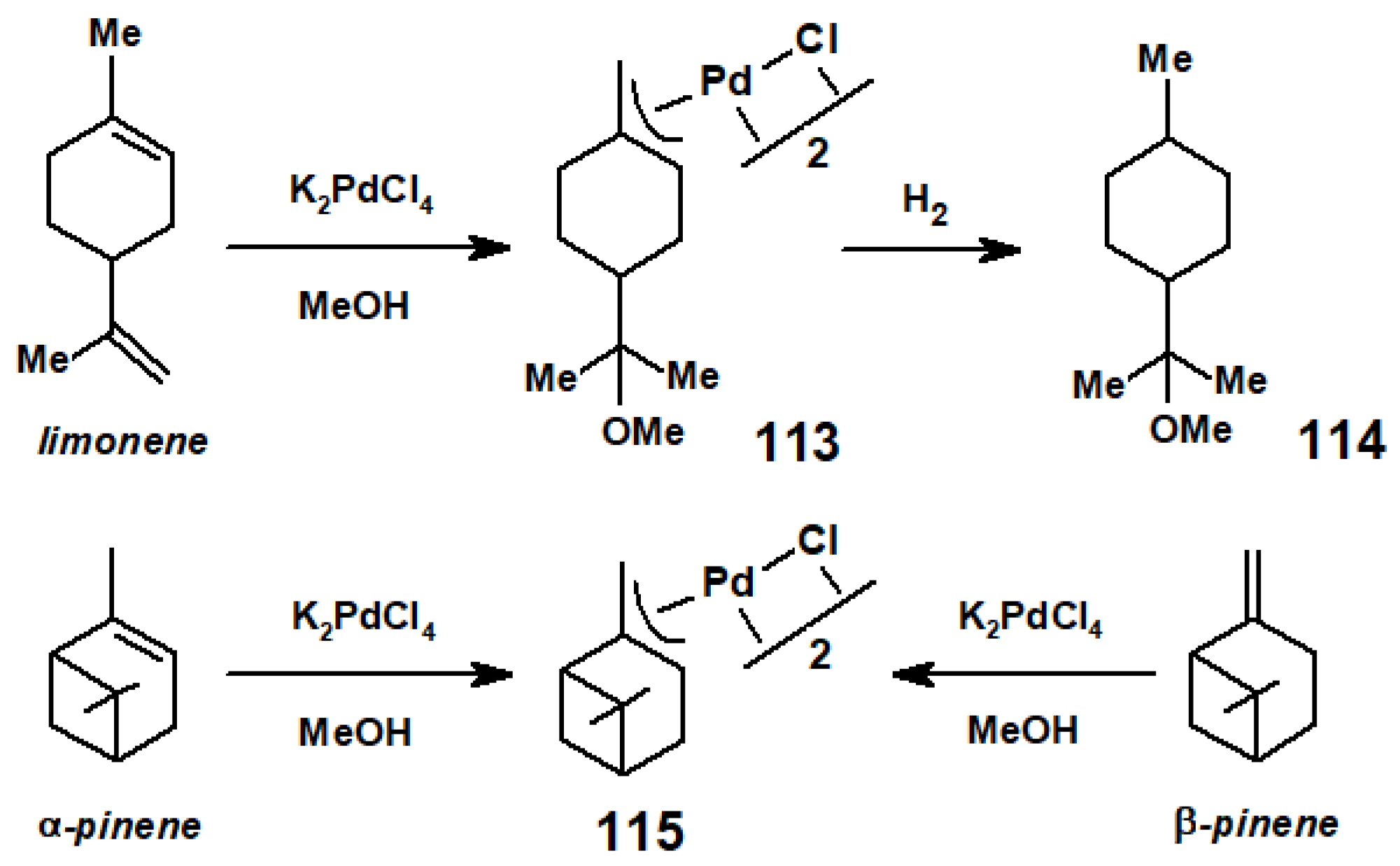
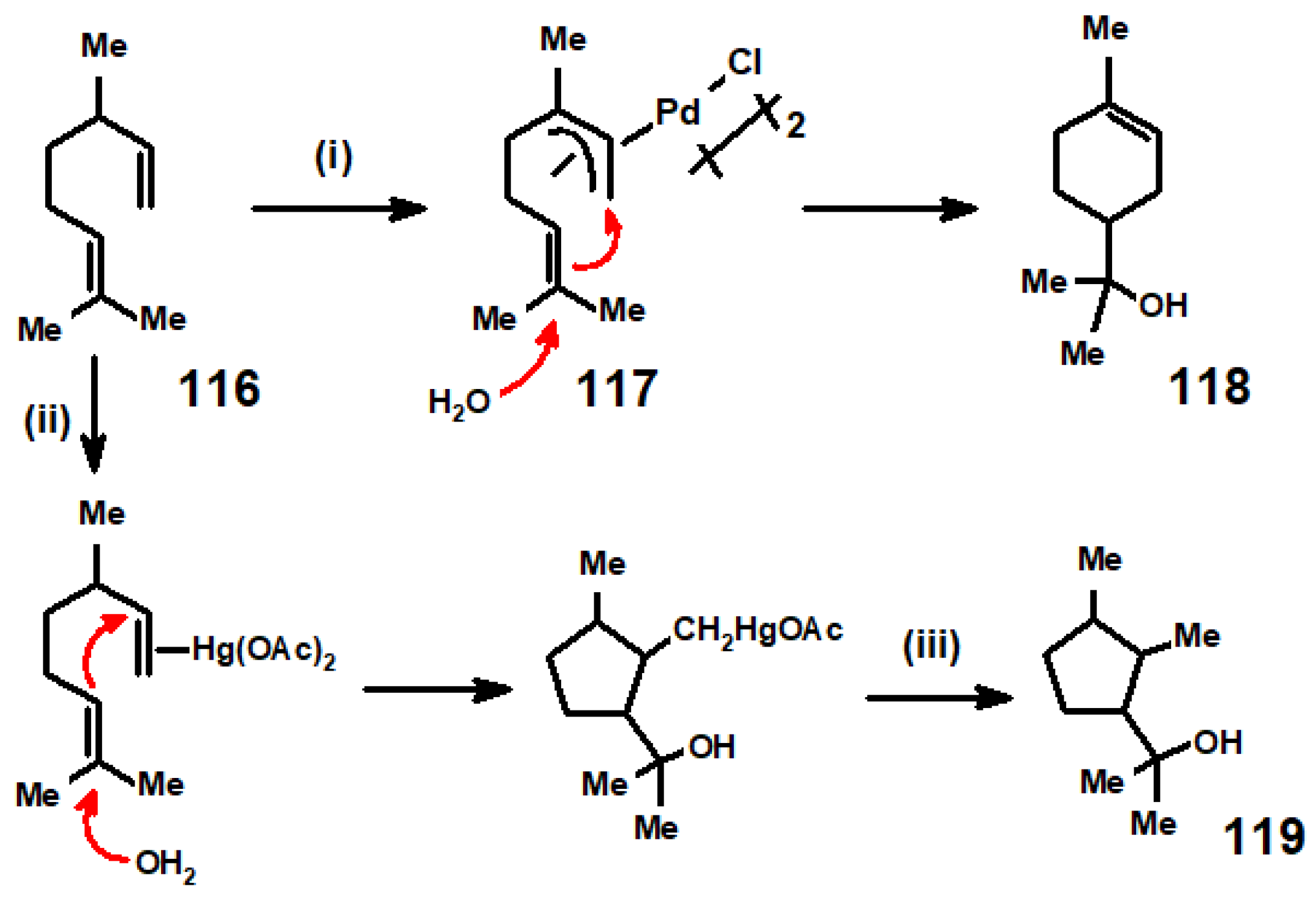
8. Reactions of Terpene—Palladium(II) Complexes with Nucleophiles
9. Reactions of Terpene—Iron Carbonyl Complexes with Electrophiles
10. Metal-Promoted Terpene Isomerisations
10.1. π-Allyl Metal Hydrides
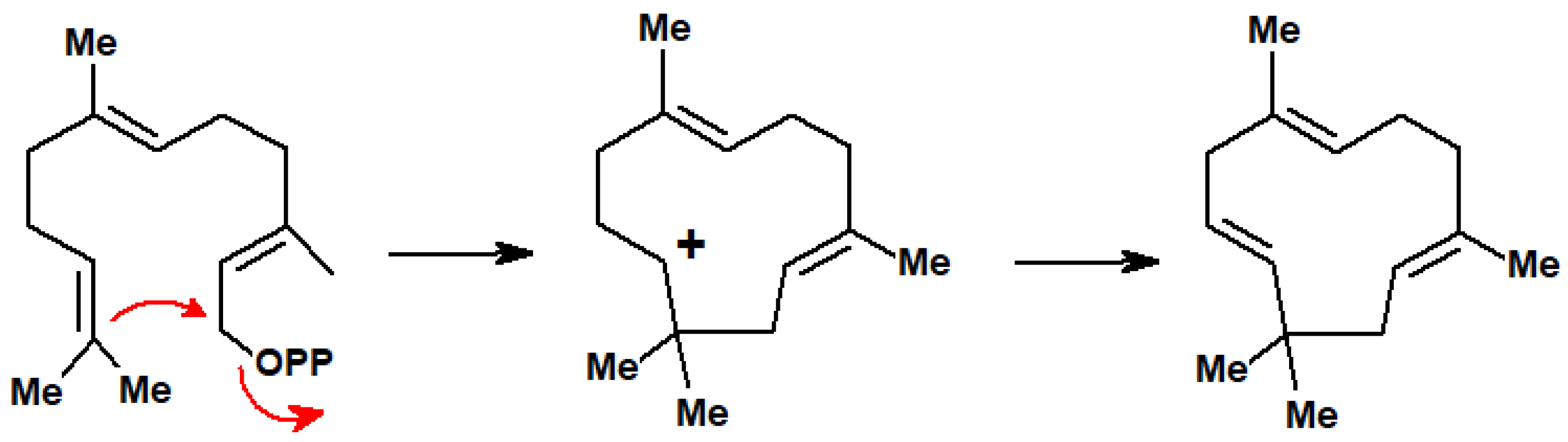
10.2. [3,3]-Sigmatropic Shifts in Terpenoids
10.3. Interactions of Ring-Strained Terpenes with Organometallic Reagents
10.4. Terpene Rearrangements Induced by Pt(II) or Rh(I) Salts
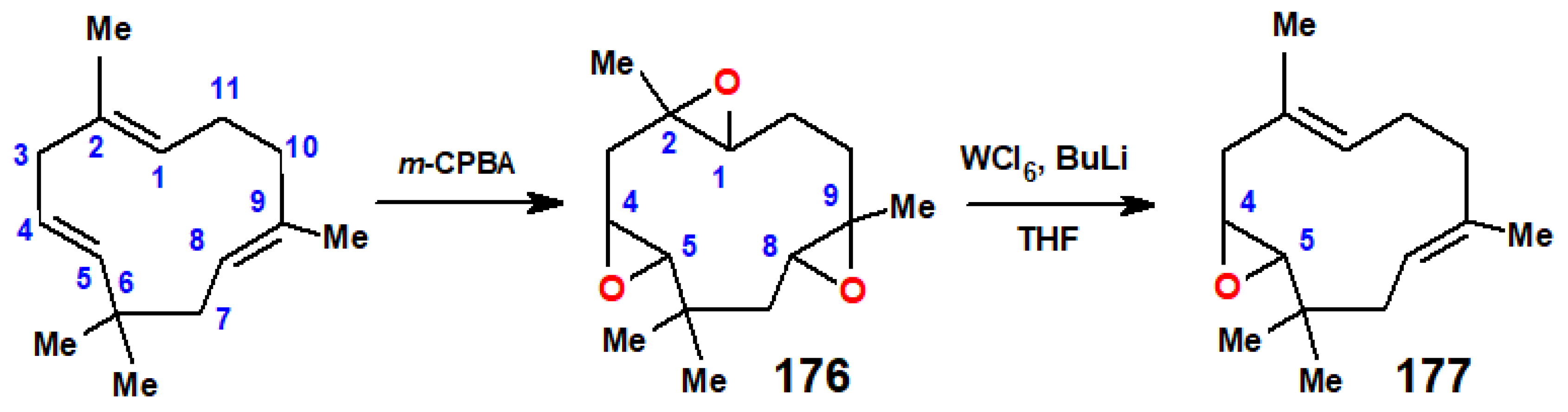
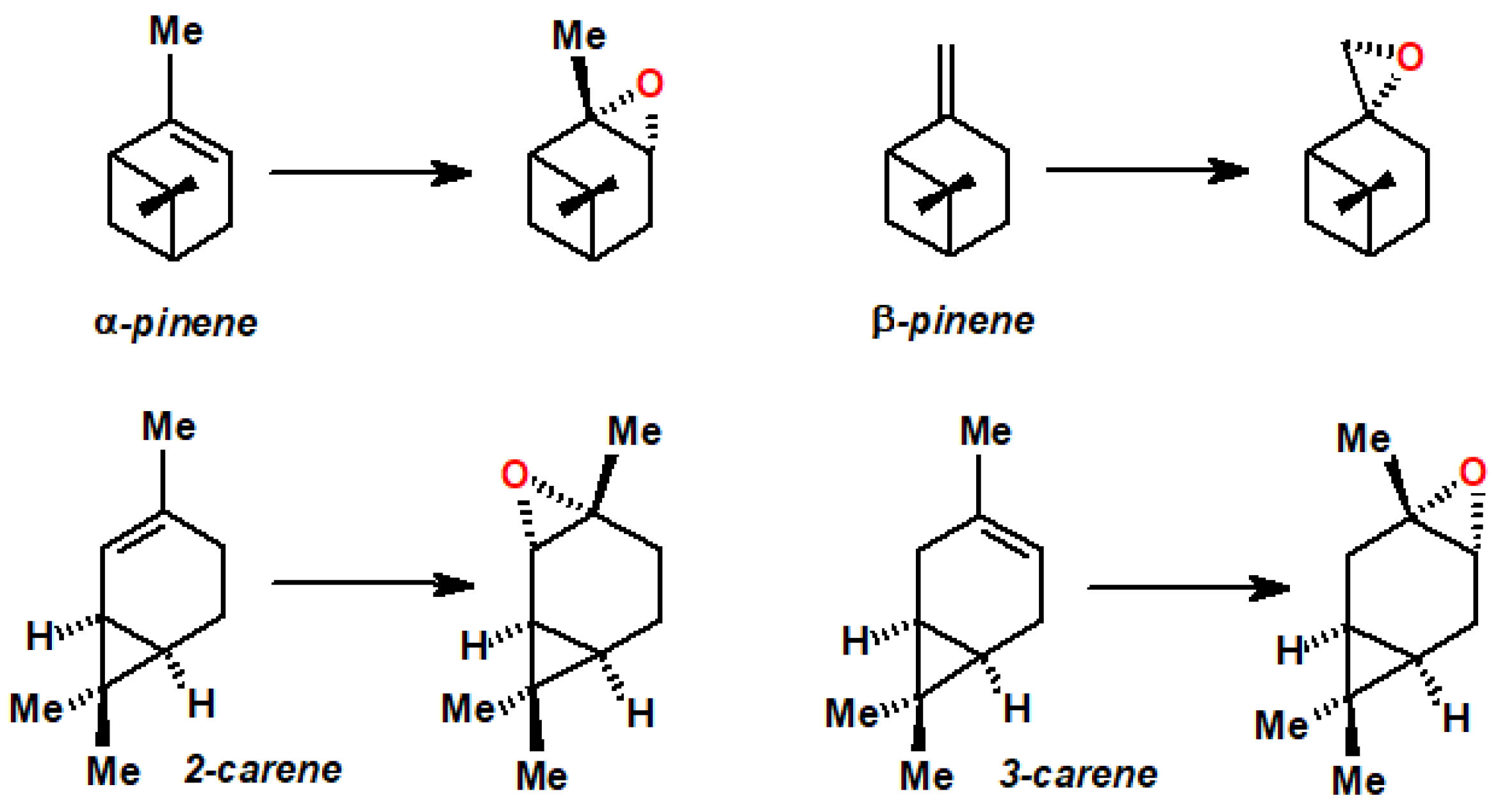
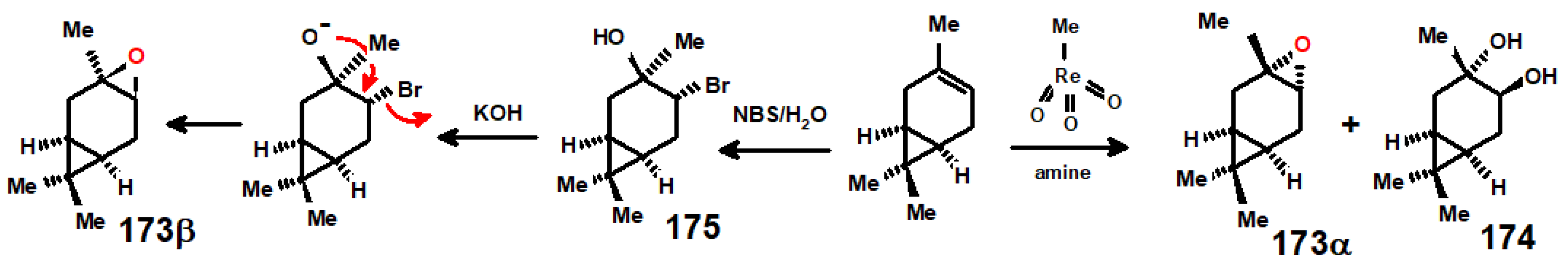
11. Regiospecific Epoxidation of Terpenes
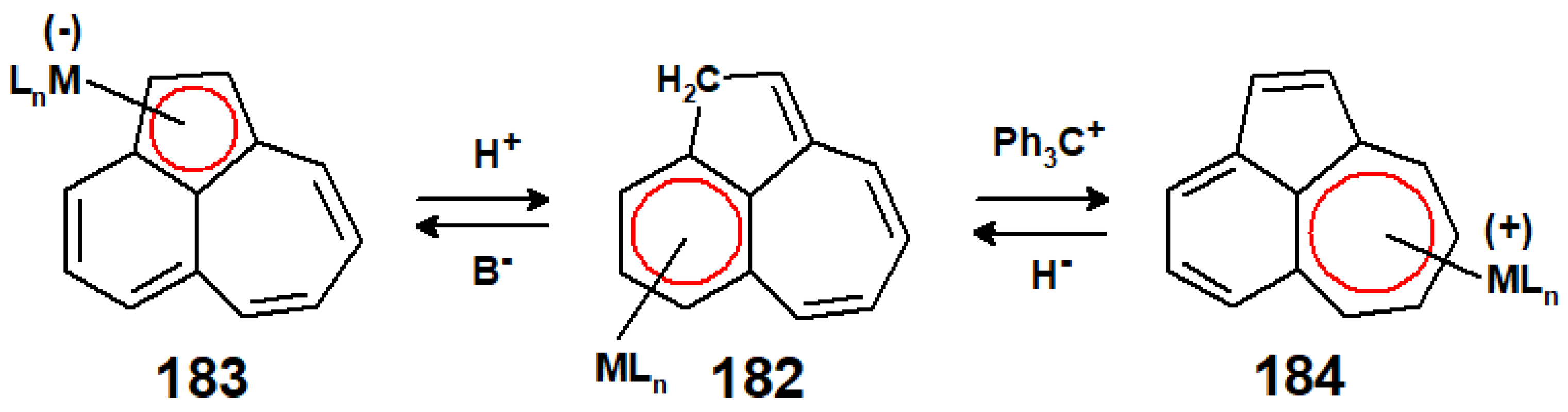
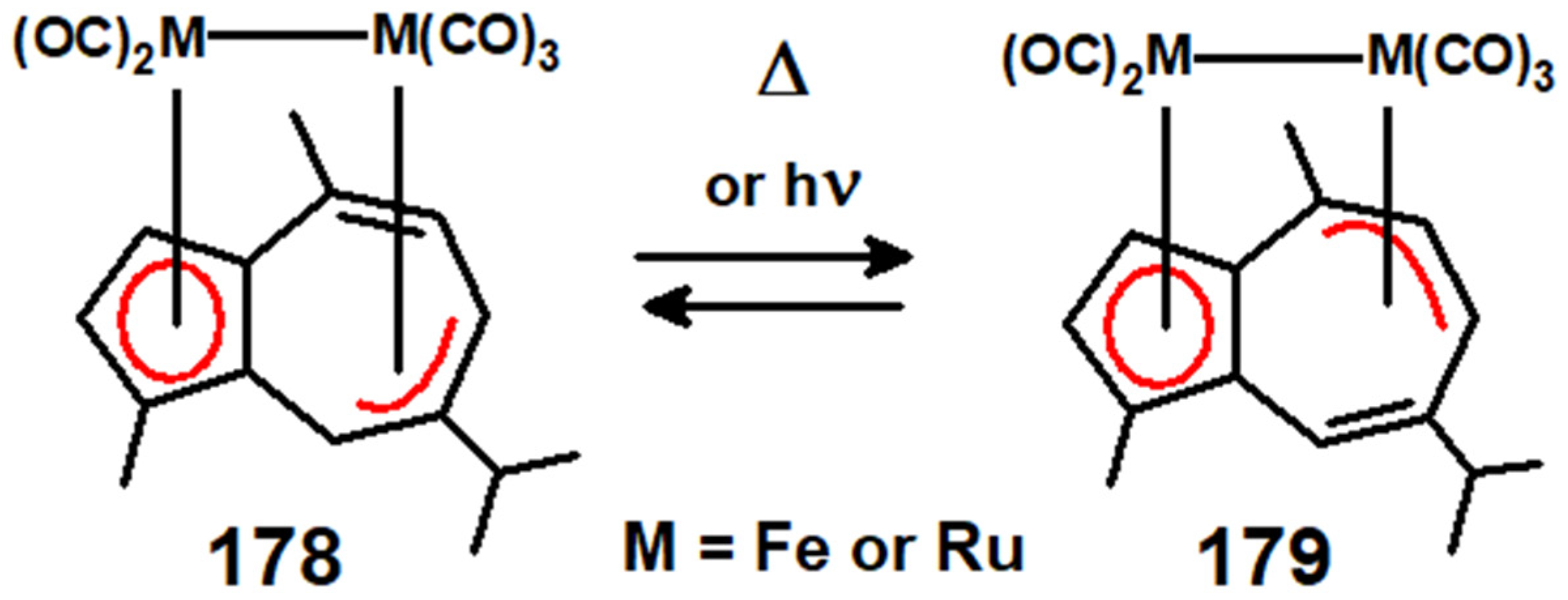
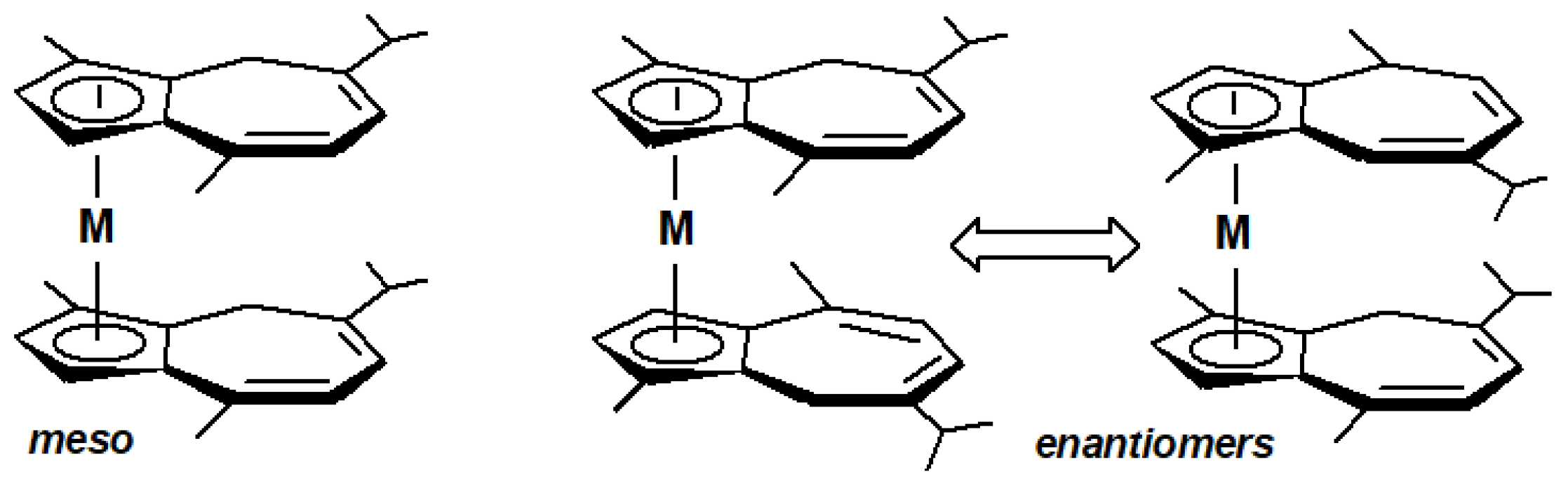
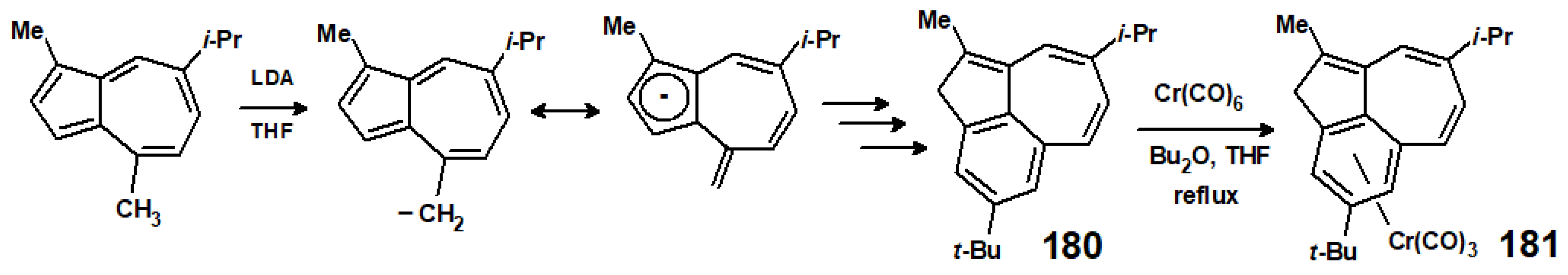
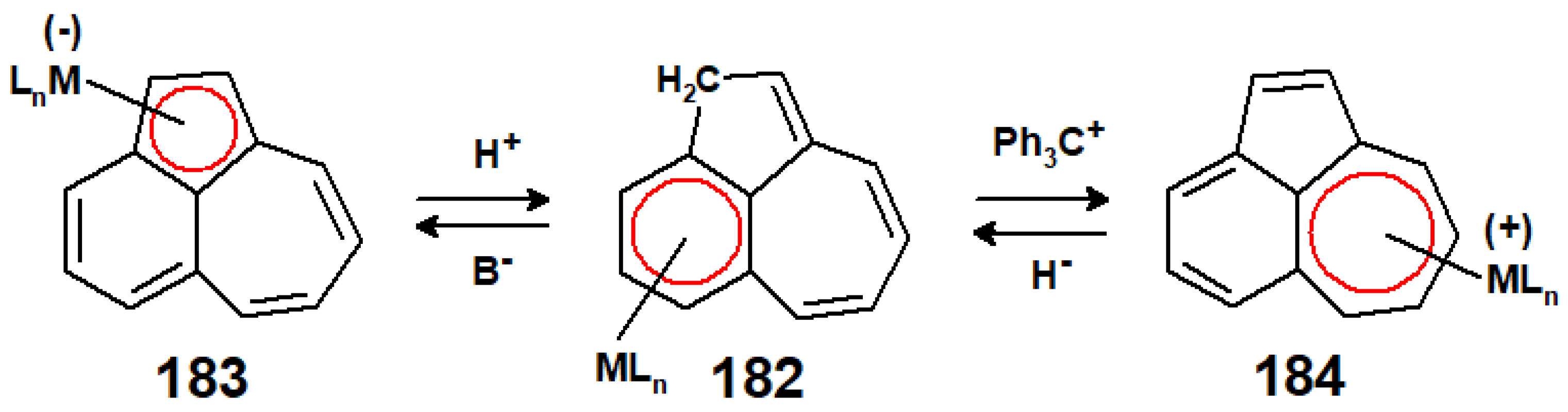
12. Organometallic Derivatives of Guaiazulene
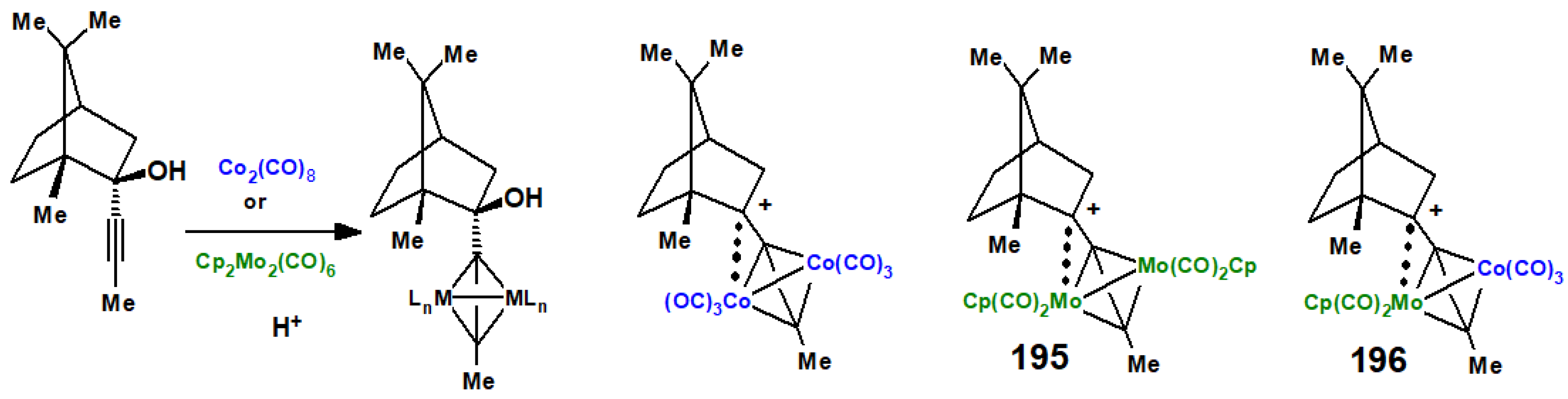
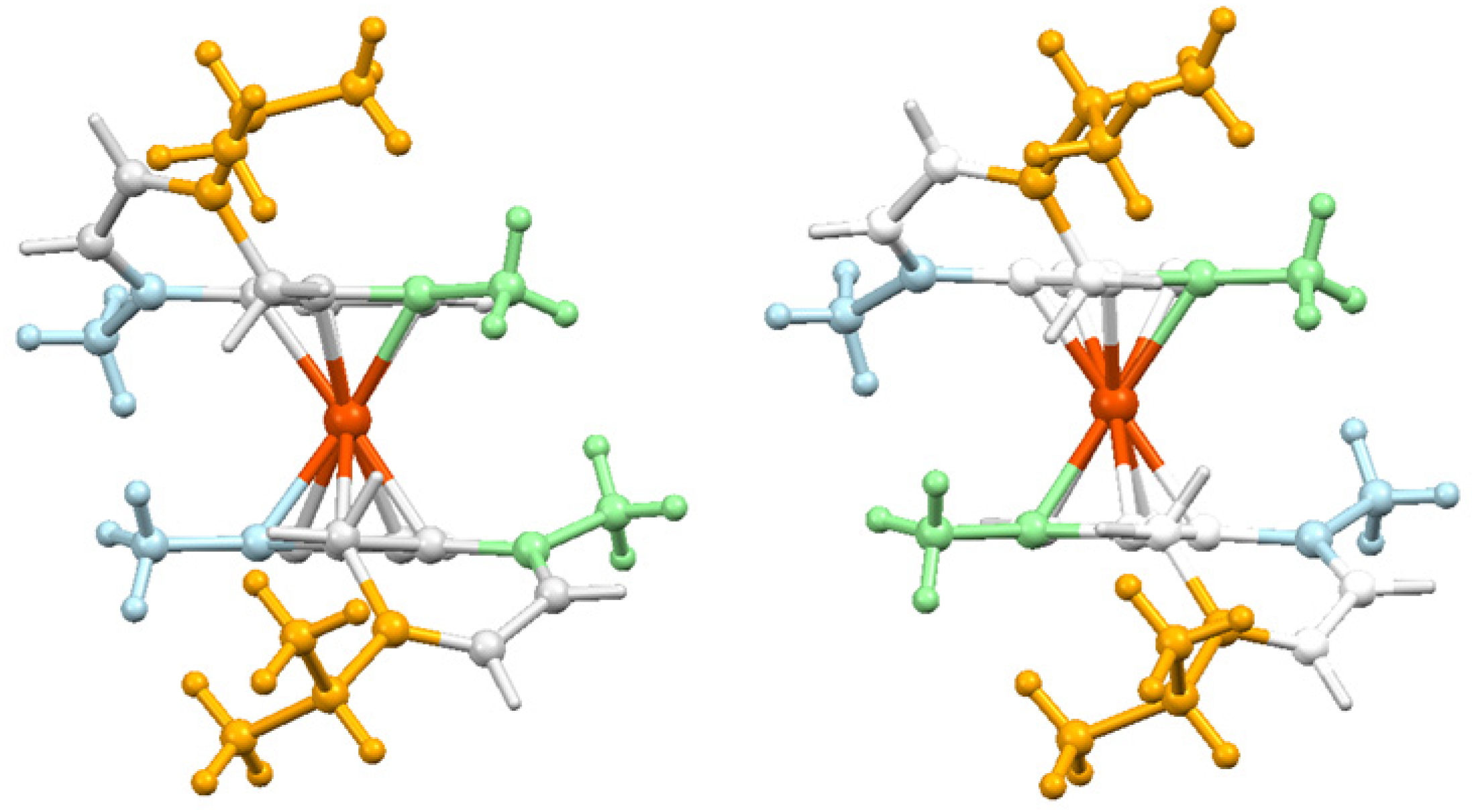
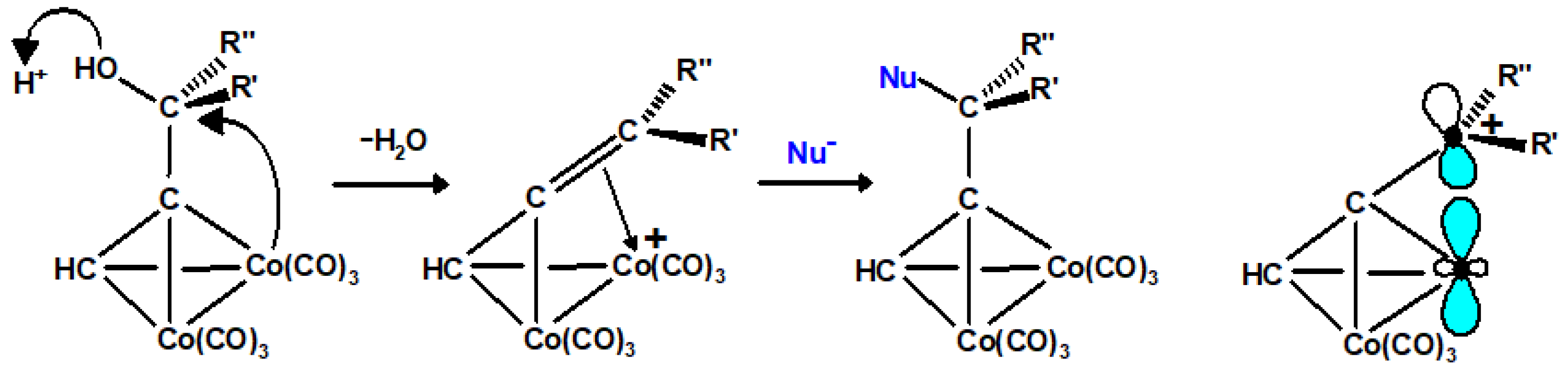
13. Metal Cluster Complexes of Terpenoids
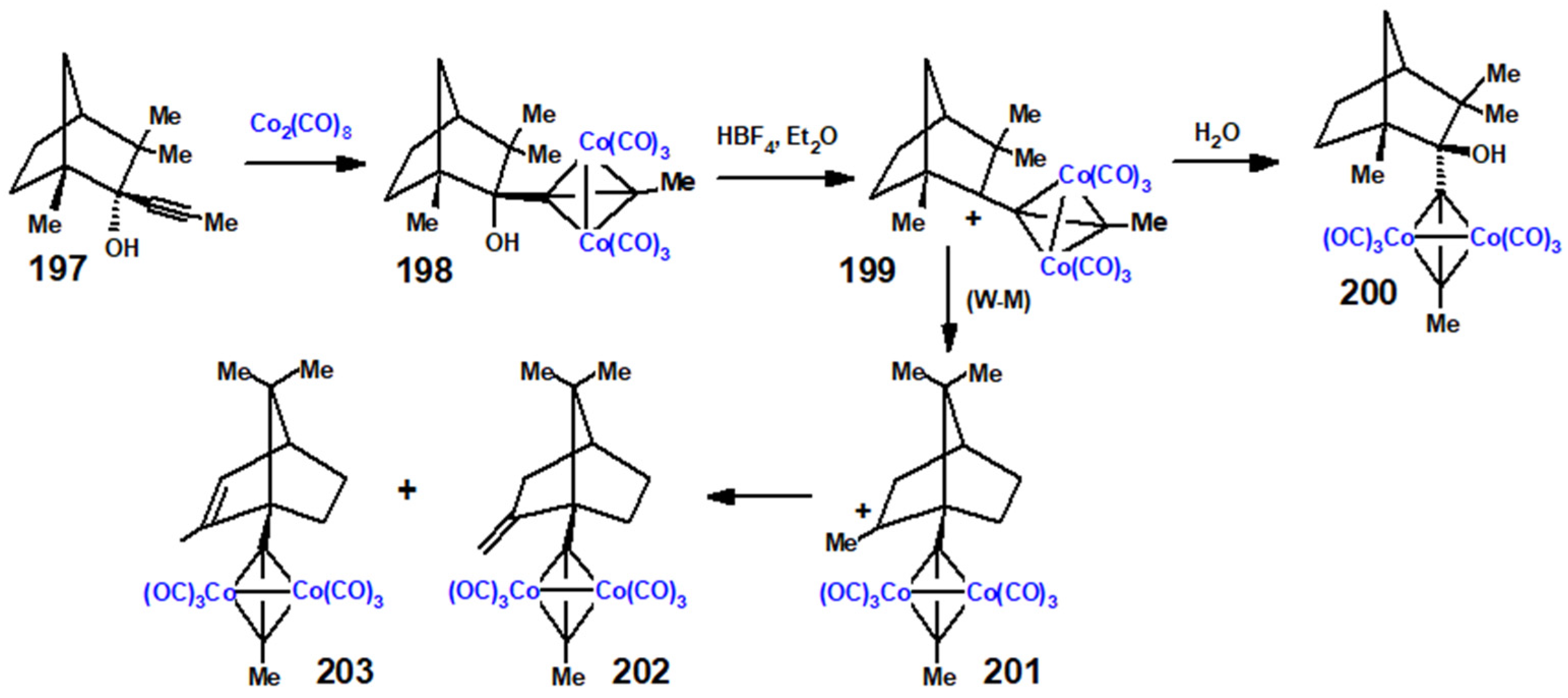
13.1. Cobalt Cluster-Stabilised Carbocations
13.2. X-ray Crystallographic Characterisation of Metal-Stabilised Bornyl Carbocations
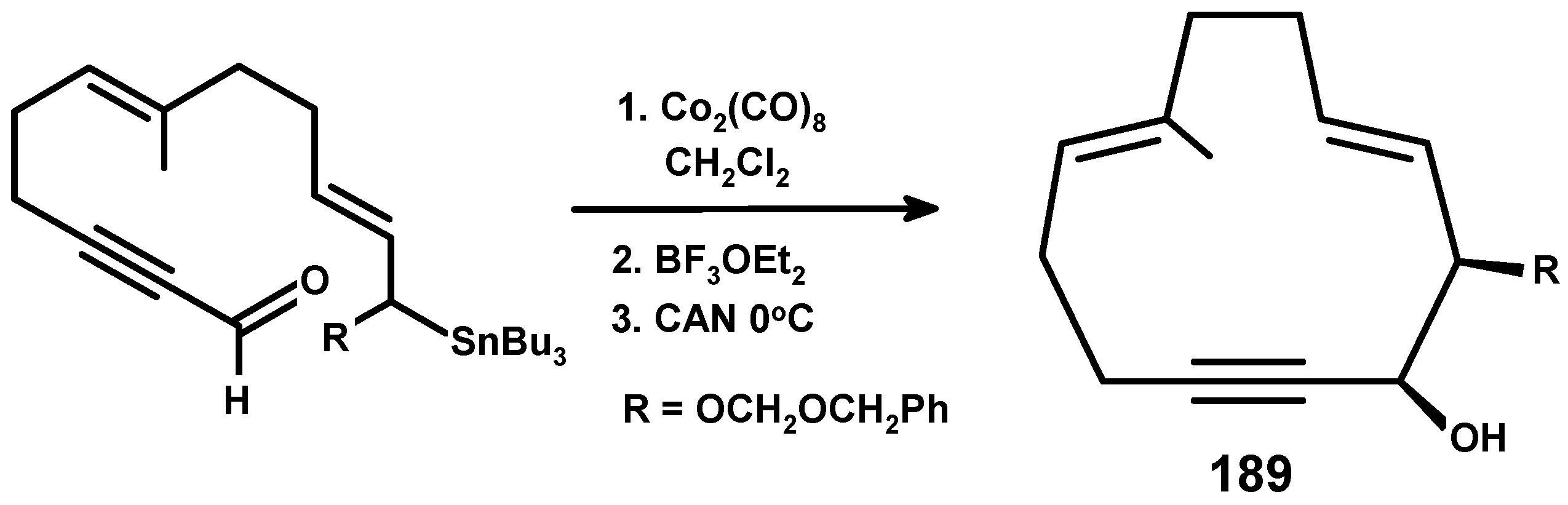
13.3. Metal Cluster Complexes in the Fenchyl, Verbenyl, Menthyl and Geranyl Series
14. Concluding Remarks
Funding
Acknowledgments
Conflicts of Interest
References
- Mann, J. Secondary Metabolism, 2nd ed.; Oxford University Press: Oxford, UK, 1987; Chapter 7; pp. 303–328. [Google Scholar]
- Stolle, A.; Ondruschka, B.; Hopf, H. Thermal rearrangements of monoterpenes and monoterpenoids. Helv. Chim. Acta 2009, 92, 1673–1719. [Google Scholar] [CrossRef]
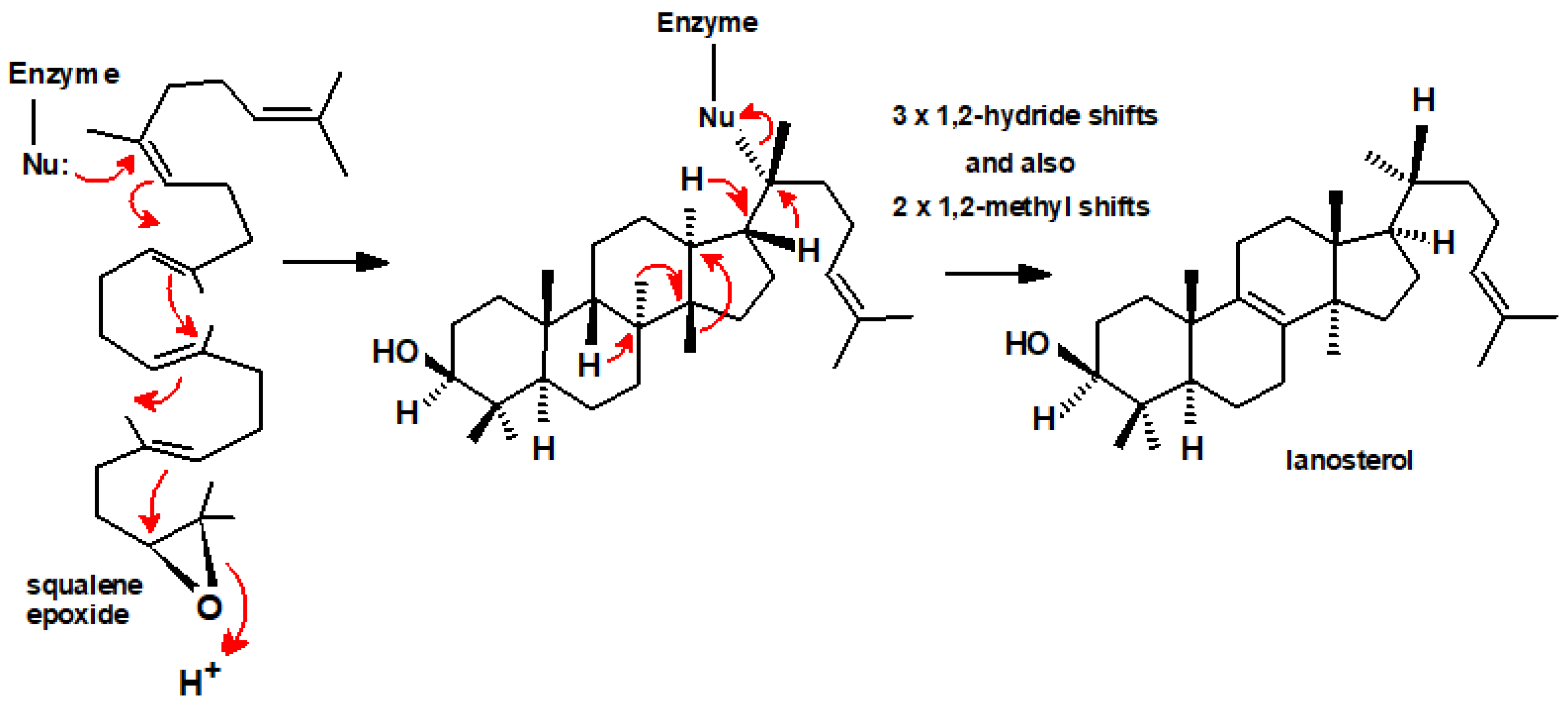
- Willett, J.D.; Sharpless, K.B.; Lord, K.E.; Van Tamelen, E.E.; Clayton, R.B. Squalene-2,3-oxide an intermediate in enzymatic conversion of squalene to lanosterol and cholesterol. J. Biol. Chem. 1967, 242, 4182–4191. [Google Scholar] [CrossRef] [PubMed]
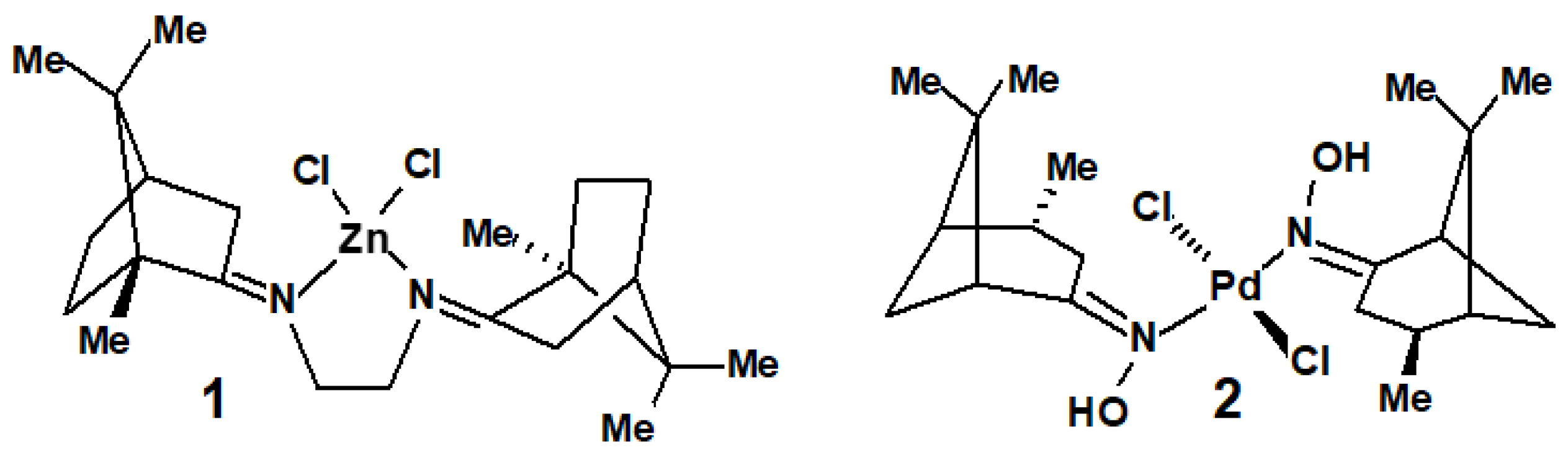
- Zalevskaya, O.A.; Gur’eva, Y.A.; Kutchin, A.V. Terpene ligands in the coordination chemistry: Synthesis of metal complexes, stereochemistry, catalytic properties and biological activity. Russ. Chem. Rev. 2019, 88, 979–1012. [Google Scholar] [CrossRef]
- Saucy, G.; Marbet, R.; Lindlar, H.; Isler, O. Über eine neue Synthese von Citral und verwandten Verbindungen. Helv. Chim. Acta 1959, 42, 1945–1955. [Google Scholar] [CrossRef]
- Zweig, J.E.; Kim, D.E.; Newhouse, T.R. Methods utilizing first-row transition metals in natural product total synthesis. Chem. Rev. 2017, 117, 11680–11752. [Google Scholar] [CrossRef] [PubMed]
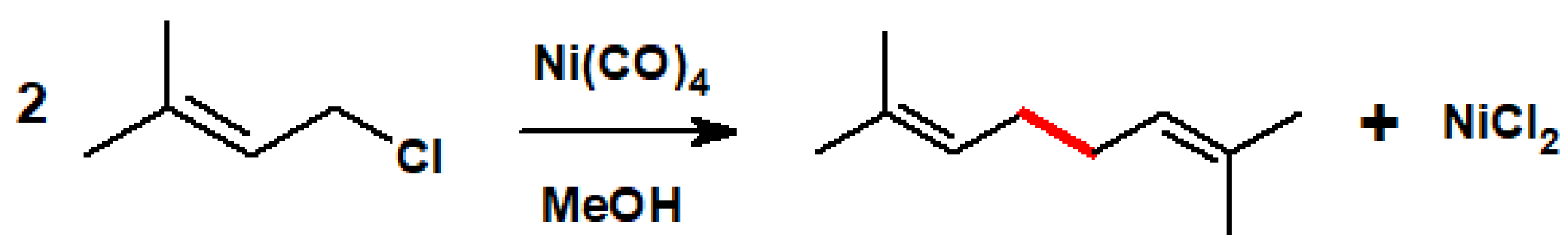
- I.G. Farbenindustrie Aktiengesellschaft. Procédé de condensation d’hydrocarbures halogénés. Belgian Patent 448,884. February 27 1943. Chem. Abstr. 1947, 41, 6576. [Google Scholar]
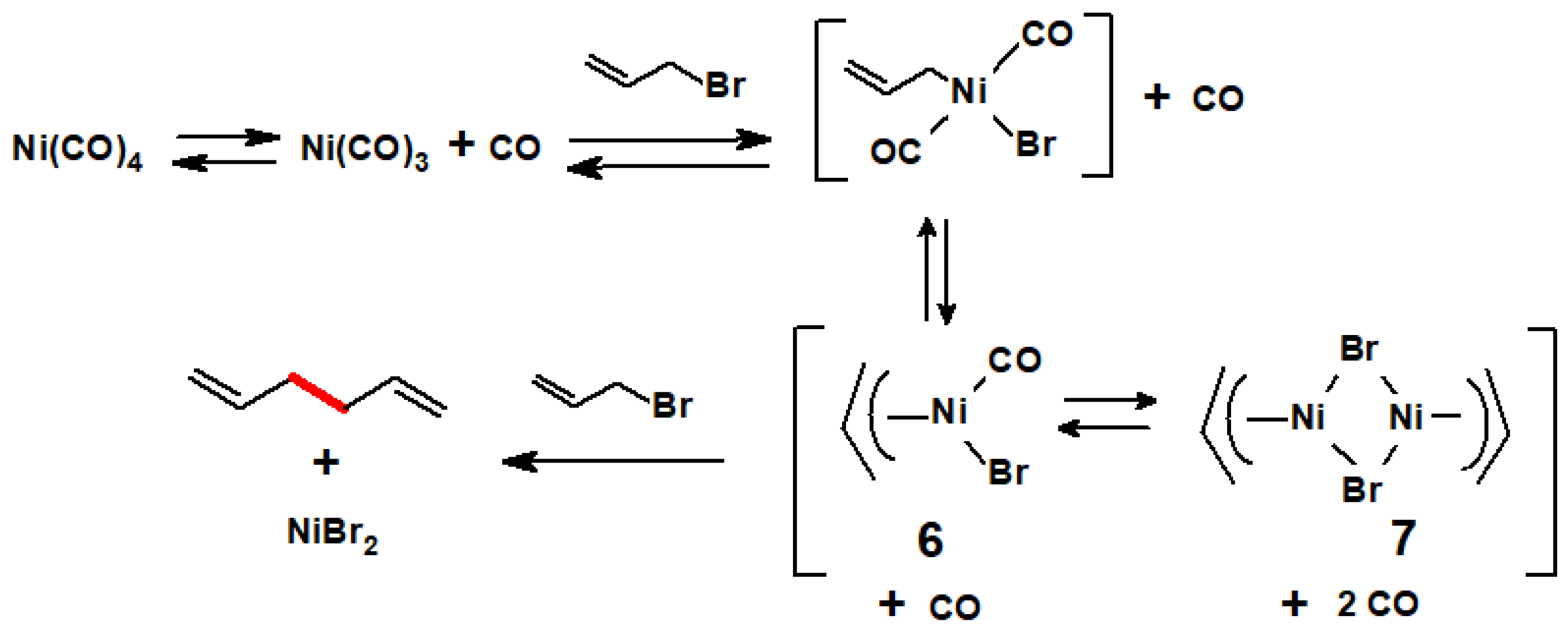
- Webb, I.D.; Borcherdt, G.T. Coupling of allylic halides by nickel carbonyl. J. Am. Chem. Soc. 1951, 73, 2654–2655. [Google Scholar] [CrossRef]
- Corey, E.J.; Hamanaka, E. A new synthetic approach to medium-size carbocyclic systems. J. Am. Chem. Soc. 1964, 85, 1641–1642. [Google Scholar] [CrossRef]
- Corey, E.J.; Semmelhack, M.F. Organonickel compounds as reagents for selective carbon-carbon bond formation between unlike groups. J. Am. Chem. Soc. 1967, 89, 2755–2757. [Google Scholar] [CrossRef]
- Corey, E.J.; Watt, E.W.K. The synthesis of large-ring 1,5-dienes by cyclization of allylic dibromides with nickel carbonyl. J. Am. Chem. Soc. 1967, 89, 2757–2758. [Google Scholar] [CrossRef]
- Corey, E.J.; Hamanaka, E. Total synthesis of humulene. J. Am. Chem. Soc. 1967, 89, 2758–2759. [Google Scholar] [CrossRef]
- Vig, O.P.; Ram, B.; Atwal, K.S.; Bari, S.S. Terpenoids 125. New synthesis of humulene (2,6,6,9-tetramethylcycloundeca-1,4,8-triene). Indian J. Chem. 1976, 14, 855–857. [Google Scholar] [CrossRef]
- Dauben, W.G.; Beasley, G.H.; Broadhurst Muller, B.; Peppard, D.J.; Pesnelle, P.; Suter, C. A synthesis of cembrene. A 14-membered ring diterpene. J. Am. Chem. Soc. 1974, 96, 4724–4726. [Google Scholar] [CrossRef]
- Drew, M.G.B.; Templeton, D.H.; Zalkin, A. The crystal and molecular structure of cembrene. Acta Cryst. 1969, B25, 261–267. [Google Scholar] [CrossRef]
- Sato, K.; Inoue, S.; Ota, S.; Fujita, Y. Reactions of π-allylic nickel(II) bromide with organic halides. A novel synthesis of monoterpenoid compounds. J. Org. Chem. 1972, 37, 462–466. [Google Scholar] [CrossRef]
- Wilke, G. Contributions to organo-nickel chemistry. Angew. Chem. Int. Ed. 1988, 27, 185–206. [Google Scholar] [CrossRef]
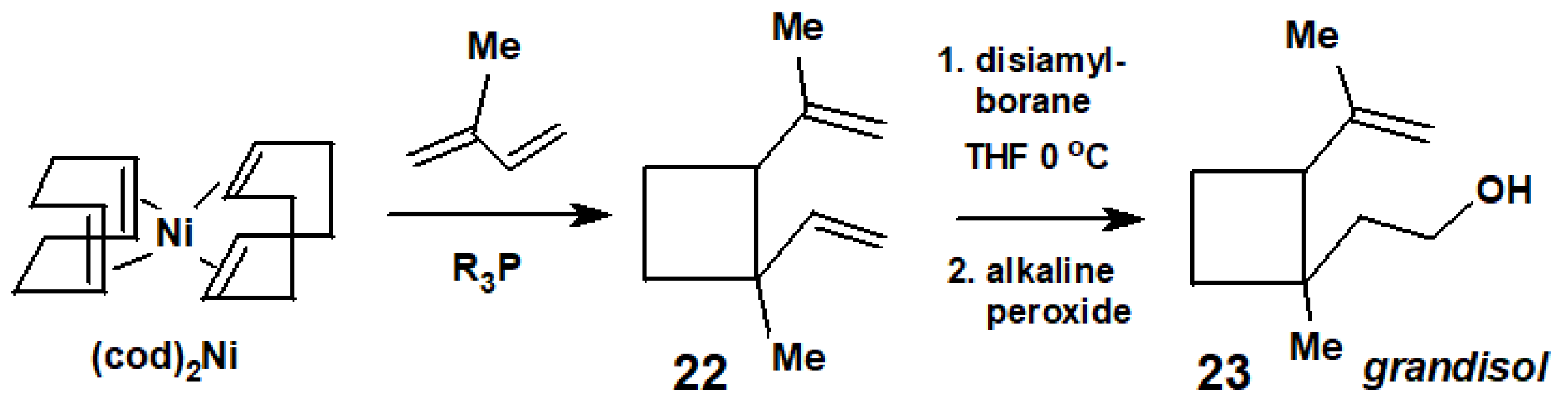
- Billups, W.E.; Cross, J.H.; Smith, C.V. Synthesis of (+/−)-grandisol. J. Am. Chem. Soc. 1973, 95, 3438–3439. [Google Scholar] [CrossRef]
- Baker, R.; Blackett, B.N.; Cookson, R.C. Reaction of dodecatrienylnickel; with allene—Formation of muscone. J. Chem. Soc. Chem. Commun. 1972, 802–803. [Google Scholar] [CrossRef]
- Baker, R.; Cookson, R.C.; Vinson, J.R. Insertion of isocyanides into bis-pi-allylnickel complexes and formation of (+/−)-muscone. J. Chem. Soc. Chem. Commun. 1974, 515–516. [Google Scholar] [CrossRef]
- Momose, D.-I.; Iguchi, K.; Sugiyama, T.; Yamada, Y. Reductive coupling of allylic halides by chlorotris(triphenylphosphine)cobalt(I). Tetrahedron Lett. 1983, 24, 921–924. [Google Scholar] [CrossRef]
- Hegedus, L.S.; Perry, R.J. Phosphinecarbonylnitrosylacylcobaltate complexes as acyl transfer reagents. Acylation of allylic halides, conjugated enones, and quinones. J. Org. Chem. 1985, 50, 4960–4995. [Google Scholar] [CrossRef]
- Hayakawa, Y.; Yokoyama, K.; Noyori, R. Carbon-carbon bond formation promoted by transition metal carbonyls. 20. Iron carbonyl promoted reaction of α,α′-dibromo ketones and aromatic olefins leading to 3-arylcyclopentanones. The [3+2) cycloaddition involving an allylic cation. J. Am. Chem. Soc. 1978, 100, 1791–1799. [Google Scholar] [CrossRef]
- Noyori, R.; Hayakawa, Y.; Takaya, H.; Murai, S.; Kobayashi, R.; Sonoda, N. Reaction of α,α′-dibromo ketones and iron carbonyls. Mechanistic aspects. J. Am. Chem. Soc. 1978, 100, 1759–1765. [Google Scholar] [CrossRef]
- Noyori, R.; Yokoyama, K.; Hayakawa, Y. Reaction of α,α′-dibromo ketones and aromatic olefins promoted by iron carbonyl. A cationic 3 + 2 → 5 cycloaddition. J. Am. Chem. Soc. 1973, 95, 2722–2723. [Google Scholar] [CrossRef]
- Noyori, R.; Hayakawa, Y. Natural product syntheses via the polybromo ketone-iron carbonyl reaction. Tetrahedron 1985, 41, 5879–5886. [Google Scholar] [CrossRef]
- Celebuski, J.; Rosenblum, M. Carbon-carbon bond formation employing organoiron reagents: Syntheses of lavandulol and red scale pheromone. Tetrahedron 1985, 41, 5741–5746. [Google Scholar] [CrossRef]
- Heck, R.F. The allylation of aromatic compounds with organopalladium salts. J. Am. Chem. Soc. 1968, 90, 5531–5534. [Google Scholar] [CrossRef]
- Dunne, K.; McQuillin, F.J. Complexes of terpenes with transition metals. Part III. Dimerisation by means of tetrakis(triphenylphosphine)palladium. J. Chem. Soc. C 1970, 2203–2206. [Google Scholar] [CrossRef]
- Neilan, J.P.; Laine, R.M.; Cortese, N.; Heck, R.F. Monoterpene syntheses via a palladium catalyzed isoprene dimerization. J. Org. Chem. 1976, 41, 3455–3460. [Google Scholar] [CrossRef]
- Patel, B.A.; Kao, L.-C.; Cortese, N.A.; Minkiewicz, J.V.; Heck, R.F. Palladium-catalyzed vinylation of conjugated dienes. J. Org. Chem. 1979, 44, 918–921. [Google Scholar] [CrossRef]
- Tsuji, J. Carbon-carbon bond formation via palladium complexes. Acc. Chem. Res. 1969, 2, 144–152. [Google Scholar] [CrossRef]
- Trost, B.M.; Weber, L. New synthetic reactions. Stereochemistry of allylic alkylation. J. Am. Chem. Soc. 1975, 97, 1611–1612. [Google Scholar] [CrossRef]
- Trost, B.M.; Weber, L.; Strege, P.; Fullerton, T.J.; Dietsche, T.J. Allylic alkylation: Nucleophilic attack on π-allylpalladium complexes. J. Am. Chem. Soc. 1978, 100, 3416–3426. [Google Scholar] [CrossRef]
- Ronson, T.O.; Taylor, R.J.K.; Fairlamb, I.J.S. Palladium-catalysed macrocyclisations in the total synthesis of natural products. Tetrahedron 2015, 71, 989–1009. [Google Scholar] [CrossRef]
- Trost, B.M. Cyclization via palladium-catalyzed allylic alkylations. Angew. Chem. Int. Ed. 1989, 28, 1173–1192. [Google Scholar] [CrossRef]
- Trost, B.M.; Min, C. Total synthesis of terpenes via palladium-catalysed cyclisation strategy. Nature Chem. 2020, 12, 568–573. [Google Scholar] [CrossRef]
- Trost, B.M.; Weber, L.; Strege, P.; Fullerton, T.J.; Dietsche, T.J. Allylic alkylation: Nature of the nucleophile and application to prenylation. J. Am. Chem. Soc. 1978, 100, 3426–3435. [Google Scholar] [CrossRef]
- Trost, B.M.; Malhotra, S.; Chan, W.H. Exercising regiocontrol in palladium-catalyzed asymmetric prenylations and geranylation: Unifying strategy toward flustramines A and B. J. Am. Chem. Soc. 2011, 133, 7328–7331. [Google Scholar] [CrossRef] [PubMed]
- Goliaszewski, A.; Schwartz, J. Specific allylic-allylic coupling procedures effected by ligand-induced elimination from di(allylic)palladium species. Tetrahedron 1985, 41, 5779–5789. [Google Scholar] [CrossRef]
- Kitagawa, Y.; Itoh, A.; Hashimoto, S.; Yamamoto, H.; Nozaki, H. Total synthesis of humulene. A stereoselective approach. J. Am. Chem. Soc. 1977, 99, 3864–3867. [Google Scholar] [CrossRef]
- Miyaura, N.; Suginome, H.; Suzuki, A. New stereo- and regiospecific synthesis of humulene by means of the palladium-catalyzed cyclization of haloalkenylboranes. Tetrahedron Lett. 1984, 25, 761–764. [Google Scholar] [CrossRef]
- Hu, T.; Corey, E.J. Short syntheses of (±)-δ-araneosene and humulene utilizing a combination of four-component assembly and palladium-mediated cyclization. Org. Lett. 2002, 4, 2441–2443. [Google Scholar] [CrossRef]
- Han, Y.T.; Kim, N.-J.; Jung, J.-W.; Yun, H.; Lee, S.; Suh, Y.-G. A versatile synthetic approach to grandisol monoterpene pheromone. Arch. Pharm. Res. 2011, 34, 1437–1442. [Google Scholar] [CrossRef]
- Manchand, P.S.; Wong, H.S.; Blount, J.F. Synthesis of vitamin A and related compounds via a π-allyl palladium complex. J. Org. Chem. 1978, 43, 4669–4774. [Google Scholar] [CrossRef]
- Negishi, E.; Valente, L.F.; Kobayashi, M. Palladium-catalyzed cross-coupling reaction of homoallylic or homopropargylic organozincs with alkenyl halides as a new selective route to 1,5-dienes and 1,5-enynes. J. Am. Chem. Soc. 1980, 102, 3298–3299. [Google Scholar] [CrossRef]
- Sum, F.W.; Weiler, L. Synthesis of mokupalide. J. Am. Chem. Soc. 1979, 101, 4401–4403. [Google Scholar] [CrossRef]
- Hart, D.W.; Blackburn, T.F.; Schwartz, J. Hydrozirconation. III Stereospecific and regioselective functionalization of alkylacetylenes via vinylzirconium(IV) intermediates. J. Am. Chem. Soc. 1975, 97, 679–680. [Google Scholar] [CrossRef]
- McMurry, J.E. Titanium-induced dicarbonyl-coupling reactions. Acc. Chem. Res. 1983, 16, 405–411. [Google Scholar] [CrossRef]
- McMurry, J.E.; Matz, J.R. Stereospecific synthesis of humulene by titanium-induced dicarbonyl coupling. Tetrahedron Lett. 1982, 23, 2723–2724. [Google Scholar] [CrossRef]
- McMurry, J.E.; Matz, J.R.; Kees, K.L.; Bock, P.A. Synthesis of flexibilene, a naturally-occurring 15-membered-ring diterpene. Tetrahedron Lett. 1982, 23, 1777–1780. [Google Scholar] [CrossRef]
- Kamat, V.P.; Hagiwara, H.; Katsumi, T.; Susuki, T.; Ando, M. Ring closing metathesis directed synthesis of (R)-(−)-muscone from (+)-citronellal. Tetrahedron 2000, 56, 4397–4403. [Google Scholar] [CrossRef]
- Barrero, A.F.; Herrador, M.M.; Quilez del Moral, J.F.; Arteaga, P.; Arteaga, J.F.; Diéguez, H.R.; Sánchez, E.M. Mild Ti(III)- and Mn/Zr(IV)-catalytic reductive coupling of allylic halides: Efficient synthesis of symmetric terpenes. J. Org. Chem. 2007, 72, 2988–2995. [Google Scholar] [CrossRef]
- Fürstner, A.; Hannen, P. Carene terpenoids by gold-catalyzed cycloisomerization reactions. Chem. Commun. 2004, 2546–2547. [Google Scholar] [CrossRef]
- Dunne, K.; McQuillin, F.J. Complexes of terpenes with transition metals. Part I. The reactions of cis-ocimene and trans-ocimene and of myrcene with palladium(II). J. Chem. Soc. C 1970, 2196–2200. [Google Scholar] [CrossRef]
- Takahashi, M.; Suzuki, H.; Moro-Oka, Y.; Ikawa, T. Regioselective hydroxylation of β-myrcene using palladium(II) complexes. Chem. Lett. 1979, 8, 53–56. [Google Scholar] [CrossRef]
- Takahashi, M.; Urata, H.; Suzuki, H.; Moro-Oka, Y.; Ikawa, T. Regioselective introduction of O-nucleophiles into β-myrcene and trans-ocimene using palladium(II) complexes. J. Organomet. Chem. 1984, 266, 327–336. [Google Scholar] [CrossRef]
- Dunne, K.; McQuillin, F.J. Complexes of terpenes with transition metals. Part II. Reactions of linalyl and nerolidyl acetates, limonene and the pinenes with disodium tetrachloropalladate(II). J. Chem. Soc. C 1970, 2200–2203. [Google Scholar] [CrossRef]
- McQuillin, F.J.; Parker, D.G. Complexing of terpenes with transition metals. Part IV. A comparison of the reactions of (+)-3,7-dimethylocta-1,6-diene with palladium(II) and mercury(II). J. Chem. Soc. Perkin Trans. 1 1974, 809–815. [Google Scholar] [CrossRef]
- Rienacker, R.; Ohloff, G. Optisch aktives β-Citronellol aus (+) or (−) pinane. Angew. Chemie 1961, 73, 240. [Google Scholar] [CrossRef]
- Strickler, H.; Ohloff, G.; Kováts, E. The thermal cyclisation of (−)-(R)-linalool. The structure of the plinoles and some derivates with an iridan framework. Helv. Chim. Acta 1967, 50, 759–797. [Google Scholar] [CrossRef]
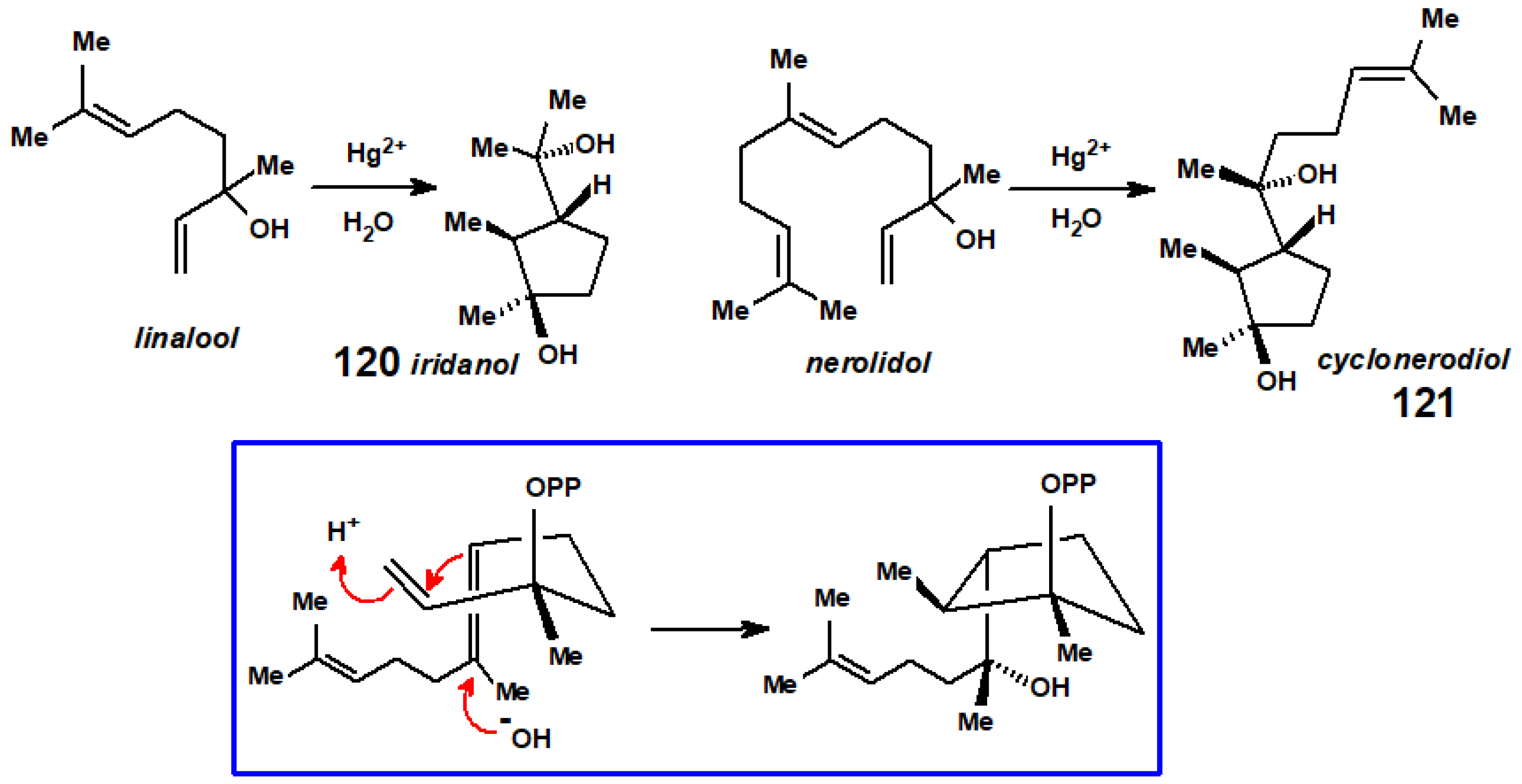
- Matsuki, Y.; Kodama, M.; Ito, S. Regioselective and stereoselective cyclization of linalool and nerolidol with mercuric salts. Synthesis of iridanols and cyclonerodiol. Tetrahedron Lett. 1979, 20, 2901–2904. [Google Scholar] [CrossRef]
- Cane, D.; Shiao, M.-S. Biosynthesis of cyclonerodiol. J. Am. Chem. Soc. 1978, 100, 3203–3207. [Google Scholar] [CrossRef]
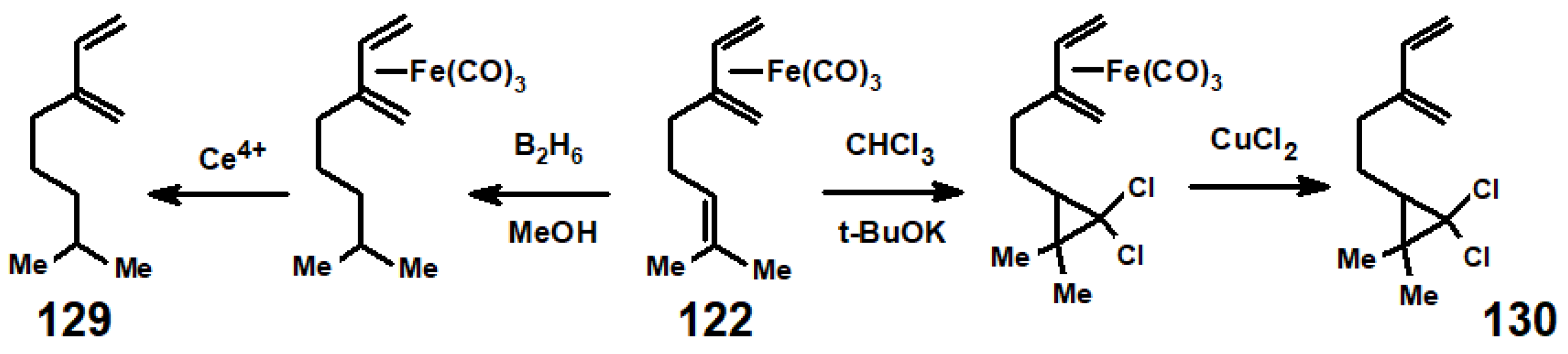
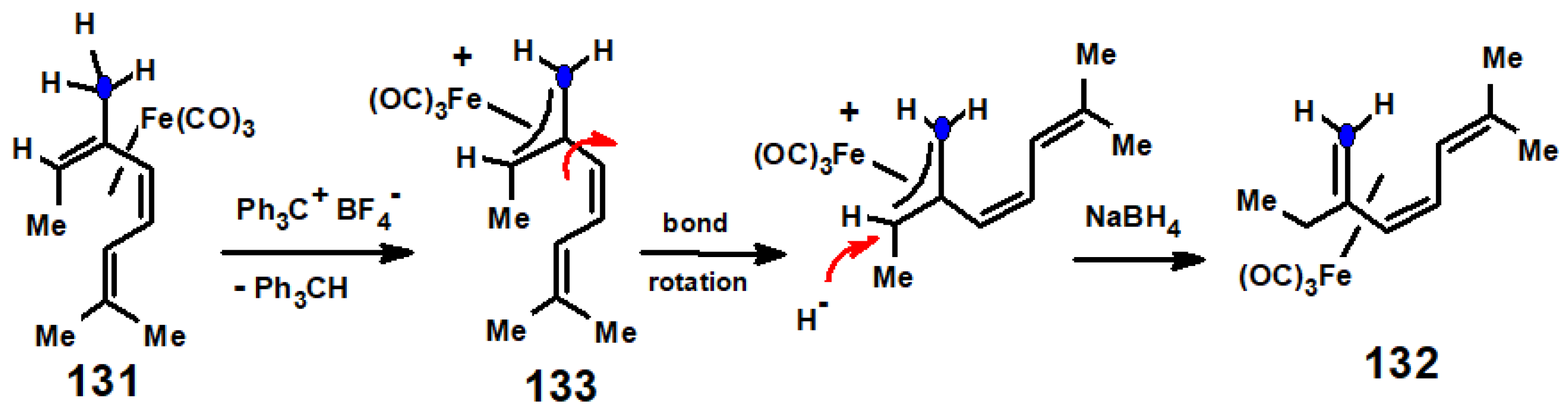
- Pearson, A.J. Protonation of tricarbonyliron complexes: Acid-catalyzed cyclization of tricarbonylmyrceneiron. Aust. J. Chem. 1976, 29, 1841–1844. [Google Scholar] [CrossRef]
- Birch, A.J.; Pearson, A.J. Friedel-Crafts chemistry of tricarbonyldieneiron complexes: Carbonylative annulation of tricarbonylmyrceneiron. J. Chem. Soc. Chem. Commun. 1976, 601–602. [Google Scholar] [CrossRef]
- Banthorpe, D.V.; Fitton, H.; Lewis, J. Isomerisation and addition reactions of some monoterpene-tricarbonyliron complexes. J. Chem. Soc. Perkin Trans. 1 1973, 2051–2057. [Google Scholar] [CrossRef]
- Taylor, G.A. Dichlorocarbene addition to tricarbonyliron complexes of polyenes. J. Chem. Soc. Perkin I 1979, 1716–1719. [Google Scholar] [CrossRef]
- Hubert, A.J.; Georis, A.; Warin, R.; Tessié, P. Base-catalysed prototropic rearrangement. Part 1. Comparison of the base-catalysed and the metal carbonyl-catalysed isomerisation of allyl ethers. J. Chem. Soc. Perkin II 1972, 366–370. [Google Scholar] [CrossRef]
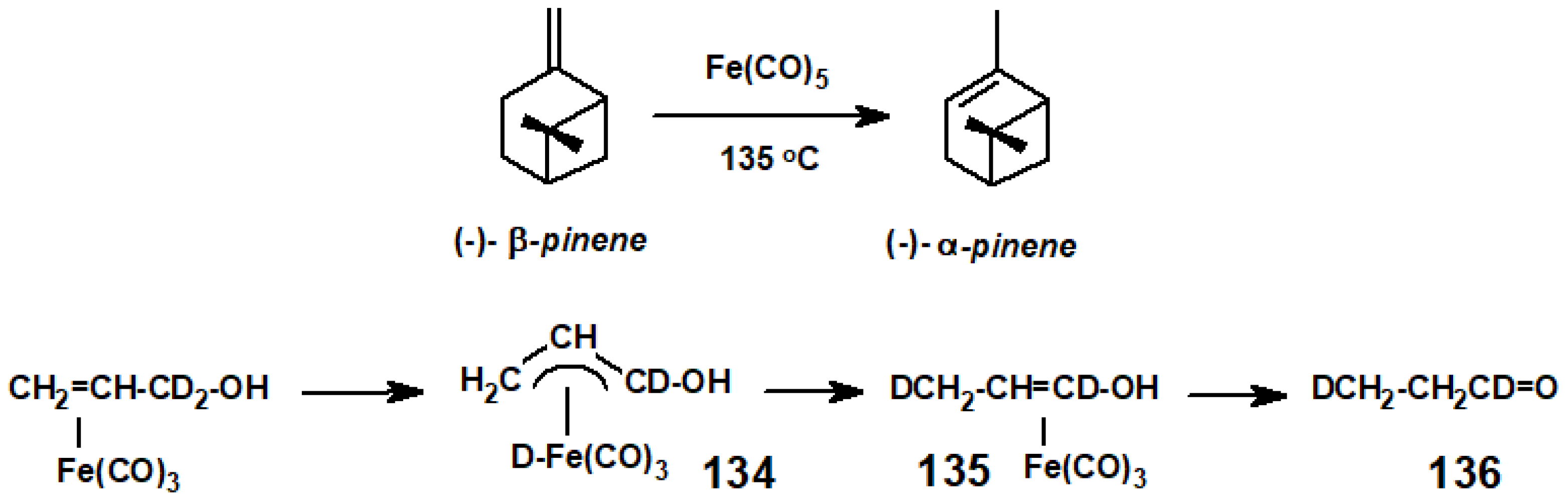
- Spanninger, P.A.; von Rosenberg, J.L. Isomerization of (−)-β-pinene to high optical purity (−)-α-pinene. J. Org. Chem. 1969, 34, 3658–3659. [Google Scholar] [CrossRef]
- Hendrix, W.Y.; Cowherd, F.G.; von Rosenberg, J.L. Mechanism of the rearrangement of allyl alcohol with iron carbonyl: Evidence for a π-allyl-hydroirontricarbonyl complex. Chem. Commun. 1968, 97–99. [Google Scholar] [CrossRef]
- Cowherd, F.G.; von Rosenberg, J.L. Mechanism of iron pentacarbonyl-catalyzed 1,3-hydrogen shifts. J. Am. Chem. Soc. 1969, 91, 2157–2158. [Google Scholar] [CrossRef]
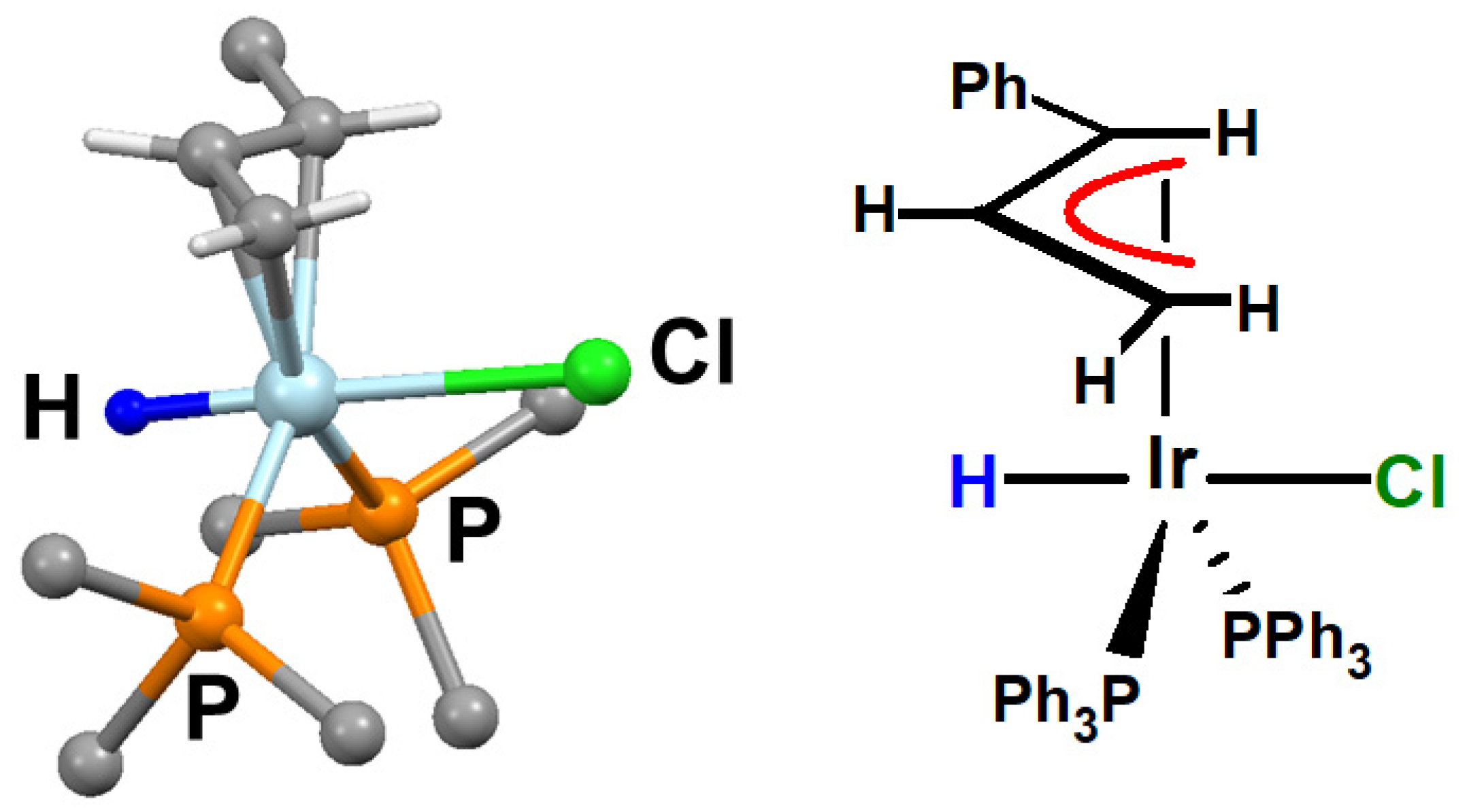
- Tulip, T.H.; Ibers, J.A. η3-Allyl metal hydride complexes. Oxidative addition of cyclopropane and olefin substrates to iridium(I) complexes. Structure of IrClH[η3-C3H4(1-C6H5)][P(C6H5)3]2. J. Am. Chem. Soc. 1979, 101, 4201–4211. [Google Scholar] [CrossRef]
- Grieco, P.A.; Takigawa, T.; Bongers, S.L.; Tanaka, H. Complete transfer of chirality in the [3.3]-sigmatropic rearrangement of allylic acetates catalyzed by palladium(II). Application to stereocontrolled syntheses of prostaglandins possessing either the C-15(S) or C-15(R) configuration. J. Am. Chem. Soc. 1980, 102, 7588–7590. [Google Scholar] [CrossRef]
- Trebellas, J.C.; Olechowski, J.R.; Jonassen, H.B. Metal olefin complexes: II. Palladium(II) and platinum(II) complexes of 1,2-divinylcyclohexane by the metal- catalyzed isomerization of 1,5-cyclodecadiene. J. Organomet. Chem. 1966, 6, 412–420. [Google Scholar] [CrossRef]
- Heimbach, P.; Molin, M. PdCl2-induced rearrangement of substituted cis,trans-cyclodeca-1,5-diene. J. Organomet. Chem. 1973, 49, 477–482. [Google Scholar] [CrossRef]
- Overman, L.E. Mercury(II) and palladium(II)-catalyzed [3,3]-sigmatropic rearrangements. Angew. Chem. Int. Ed. Engl. 1984, 23, 579–598. [Google Scholar] [CrossRef]
- Overman, L.E.; Renaldo, A.F. Palladium dichloride catalyzed Cope rearrangements of functionalized acyclic 1,5-dienes. Tetrahedron Lett. 1983, 24, 3757–3760. [Google Scholar] [CrossRef]
- Cuerva, J.M.; Gomez-Bengoa, E.; Mendez, M.; Echavarren, A.M. Synthesis of (±)-10-epi-elemol by a highly stereoselective intramolecular palladium-catalyzed coupling of an allylstannane with an allyl acetate. J. Org. Chem. 1997, 62, 7540–7541. [Google Scholar] [CrossRef]
- Brown, E.D.; Sam, T.W.; Sutherland, J.K.; Torre, A. Medium-ring 1,5-dienes. Part II. The radical and electrophile-induced cyclisation of germacra-1(10),4,7(11)-triene. J. Chem. Soc. Perkin Trans. 1 1975, 2326–2332. [Google Scholar] [CrossRef]
- Miyashita, A.; Takahashi, M.; Takaya, H. Reaction of bicyclo[1.1.0]butanes with platinum(II) complexes. Isolation and characterization of new platinacycle compounds. J. Am. Chem. Soc. 1981, 103, 5257–6259. [Google Scholar] [CrossRef]
- Victor, R.; Ben-Shoshan, R.; Sarel, S. Photochemically induced 1,5-insertion of carbon monoxide into vinylcyclopropane systems. A novel synthesis of cyclohexenones mediated by iron carbonyl. Tetrahedron Lett. 1970, 11, 4253–4256. [Google Scholar] [CrossRef]
- Aumann, R. Iron carbonyl complexes from vinylcyclopropane. J. Am. Chem. Soc. 1974, 96, 2631–2632. [Google Scholar] [CrossRef]
- Santelli-Rouvier, C.; Santelli, M.; Zahra, J.P. Carbonylation of (+)-2-carene induced by iron pentacarbonyl. Tetrahedron Lett. 1985, 26, 1213–1216. [Google Scholar] [CrossRef]
- Stockis, A.; Weissberger, E. Metal assisted ring expansions. The stereospecific expansion of pinene induced by Fe(CO)5. J. Am. Chem. Soc. 1975, 97, 4288–4292. [Google Scholar] [CrossRef]
- Jenny, T.A.; Ma, L. Synthetic use of a σ-alkyl-π-allyl iron complex obtained by stereospecific ring opening of α-pinene. Tetrahedron Lett. 1991, 32, 6101–6104. [Google Scholar] [CrossRef]
- Jenny, T.A.; Huber, V.; Ma, L.; Raemy, J.; Zeller, D.; Stoeckli-Evans, H. Synthesis, X-ray structure, and reactivity of phosphine-substituted iron carbonyl complexes containing σ-alkyl—π-allyl ligands. Organometallics 2002, 21, 2504–2513. [Google Scholar] [CrossRef]
- De Vekki, D.A.; Uvarov, V.I.; Bel’skii, V.K.; Skvortsov, N.K. Reaction of platinum complexes with (+)-α-pinene and (+)-limonene. Synthesis, molecular structure, and catalytic activity of dichloro(η4-[p-mentha-1,8{9}-diene])platinum(II). Russ. J. Gen. Chem. 2006, 76, 1288–1294. [Google Scholar] [CrossRef]
- Costes, P.; Weckesser, J.; Dechy-Cabaret, O.; Urrutigoïty, M.; Kalck, P. Rh-and Pt-catalyzed cycloisomerization of enynes derived from terpenes. Appl. Organomet. Chem. 2008, 22, 211–214. [Google Scholar] [CrossRef]
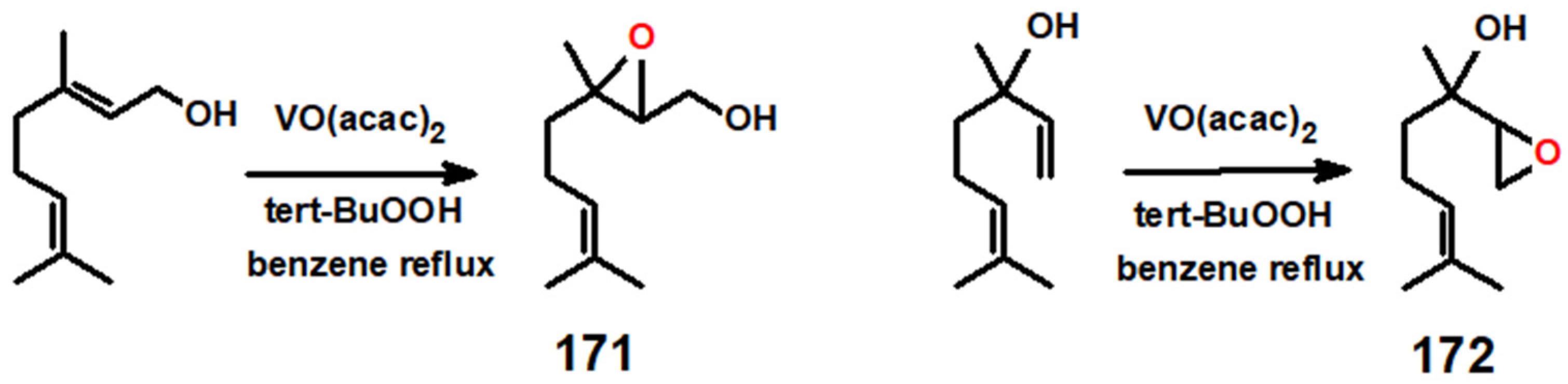
- Sharpless, K.B.; Michaelson, R.C. High stereoselectivities in the transition metal catalyzed epoxidations of olefinic alcohols by tert-butyl hydroperoxide. J. Am. Chem. Soc. 1973, 95, 6136–6137. [Google Scholar] [CrossRef]
- Pereira, C.; Silva, J.F.; Pereira, A.M.; Araújo, J.P.; Blanco, G.; Pintado, J.M.; Freire, C. [VO(acac)2] hybrid catalyst: From complex immobilization onto silica nanoparticles to catalytic application in the epoxidation of geraniol. Catal. Sci. Technol. 2011, 1, 784–793. [Google Scholar] [CrossRef]
- Yur′ev, V.P.; Gailyunas, I.A.; Isaeva, Z.G.; Tolstikov, G.A. Stereoselective epoxidation of bicyclic monoterpenes with tert-amyl hydroperoxide in presence of molybdenum compounds. Bull Acad. Sci. USSR 1974, 23, 885–886. [Google Scholar] [CrossRef]
- Curlat, S. Recent studies of (+)-3-carene transformations with the retention of the native framework. Chem. J. Mold. 2019, 14, 32–55. [Google Scholar] [CrossRef]
- Cradwick, M.E.; Cradwick, P.D.; Sim, G.A. Sesquiterpenoids. Part XV. Conformation of humulene: X-ray analysis of the crystal structure of humulene diepoxide. J. Chem. Soc. Perkin Trans. 2 1973, 4040407. [Google Scholar] [CrossRef]
- Sattar, A.; Forrester, J.; Moir, M.; Roberts, J.S.; Parker, W. Regiospecific functionalisation of humulene. Tetrahedron Lett. 1976, 17, 1403–1406. [Google Scholar] [CrossRef]
- Sharpless, K.B.; Umbreit, M.A.; Nieh, M.T.; Flood, T.C. Low valent tungsten halides. A new class of reagents for deoxygenation of organic molecules. J. Am. Chem. Soc. 1972, 94, 6538–6540. [Google Scholar] [CrossRef]
- Mlokiewicz, J.A. Chemistry of Humulene Epoxides. Ph.D. Thesis, University of Stirling, Scotland, UK, 1979; p. 101. [Google Scholar]
- Churchill, M.R. Transition metal complexes of azulene and related ligands. Prog. Inorg. Chem. 1970, 11, 53–98. [Google Scholar] [CrossRef]
- Cotton, F.A.; Hanson, B.E.; Kolb, J.R.; Lahuerta, P.; Stanley, G.G.; Stults, B.R.; White, A.W. The carbonyl scrambling processes in the isomeric pentacarbonylguaiazuleneiron and homologous ruthenium molecules; a novel mechanism for the internuclear processes. J. Am. Chem. Soc. 1977, 99, 3673–3683. [Google Scholar] [CrossRef]
- Nagashima, H.; Fukahori, T.; Nobata, M.; Suzuki, A.; Nakazawa, M.; Itoh, K. Haptotropic rearrangement between two isomers of (μ2:η3:η5-guaiazulene)Fe2(CO)5 revisited: Both thermal and photochemical processes induce haptotropic interconversion. Organometallics 1994, 13, 3427–3433. [Google Scholar] [CrossRef]
- Matsubara, K.; Oda, T.; Nagashima, H. Diruthenium carbonyl complexes bound to guaiazulene: Preparation and thermally reversible photoisomerisation studies of phosphine and phosphite derivatives of (μ2:η3:η5-guaiazulene)Ru2(CO)5 and iron homologues. Organometallics 2001, 20, 881–892. [Google Scholar] [CrossRef]
- Vollgraff, T.; Doppiu, A.; Sundermeyer, J. Dihydroguaiazulenide complexes and catalysts of Group 8-12 transition metals: Ligands from renewable feedstock replace, even outmatch petrochemical bases cyclopentadienyl chemistry. Chem. Eur. J. 2023, 30, e202302994. [Google Scholar] [CrossRef]
- Balduzzi, S.; Müller-Bunz, H.; McGlinchey, M.J. A convenient synthetic route to benz[cd]azulenes: Versatile ligands with the potential to bind to metal in an η5, η6, or η7 fashion. Chem. Eur. J. 2004, 10, 5398–5405. [Google Scholar] [CrossRef]
- Teobald, B.J. The Nicholas reaction: The use of dicobalt hexacarbonyl-stabilised propargylic cations in synthesis. Tetrahedron 2002, 58, 4133–4170. [Google Scholar] [CrossRef]
- Tyrrell, E.; Millet, J.; Tesfa, K.H.; Williams, N.; Mann, A.; Tillett, C.; Muller, C. A study into asymmetric Nicholas cyclisation reactions. Tetrahedron 2007, 63, 12769–122778. [Google Scholar] [CrossRef]
- Marshall, J.A.; Gung, B.W. Stereoselective cyclization of alpha-alkoxy allylstannane alkynals and their Co-complexes—A new route to cyclododecyne-1,2-diol derivatives. Tetrahedron Lett. 1989, 30, 309–312. [Google Scholar] [CrossRef]
- Morris, D.G.; Shepherd, A.G.; Walker, M.F.; Jemison, R.W. Anisochrony induced by transmission across a triple bond. Aust. J. Chem. 1982, 35, 1061–1064. [Google Scholar] [CrossRef]
- D’Agostino, M.F.; Frampton, C.S.; McGlinchey, M.J. Diastereoselective ligand and vertex substitutions in bimetallic bridged alkyne clusters: X-ray crystal structure of (µ2-endo-2-propynylborneol)hexacarbonyldicobalt. Organometallics 1990, 9, 2972–2984. [Google Scholar] [CrossRef]
- D’Agostino, M.F.; Frampton, C.S.; McGlinchey, M.J. An NMR spectroscopic and EHMO investigation of cationic mixed metal clusters: X-ray crystal structure of (1,7,7-trimethyl-µ2-2-propynylnorbornadiene)-bis(cyclopentadienyl)tetracarbonyldimolybdenum. J. Organomet. Chem. 1990, 394, 145–166. [Google Scholar] [CrossRef]
- Gruselle, M.; El Hafa, H.; Nikolski, M.; Jaouen, G.; Vaissermann, J.; Li, L.; McGlinchey, M.J. Metal cluster stabilized 2-norbornyl cations: A synthetic X-ray crystallographic and EHMO study. Organometallics 1993, 12, 4917–4925. [Google Scholar] [CrossRef]
- Gruselle, M. 2-Bornyl cations stabilized by metal-clusters—Synthetic and X-ray structural and quantum-chemical studies. Russ. Chem. Bull. 1994, 43, 538–542. [Google Scholar] [CrossRef]
- Kondratenko, M.; El Hafa, H.; Gruselle, M.; Vaissermann, J.; Jaouen, G.; McGlinchey, M.J. A synthetic and X-ray crystallographic study of cobalt and molybdenum complexes of the 2-fenchyl cation: Metal-mediated Wagner-Meerwein rearrangements. J. Am. Chem. Soc. 1995, 117, 6907–6913. [Google Scholar] [CrossRef]
- El Hafa, H.; Cordier, C.; Gruselle, M.; Besace, Y.; Jaouen, G.; McGlinchey, M.J. NMR study of the dynamic behavior of [2-propynylbornyl]Mo2(CO)4Cp2]+BF4−: Nonfluxional molybdenum-cobalt clusters as the key to understanding the mechanism of the formation of metal-stabilized cations. Organometallics 1994, 13, 5149–5156. [Google Scholar] [CrossRef]
- Moore, A.; Ostermann, J.; Müller-Bunz, H.; Ortin, Y.; McGlinchey, M.J. Metal-stabilized carbocations derived from monoterpenes: Dicobalt hexacarbonyl complexes of alkynyl-verbenols. Tetrahedron 2016, 72, 4186–4193. [Google Scholar] [CrossRef]
- Cadierno, V.; Conejero, S.; Gamasa, M.P.; Gimeno, J. Indenyl-ruthenium(II) allenylidene complexes containing terpenic substituents as precursors of optically active terminal alkynes: Scope and limitations. Dalton Trans. 2003, 3060–3066. [Google Scholar] [CrossRef]
- Phillipe, J.; Capmau, M.-L.; Chodkiewicz, W. Stereochemistry of addition of organometallic derivatives to terpenoid ketones. Bull. Soc. Chim. Fr. 1971, 2248–2255. [Google Scholar]
- Moore, A.; Ortin, Y.; Müller-Bunz, H.; McGlinchey, M.J. Alkynyl-dicobalt hexacarbonyl complexes of menthyl cations: Isolobal substitution of [Co(CO)3]+ by Fe(CO)3 as a structural model. Organometallics 2010, 29, 4882–4892. [Google Scholar] [CrossRef]
- Dunn, J.A.; Britten, J.F.; Daran, J.-C.; McGlinchey, M.J. Toward an understanding of the mechanism of metal exchange in (propargyl alcohol)Co2(CO)6 complexes: Syntheses and structures of (η5-C5Ph2R2-C≡C-TMS)(Fe(CO)2(µ-H)Co2(CO)6, R = Ph or Et. Organometallics 2001, 20, 4690–4694. [Google Scholar] [CrossRef]
- Moore, A.; Ostermann, J.; Ortin, Y.; McGlinchey, M.J. Organometallic derivatives of natural products: Dicobalt hexacarbonyl complexes of geranyl-alkynes. New J. Chem. 2016, 40, 7881–7888. [Google Scholar] [CrossRef]
- Dzhemilev, U.M.; Dzhelmileva, L.U.; D′yakonov, V.A. Metallacomplex catalysis in the synthesis of regular isoprenoids. Russ. Chem. Bull. 2023, 72, 404–414. [Google Scholar] [CrossRef]
- Dickmu, G.C.; Smoliakova, I.P. Preparation and characterization of cyclopalladated complexes derived from L-(−)-fenchone. J. Organomet. Chem. 2014, 772–773, 42–48. [Google Scholar] [CrossRef]
- Lu, K.; Zhu, J.; Wang, J.; Wa, Q.; Guo, Z.; Xiong, W.; Gao, Y.; Lei, R.; Cheng, B.; Wang, X. Syntheses of η1-alkyl-η3-allyl-η5-cyclopentadienyl cobalt(III) complexes and their use in low-temperature atomic layer deposition of cobalt-containing thin films. Chem. Eur. J. 2023, 29, e202203656. [Google Scholar] [CrossRef] [PubMed]
- Silva de Andrade, R.; Sales, K.A.; Abreu, L.S.; Campos, V.R.; Martins dos Santos, F., Jr.; Braz-Filho, R.; Scotti, M.T.; Tavares, J.F.; Sobral da Silva, M. Structure revision of the sesquiterpene Nordine based on NMR spectroscopic analysis and X-ray crystallography. J. Nat. Prod. 2020, 85, 2480–2483. [Google Scholar] [CrossRef] [PubMed]
- McPhail, A.T.; Sim, G.A. Sesquiterpenoids. Part IV. The stereochemistry of humulene: X-ray analysis of the humulene-silver nitrate adduct. J. Chem. Soc. B 1966, 112–120. [Google Scholar] [CrossRef]
- Zigon, N.; Hoshino, M.; Yoshioka, S.; Inokuma, Y.; Fujita, M. Where is the oxygen? Structural analysis of α-humulene oxidation products by the crystalline sponge method. Angew. Chem. Int. Ed. 2015, 54, 9033–9037. [Google Scholar] [CrossRef] [PubMed]
- De Poel, W.; Tinnemans, P.T.; Duchateau, A.L.L.; Honing, M.; Rutjes, F.P.J.T.; Vlieg, E.; de Gelder, R. Racemic and Enantiopure Camphene and Pinene Studied by the Crystalline Sponge Method. Cryst. Growth Des. 2018, 18, 126–132. [Google Scholar] [CrossRef]
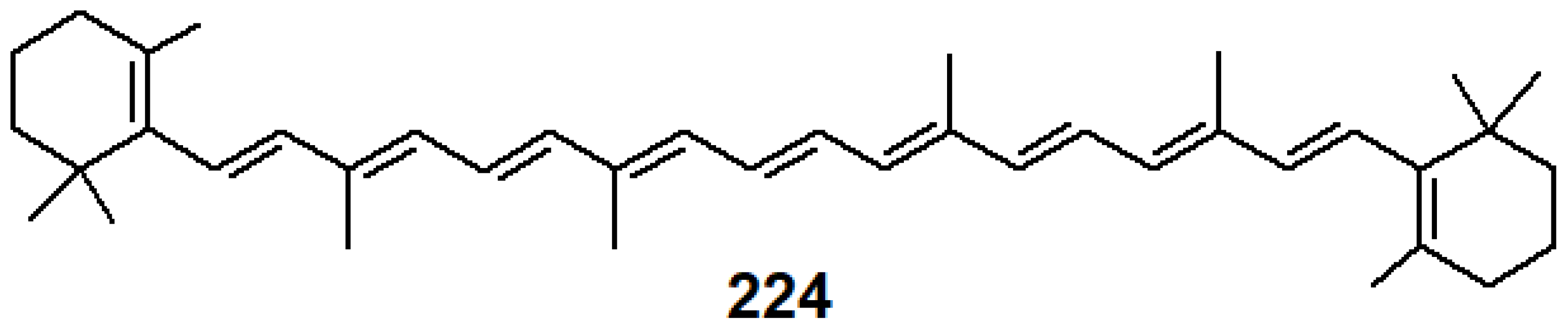
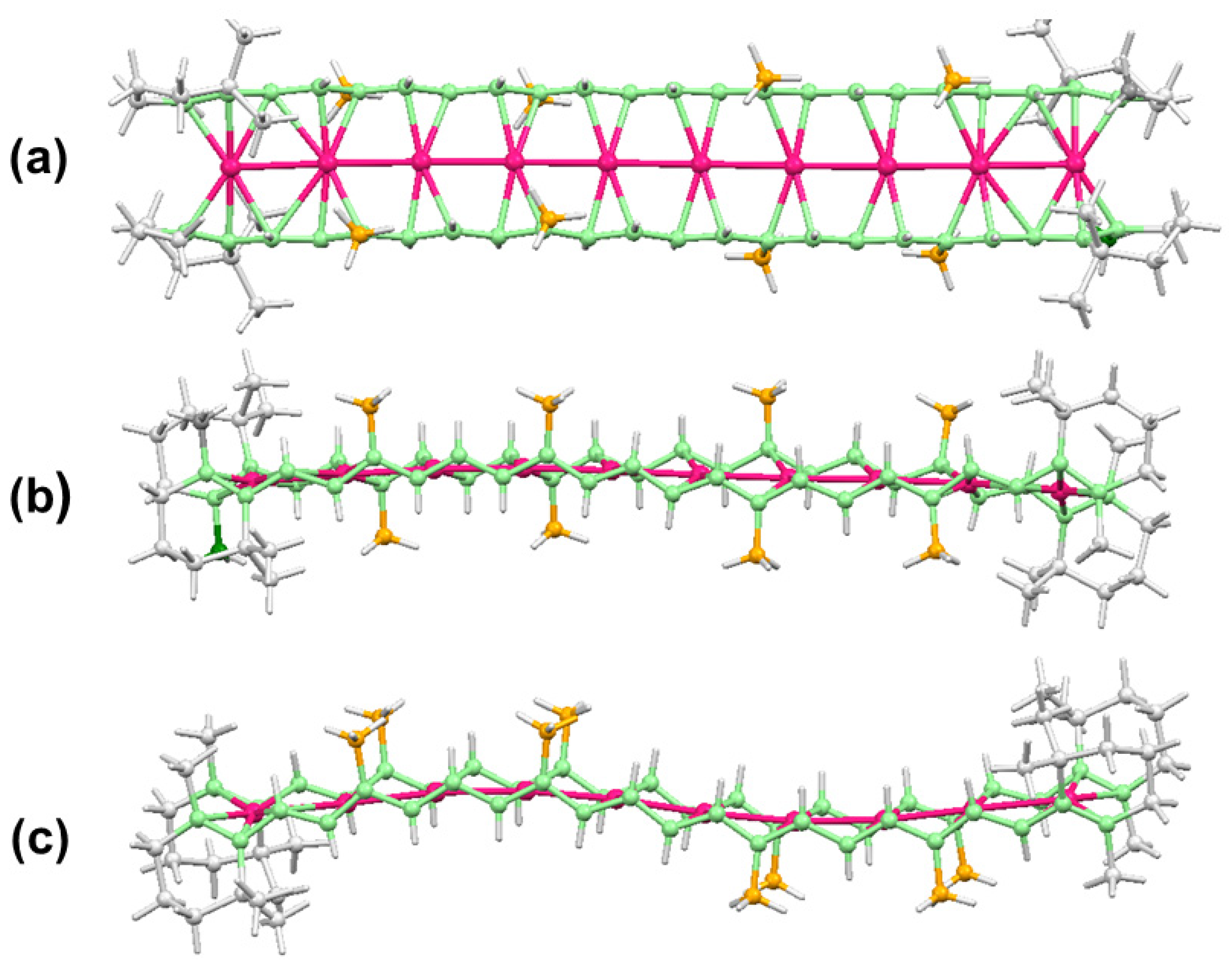
- Horiuchi, S.; Tachibana, Y.; Yamashita, M.; Yamamoto, K.; Masai, K.; Takase, K.; Matsutani, T.; Kawamata, S.; Kurashige, Y.; Yanai, T.; et al. Multinuclear metal-binding ability of a carotene. Nat. Commun. 2015, 6, 6742. [Google Scholar] [CrossRef] [PubMed]

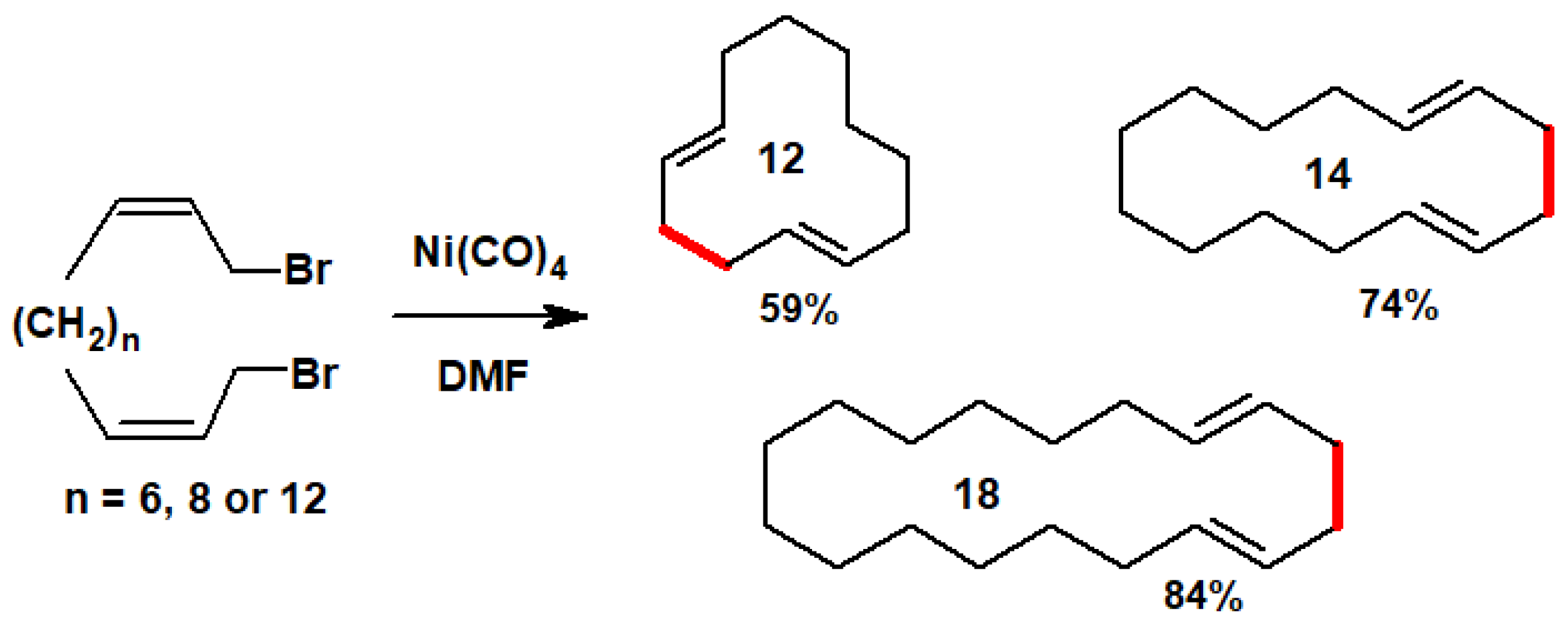

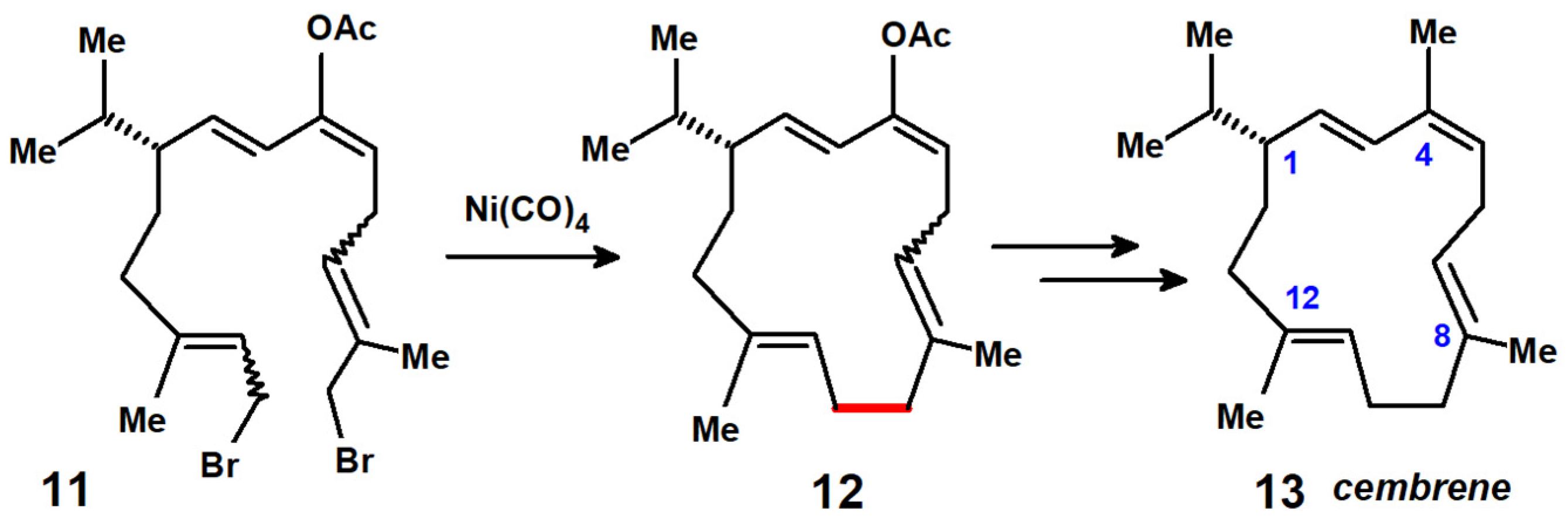
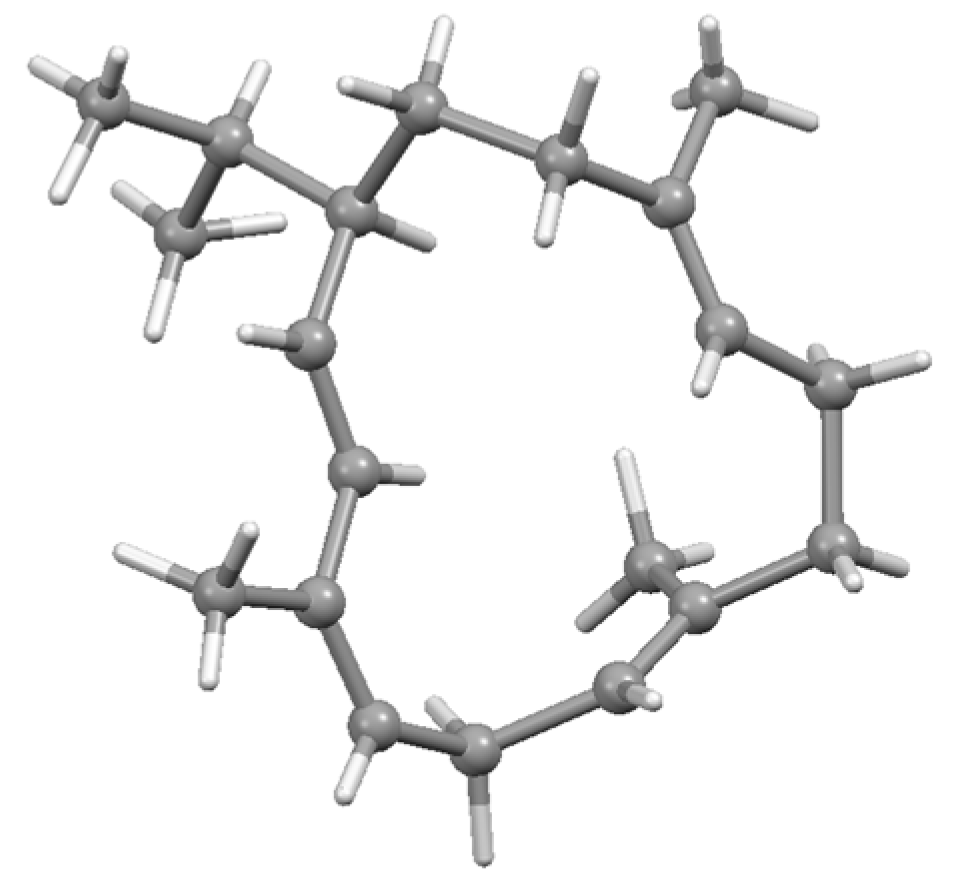
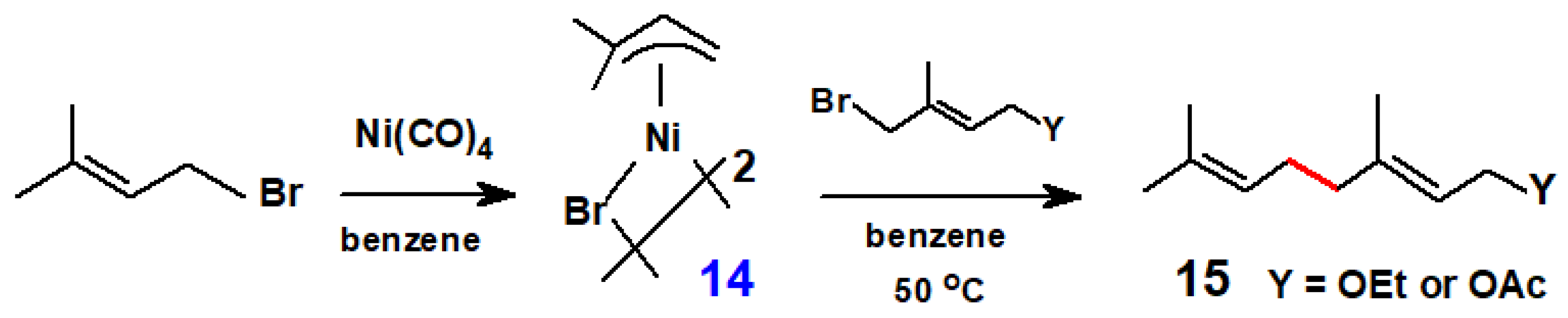



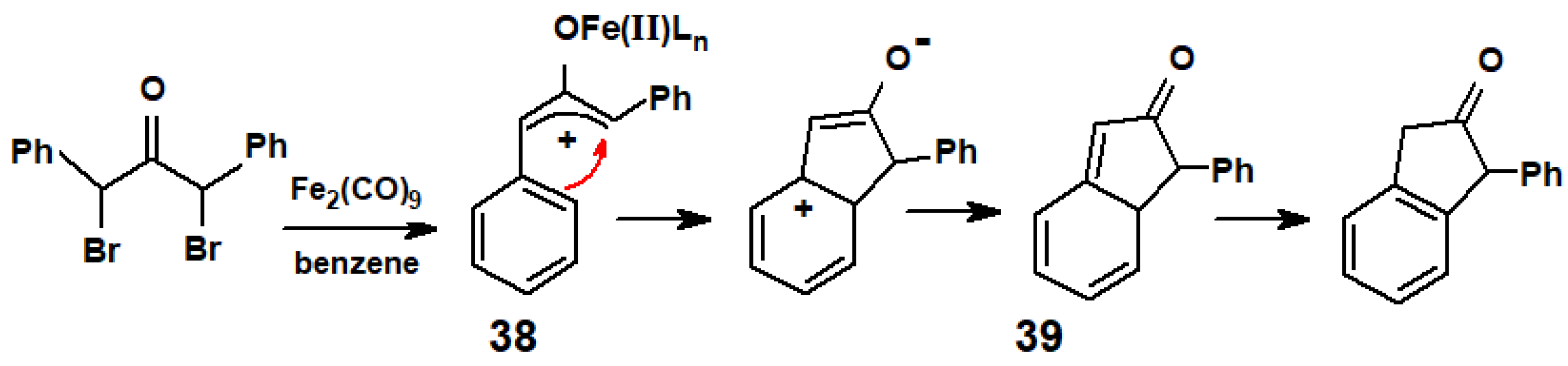

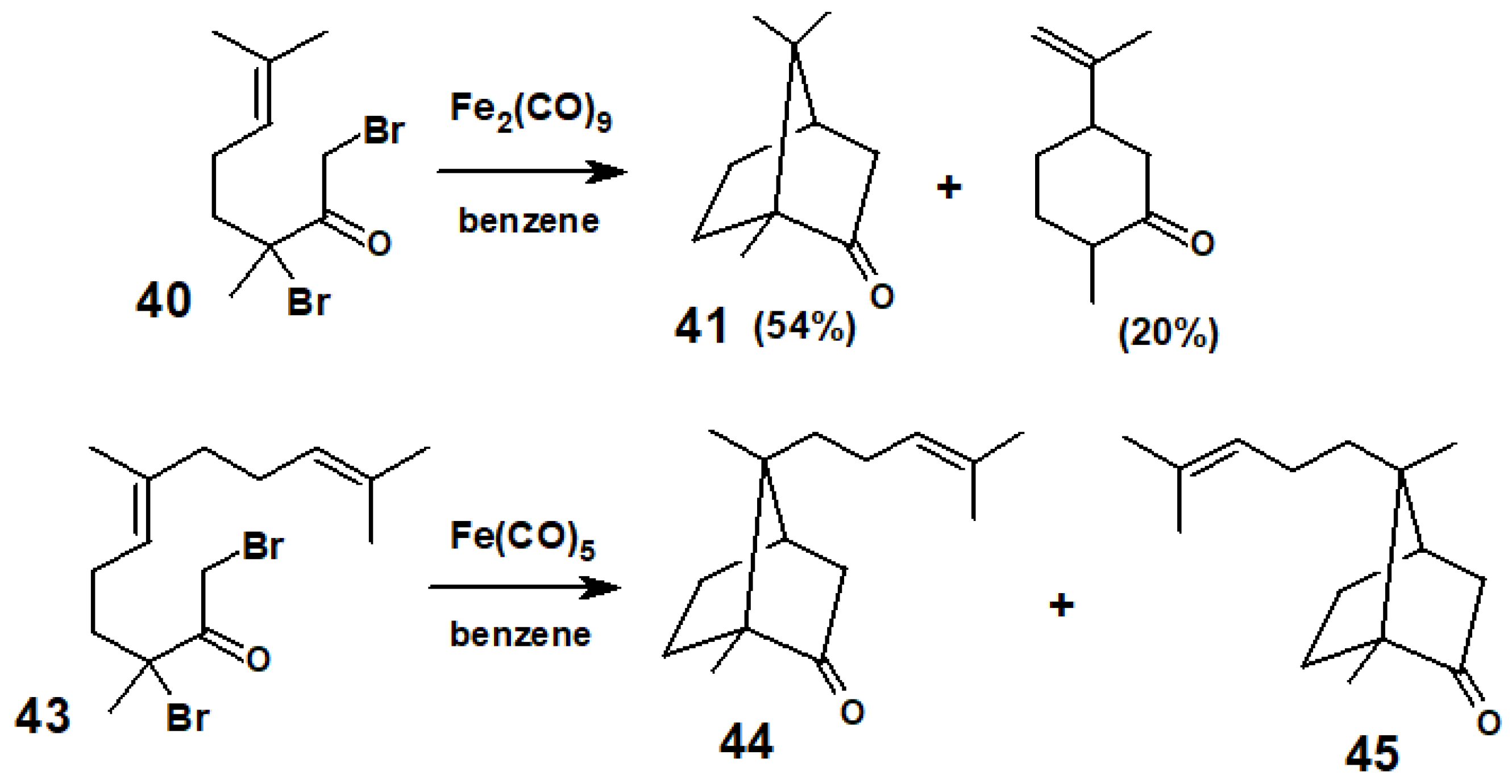
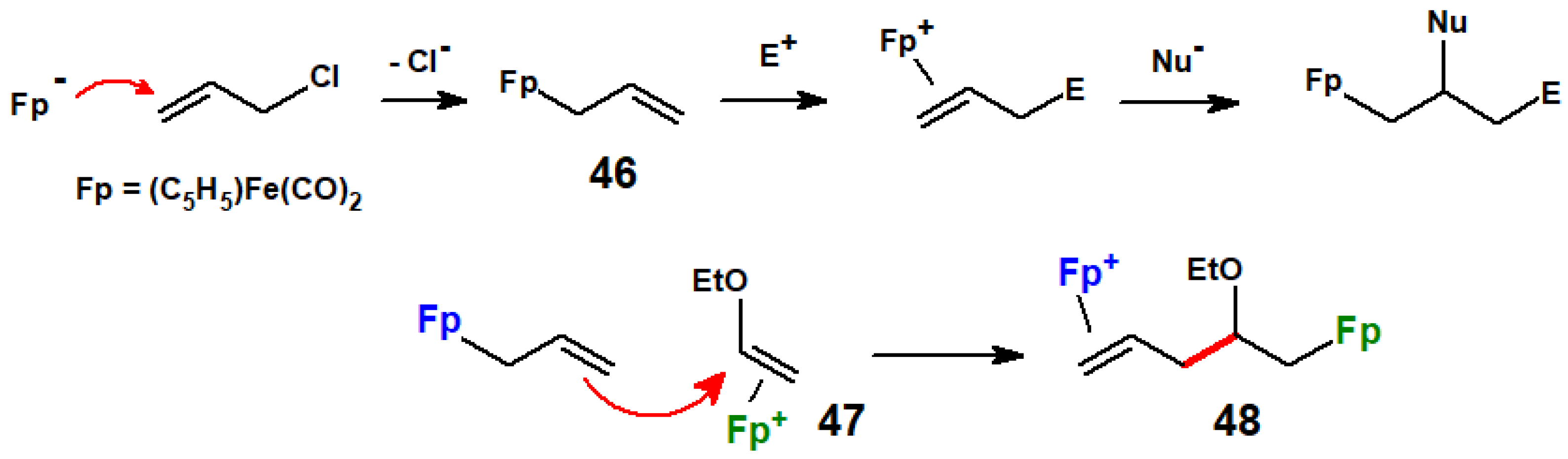
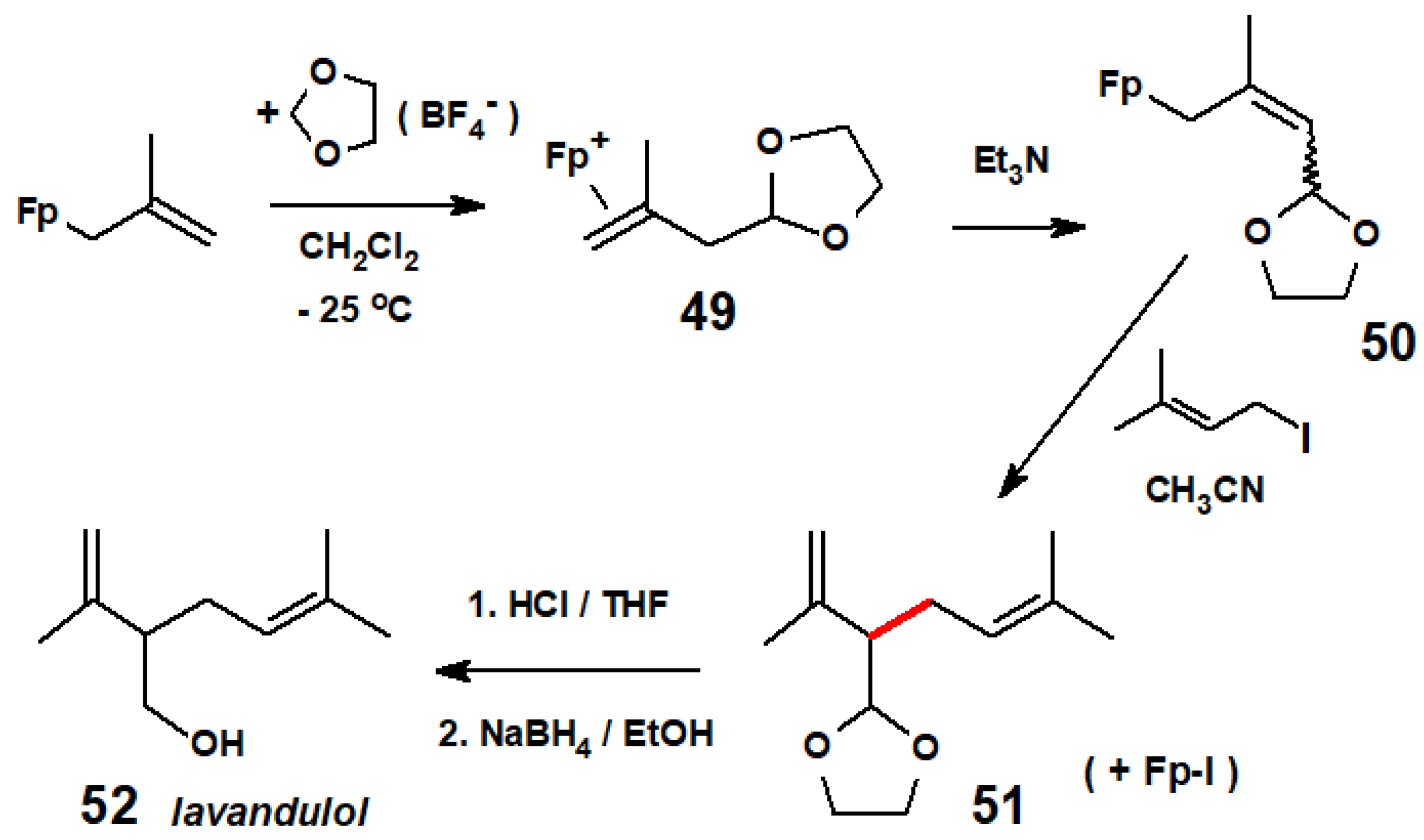

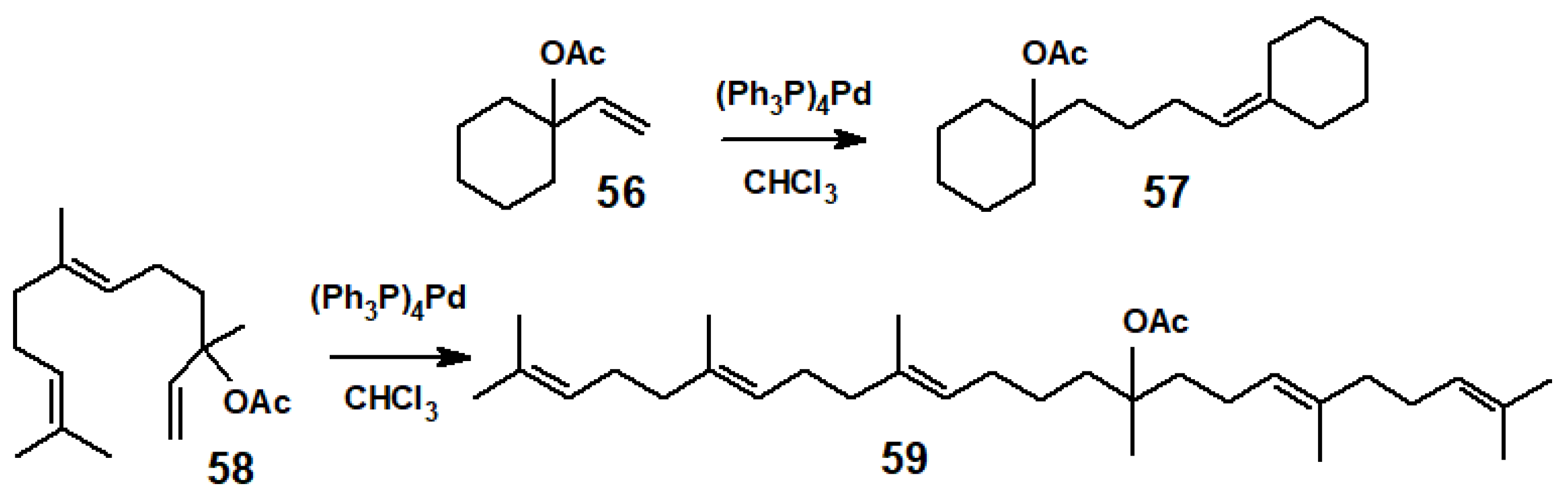
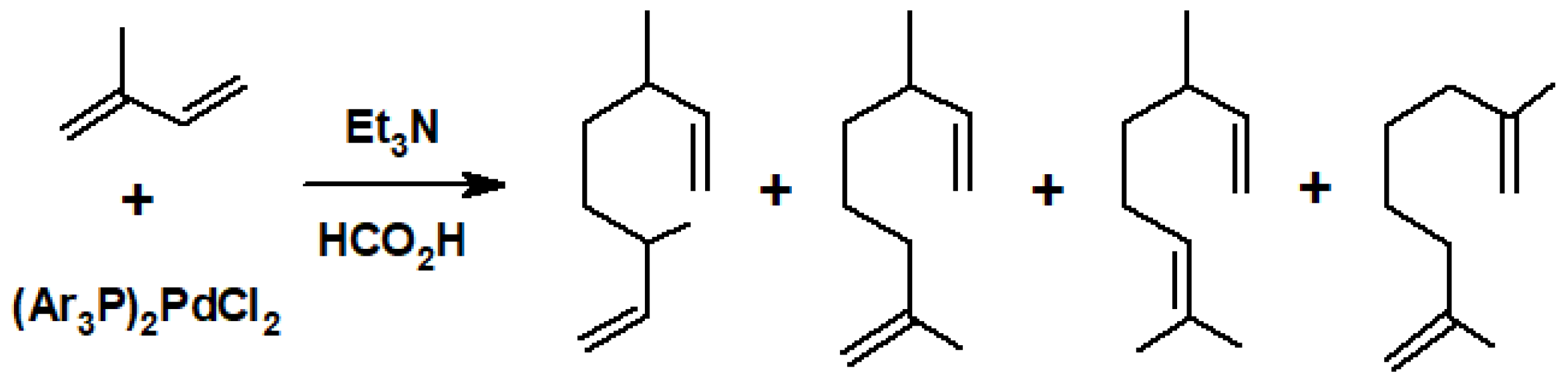
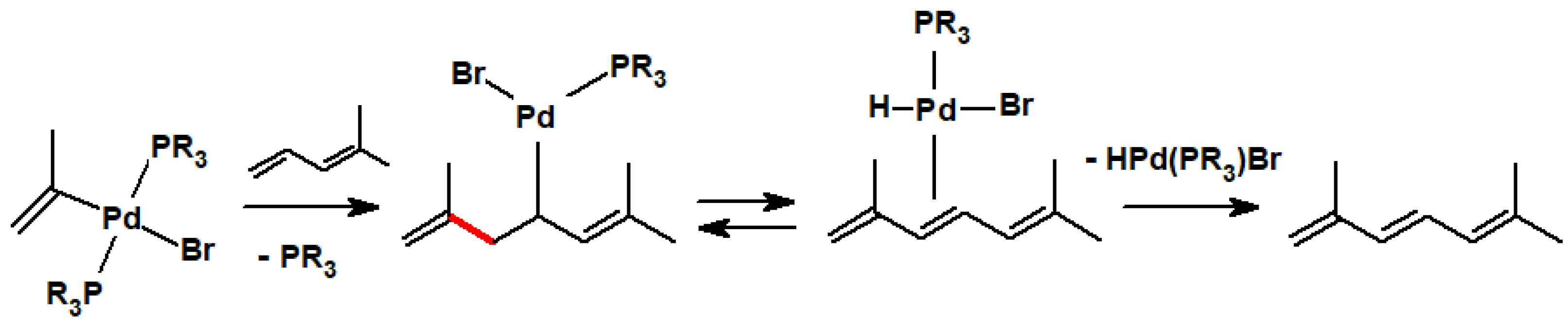
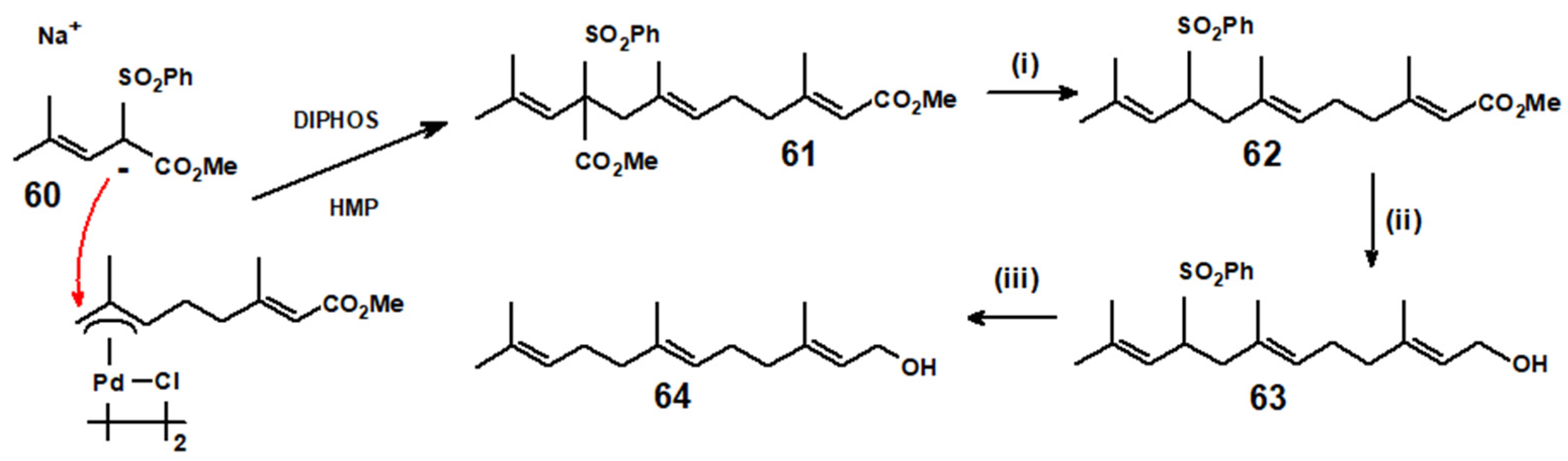
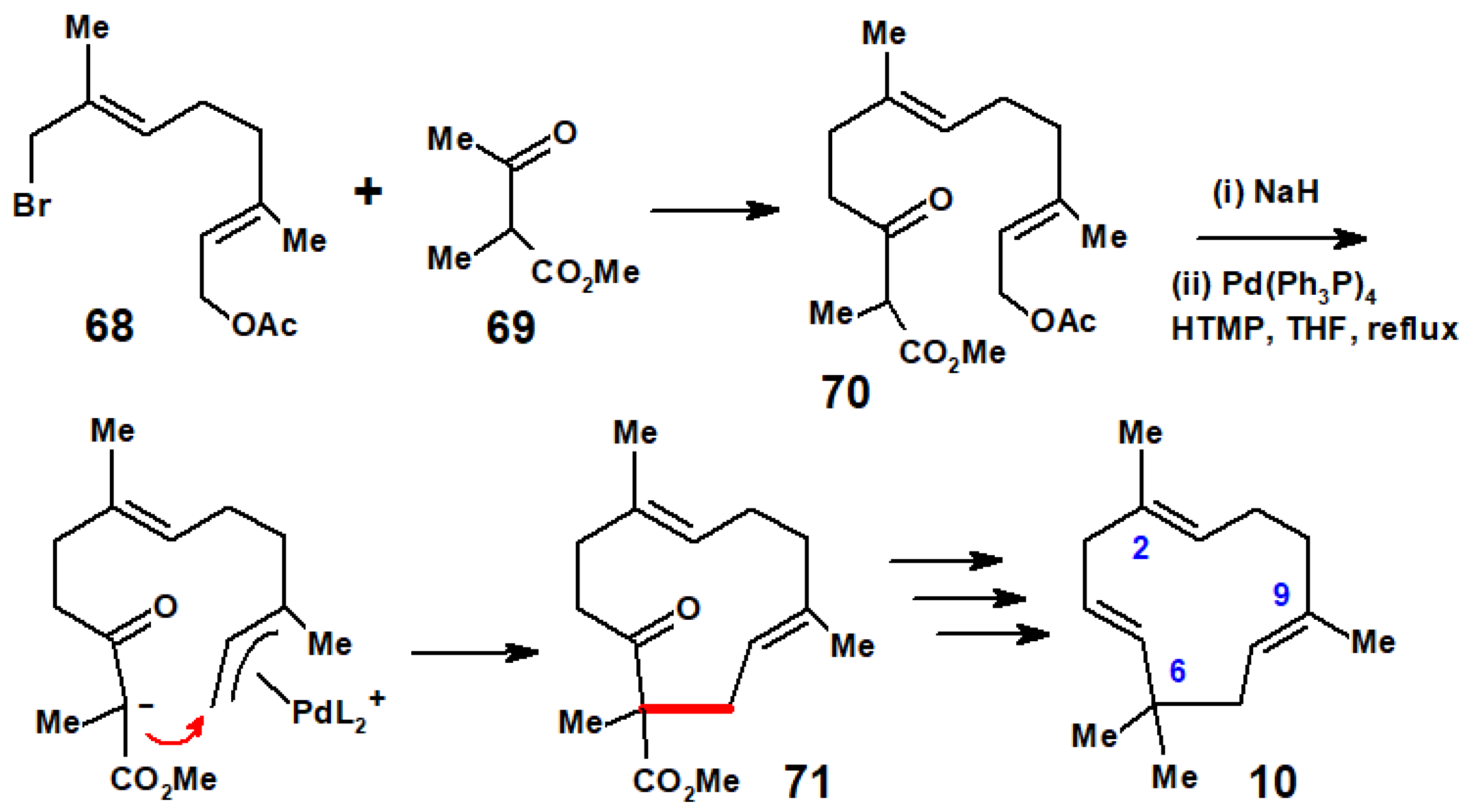
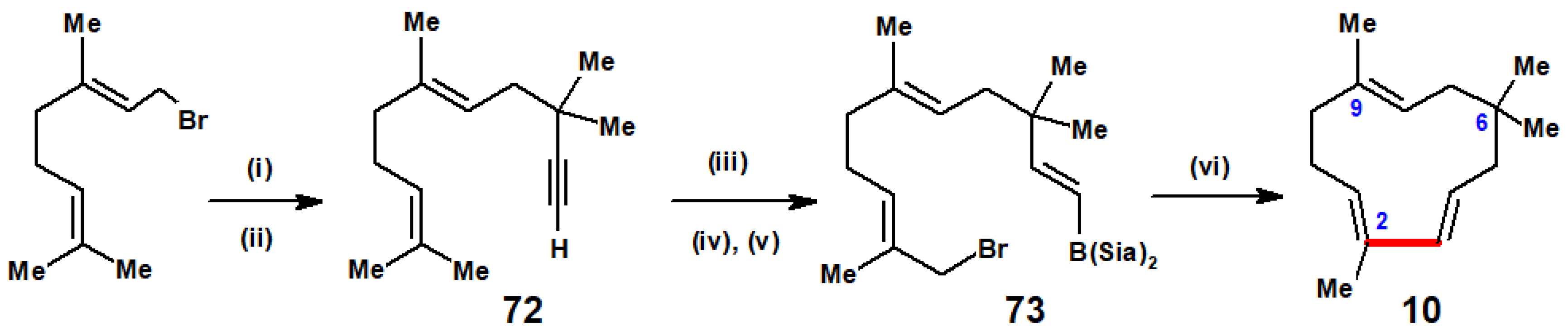
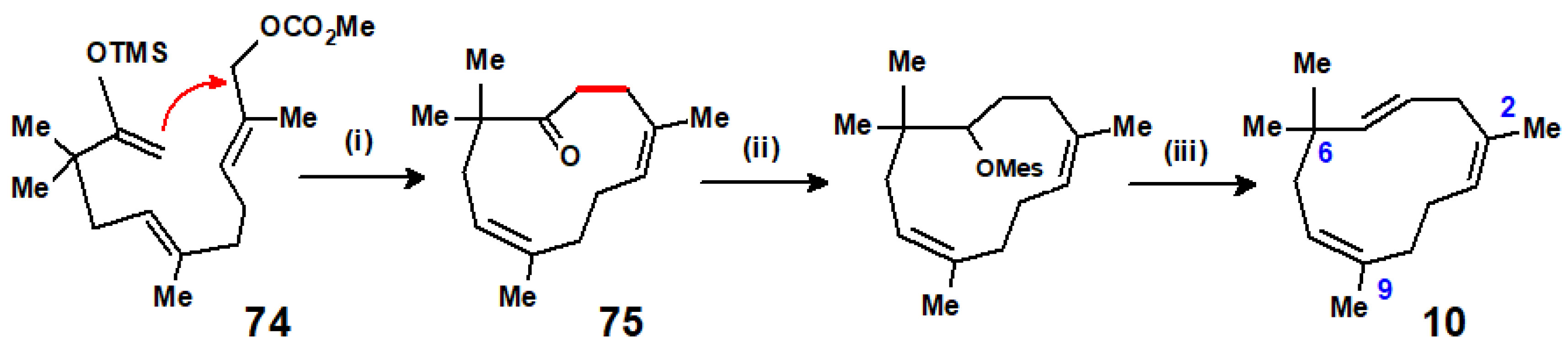
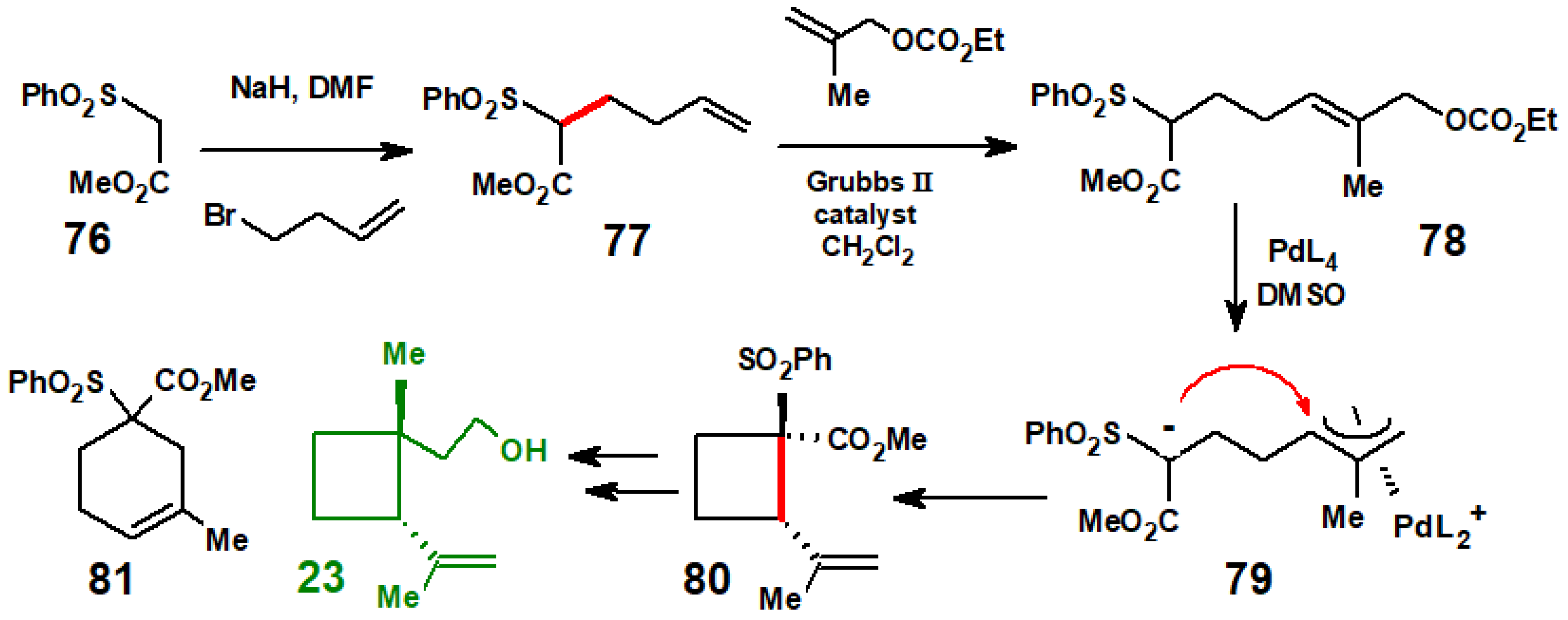

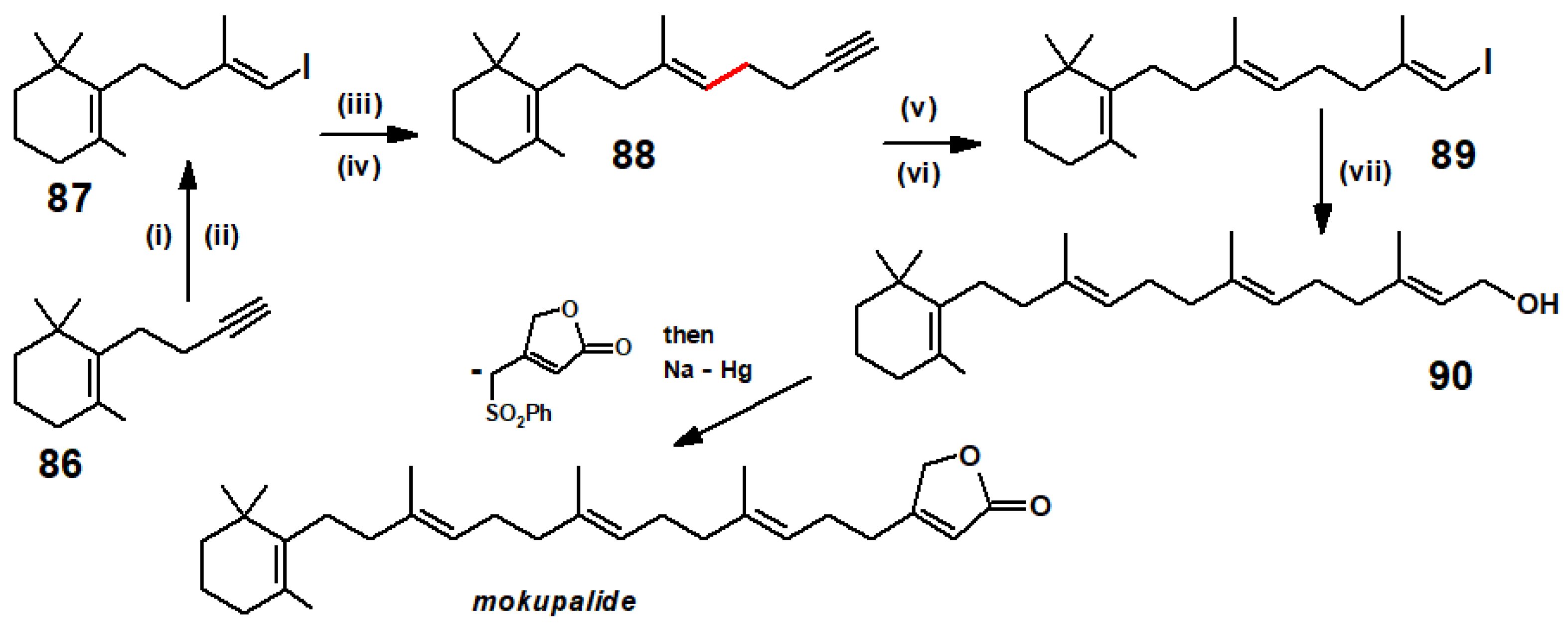
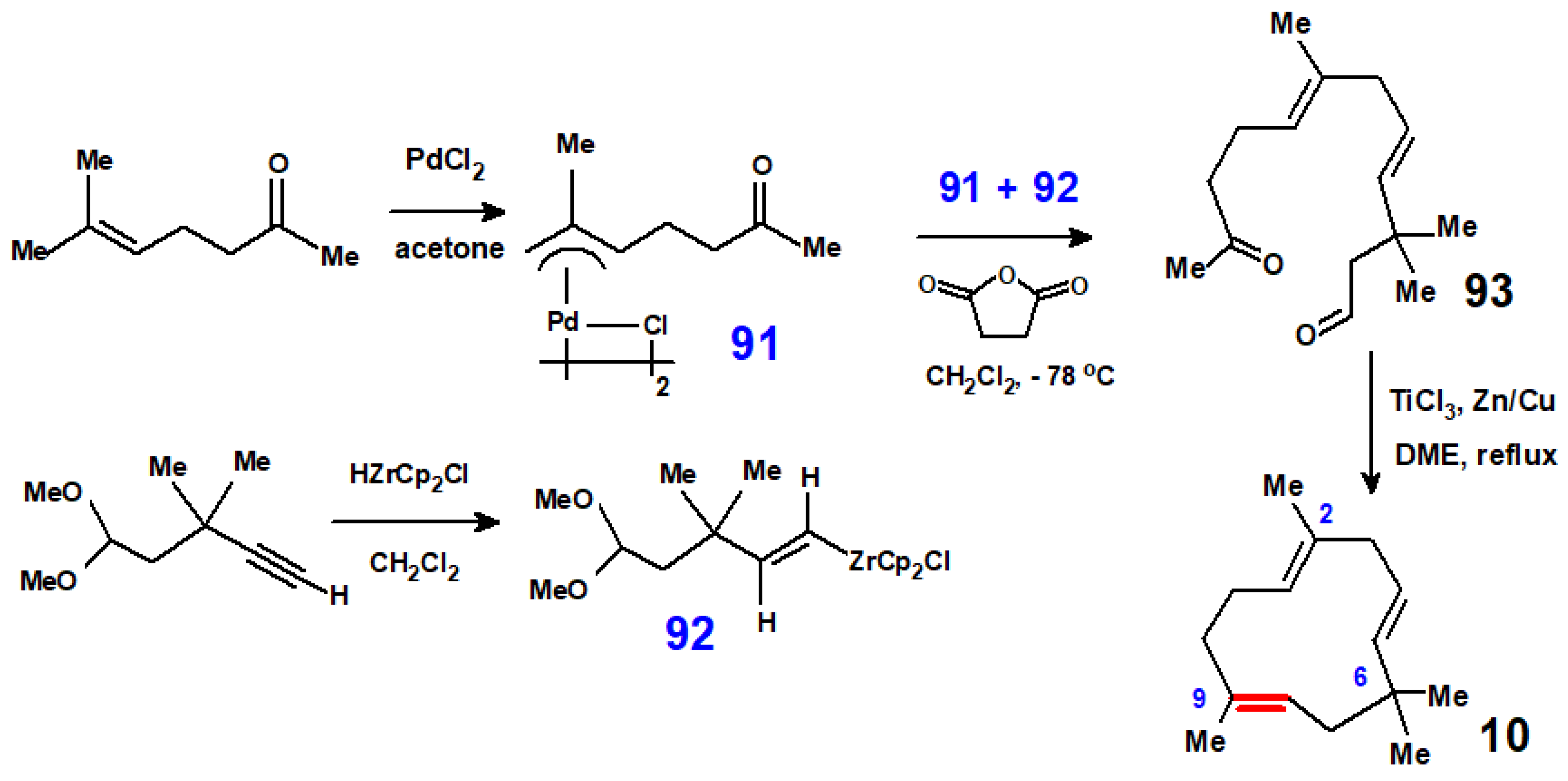
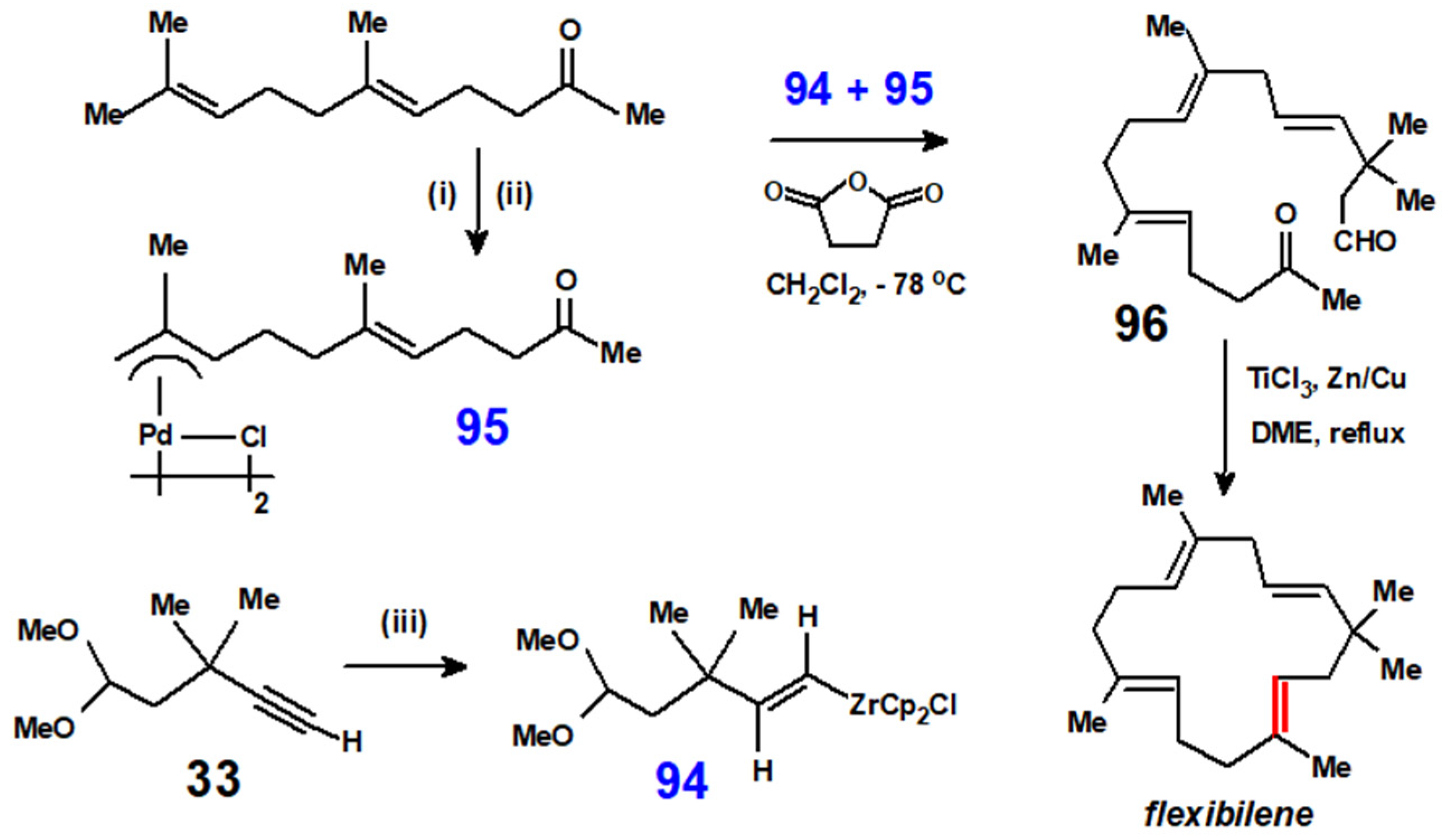
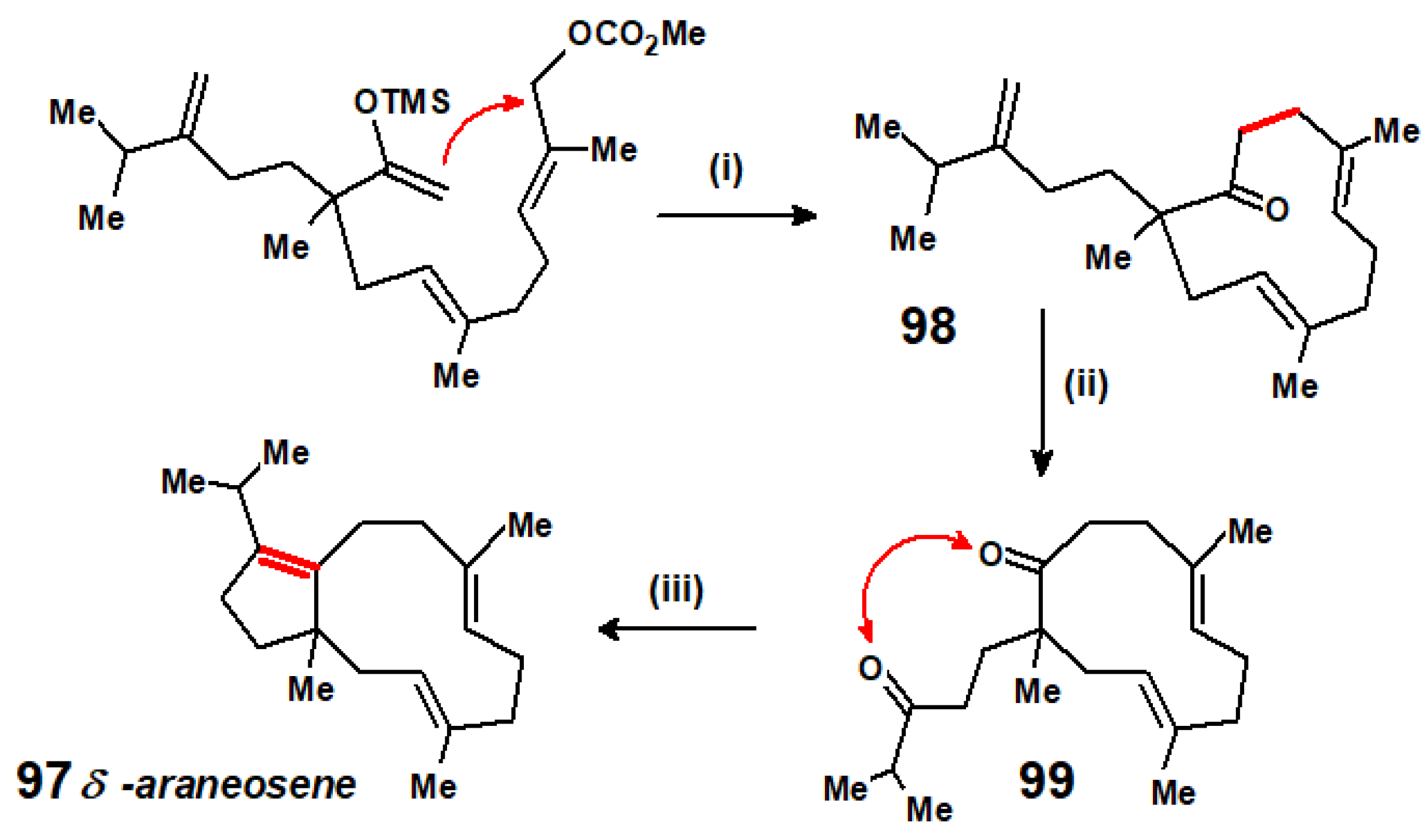






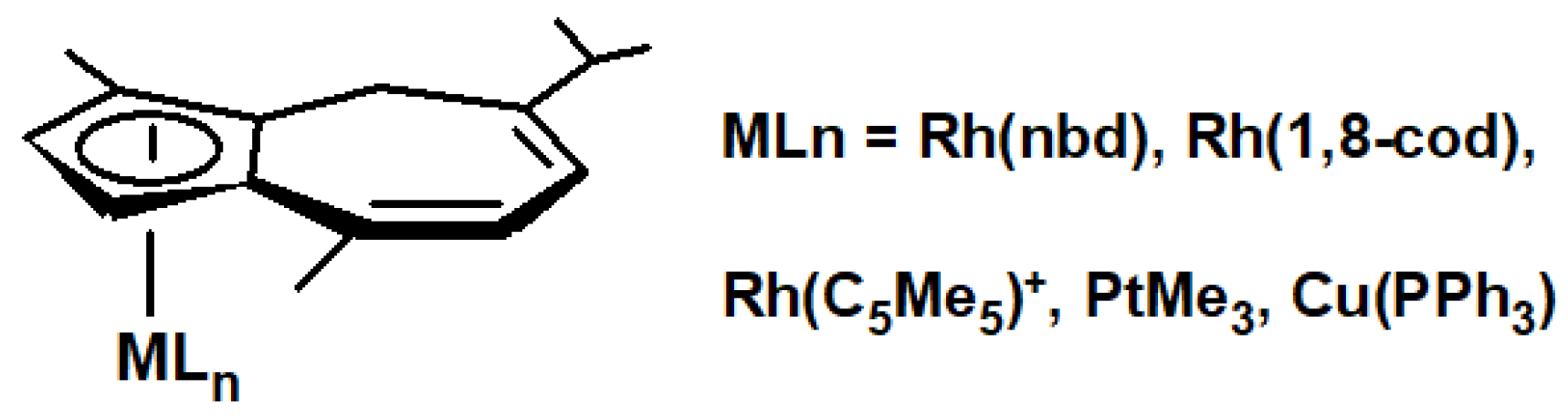

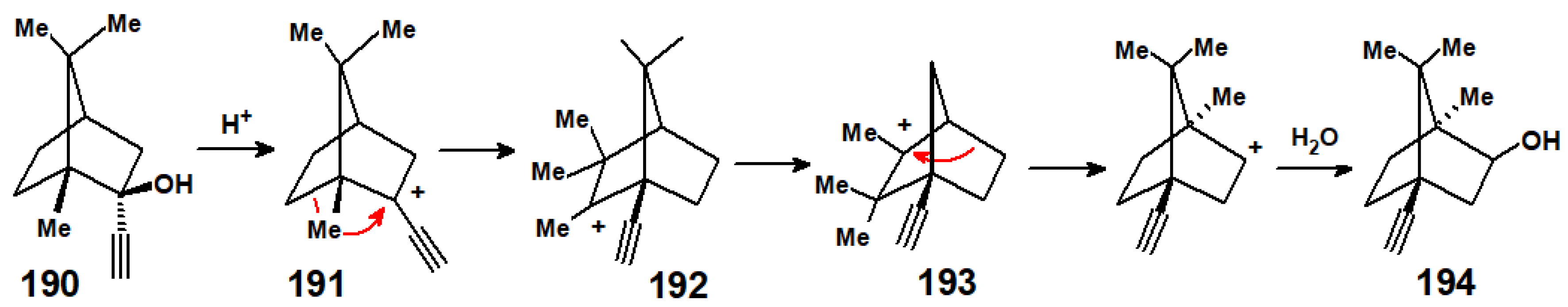

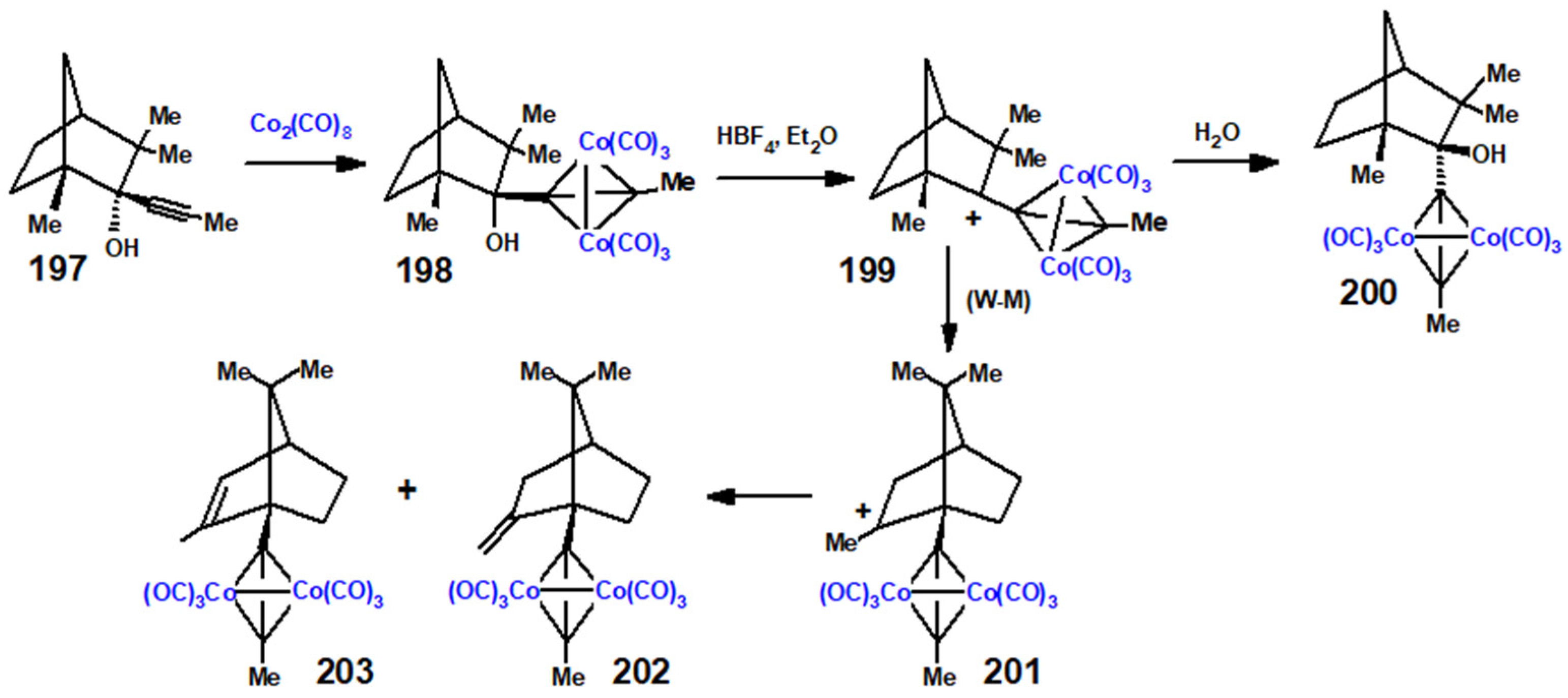
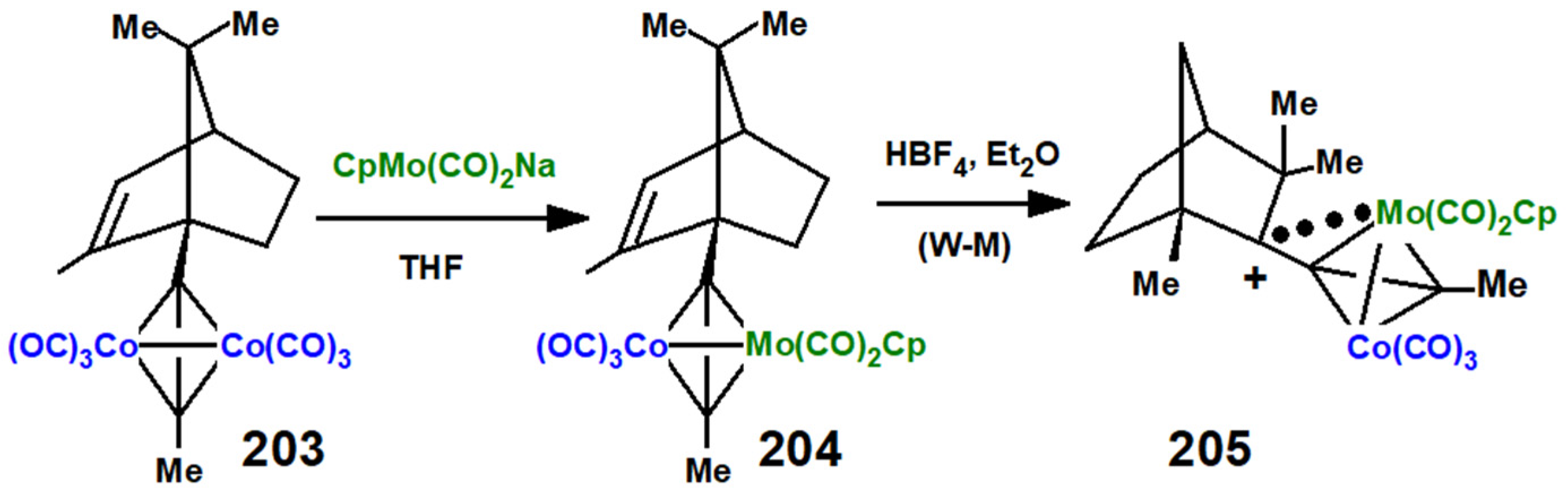
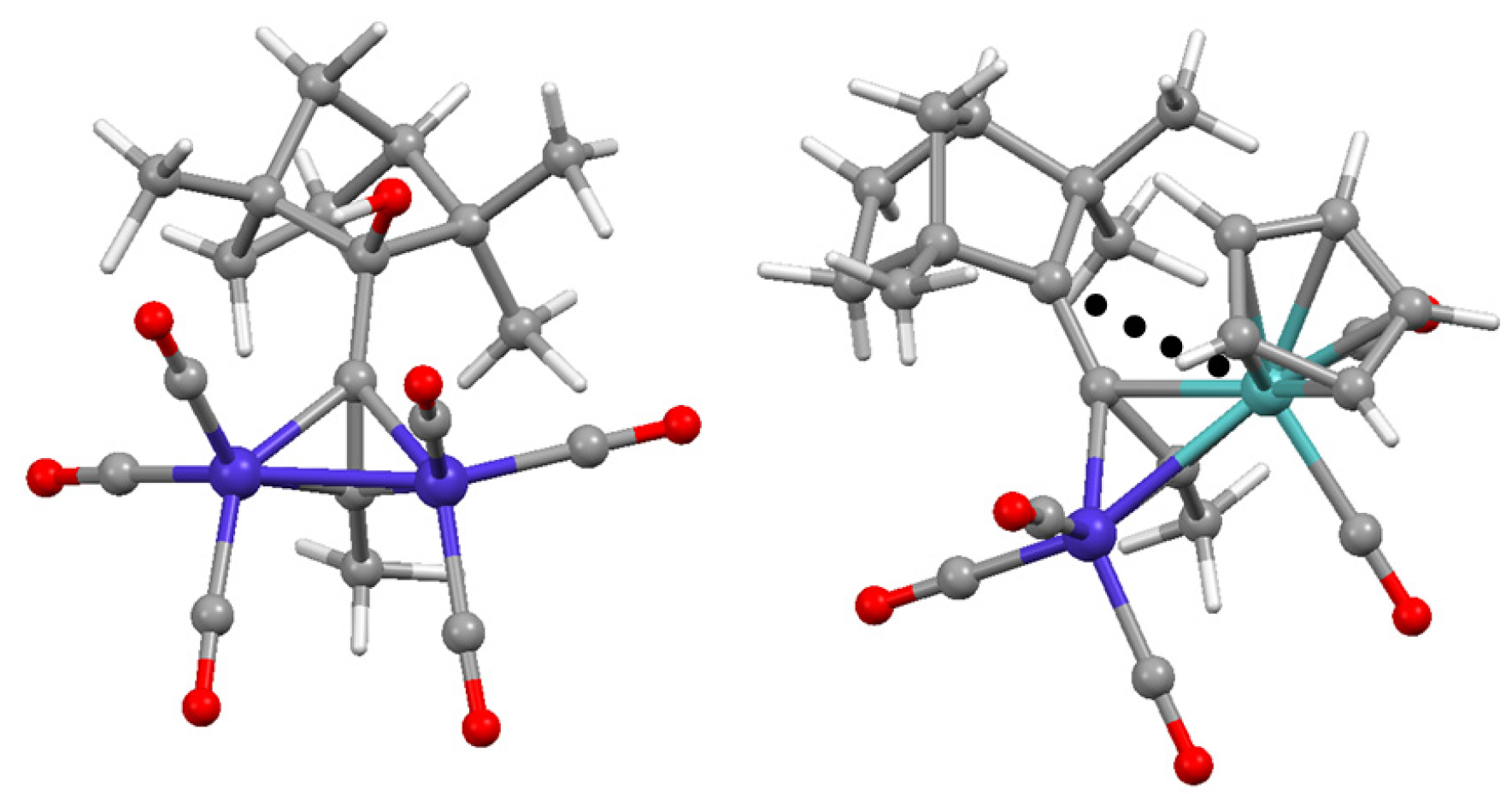
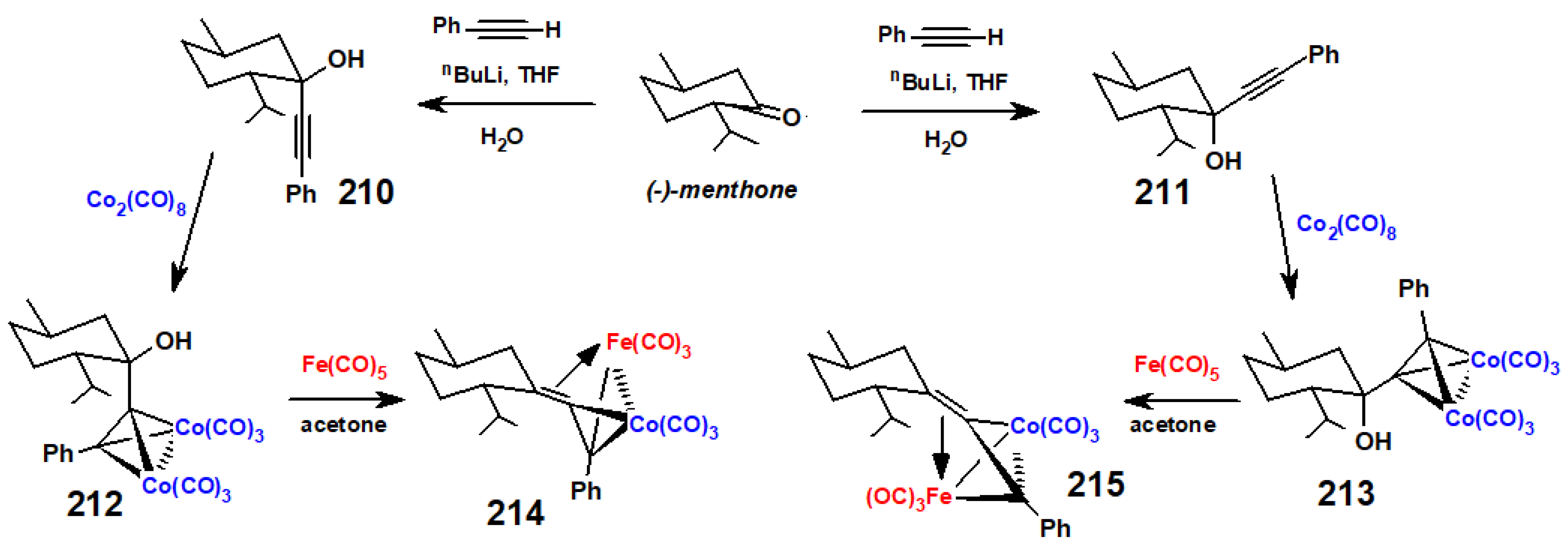
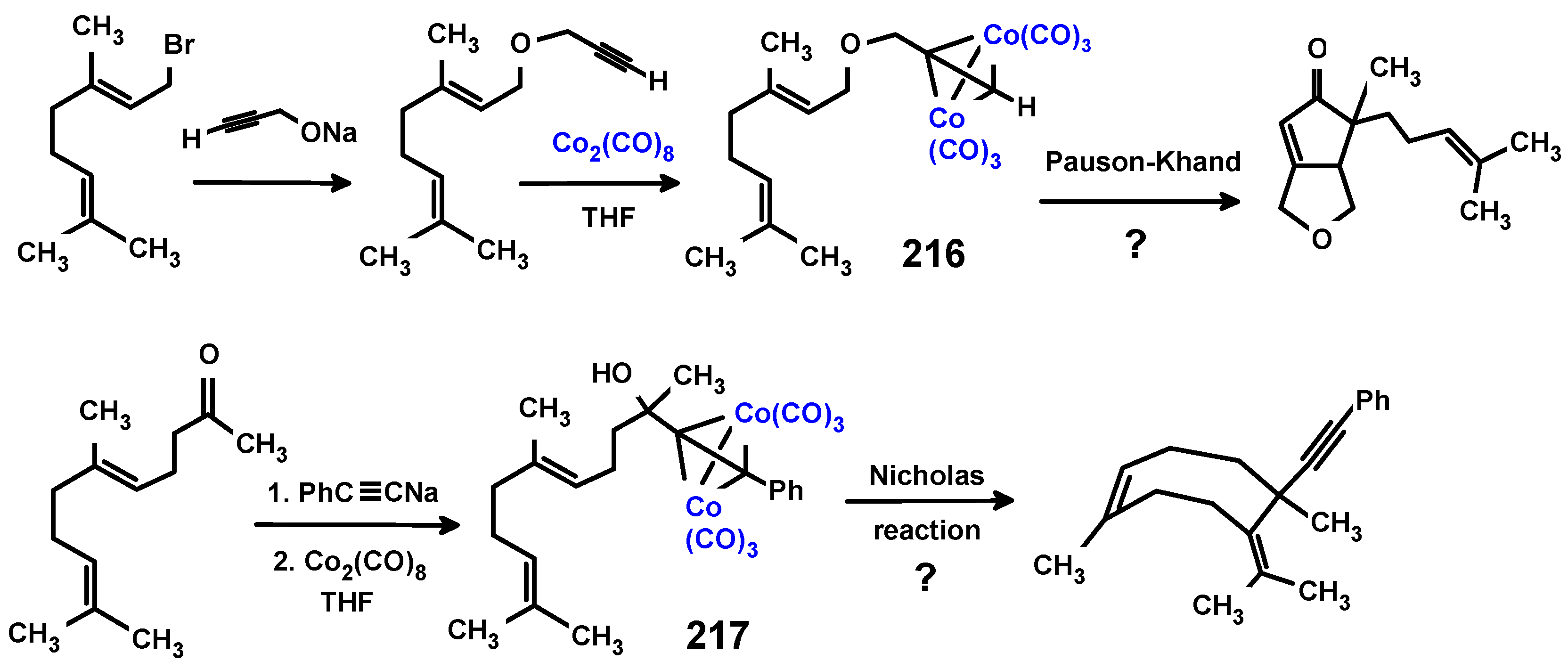
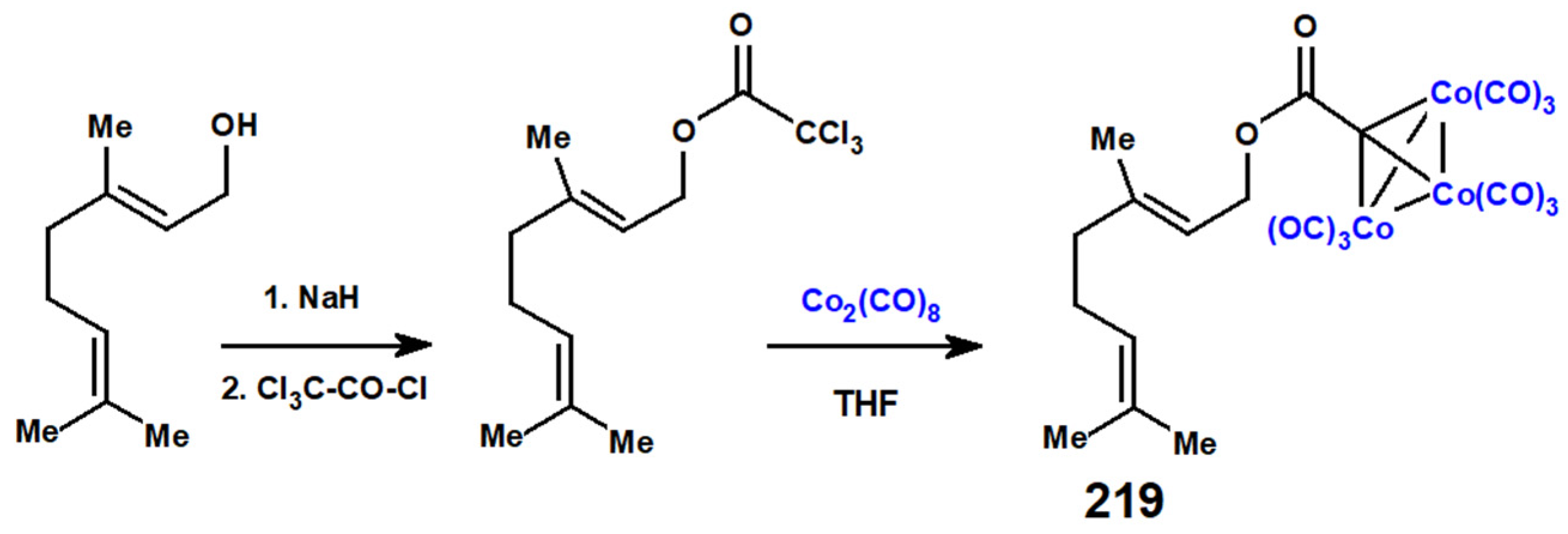
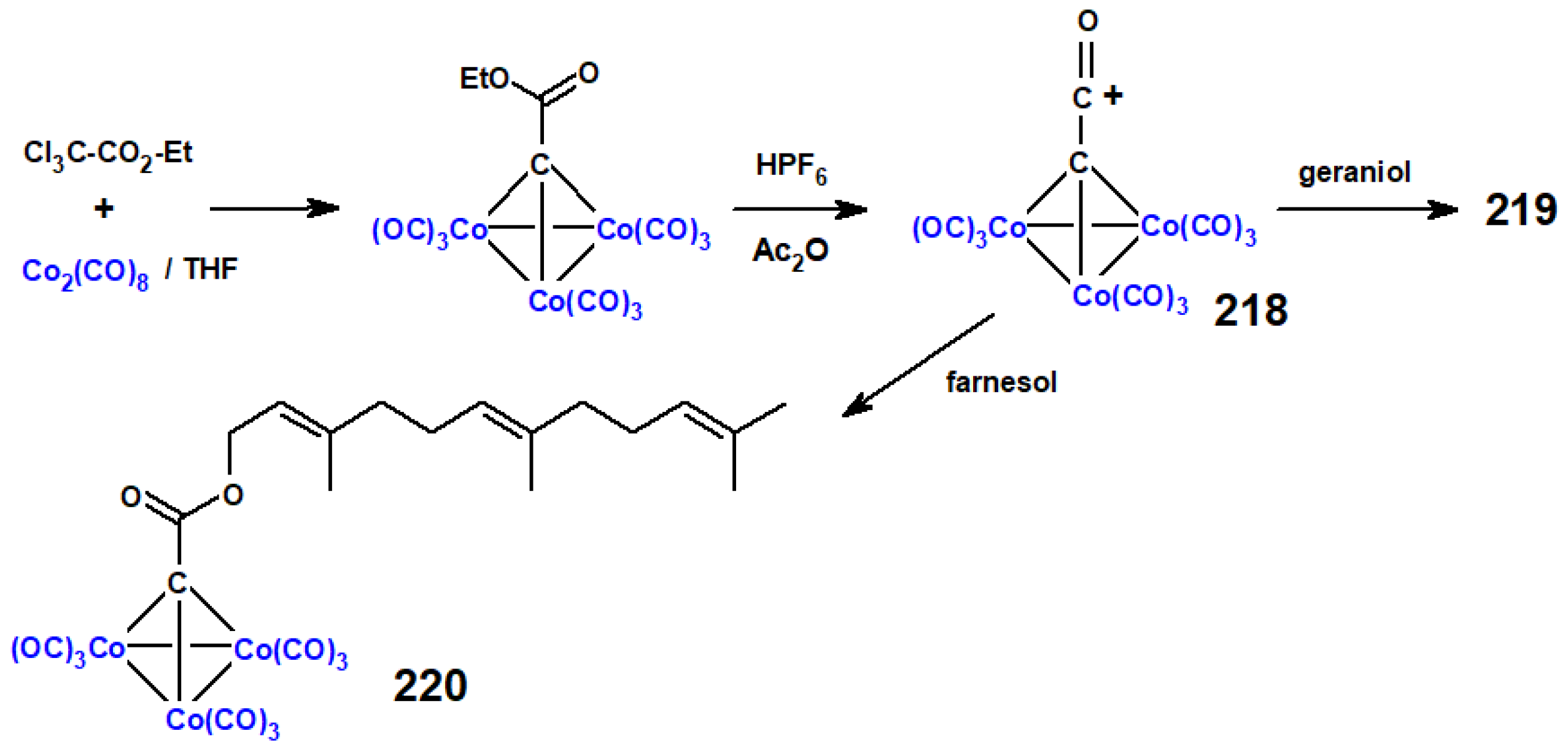
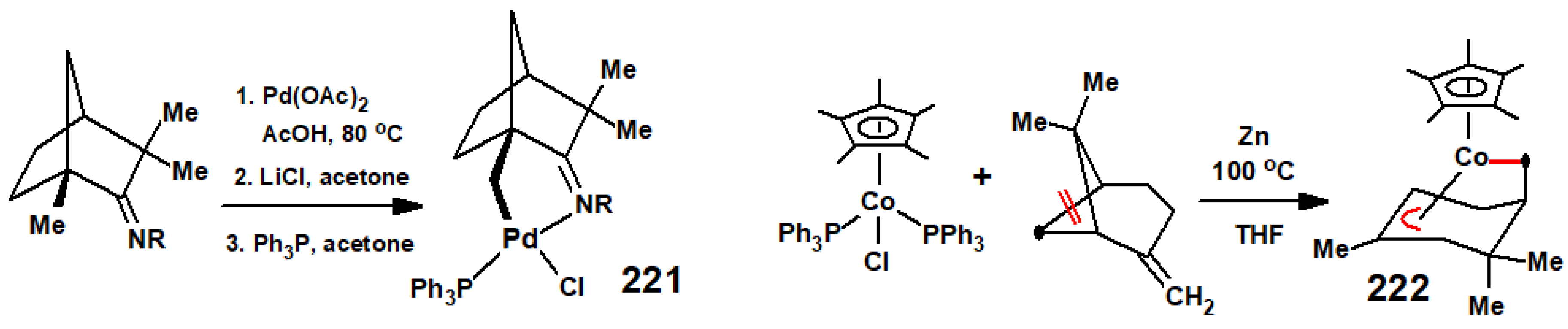
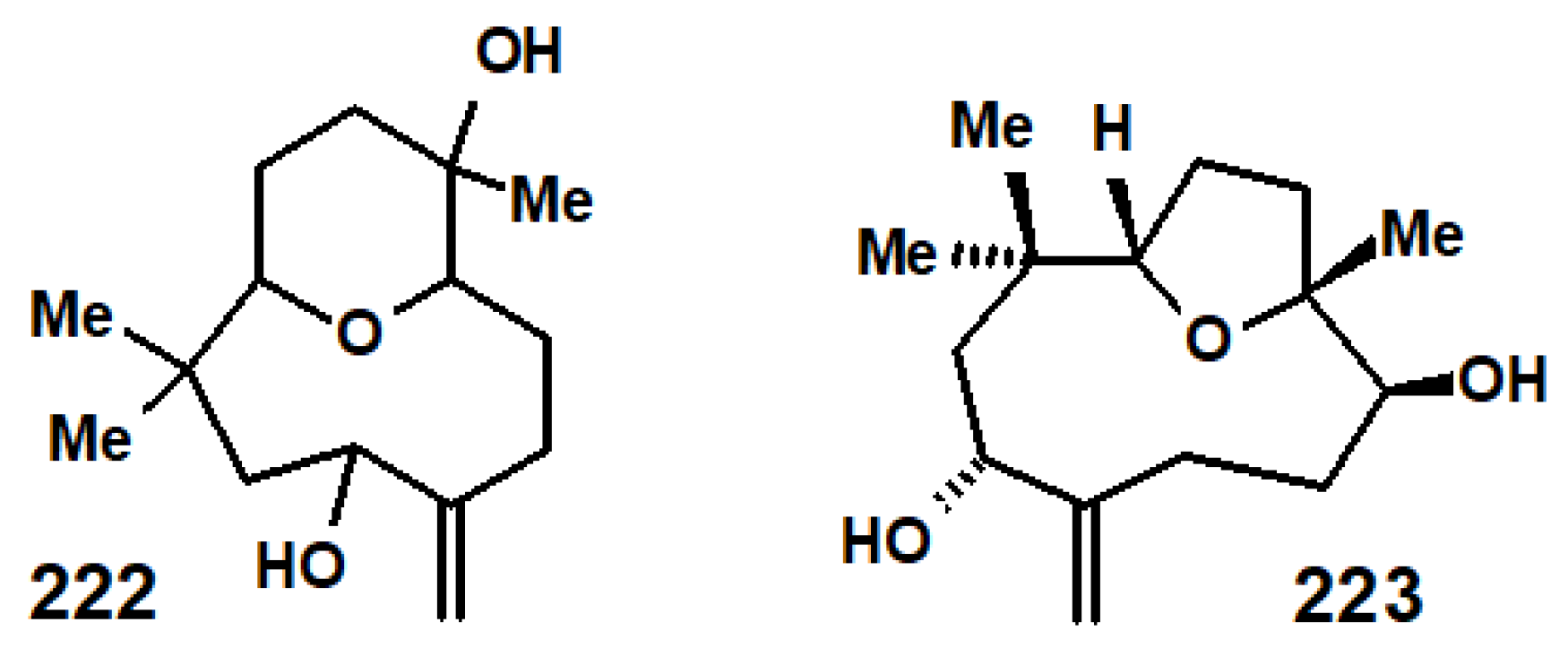
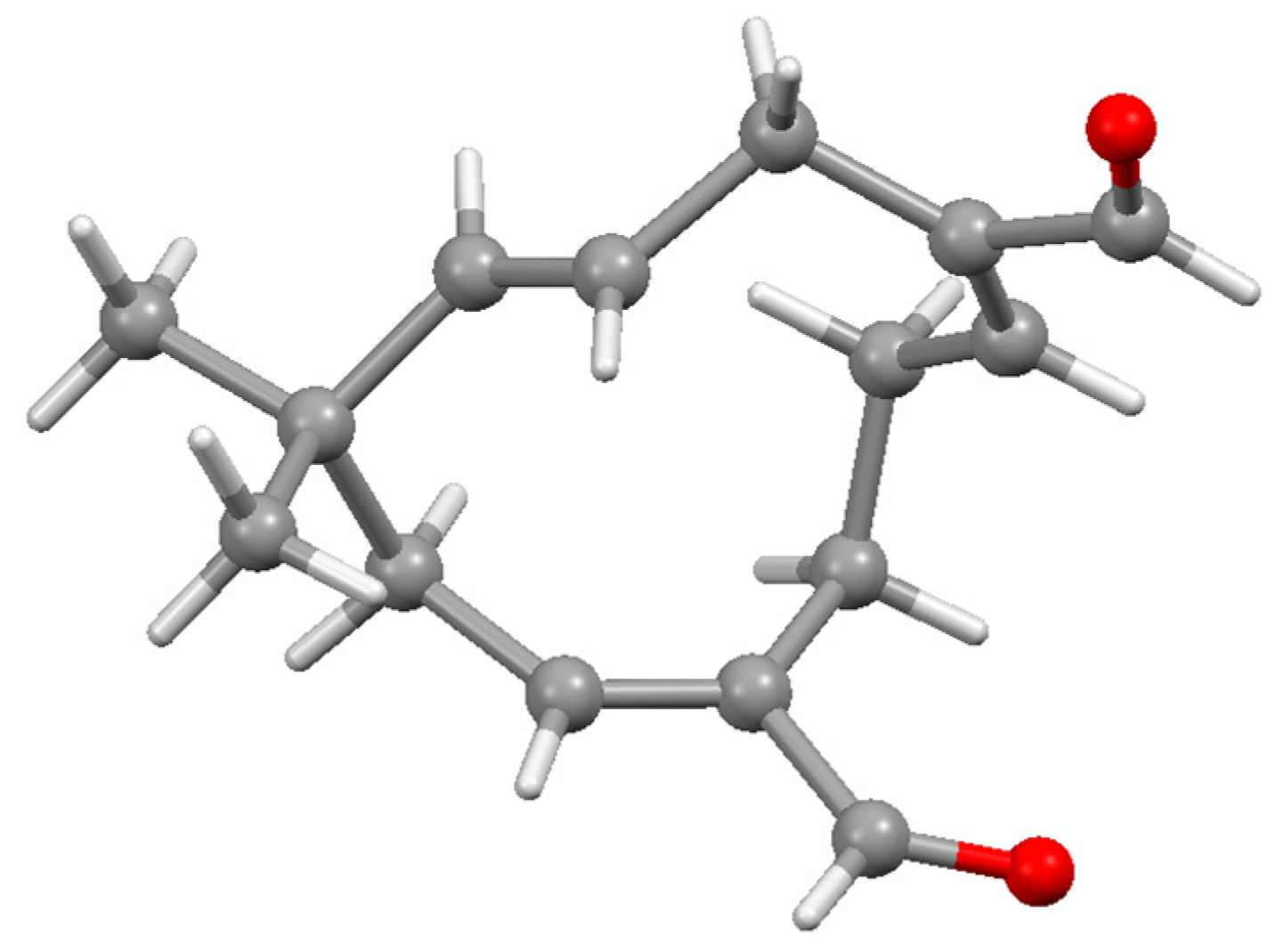
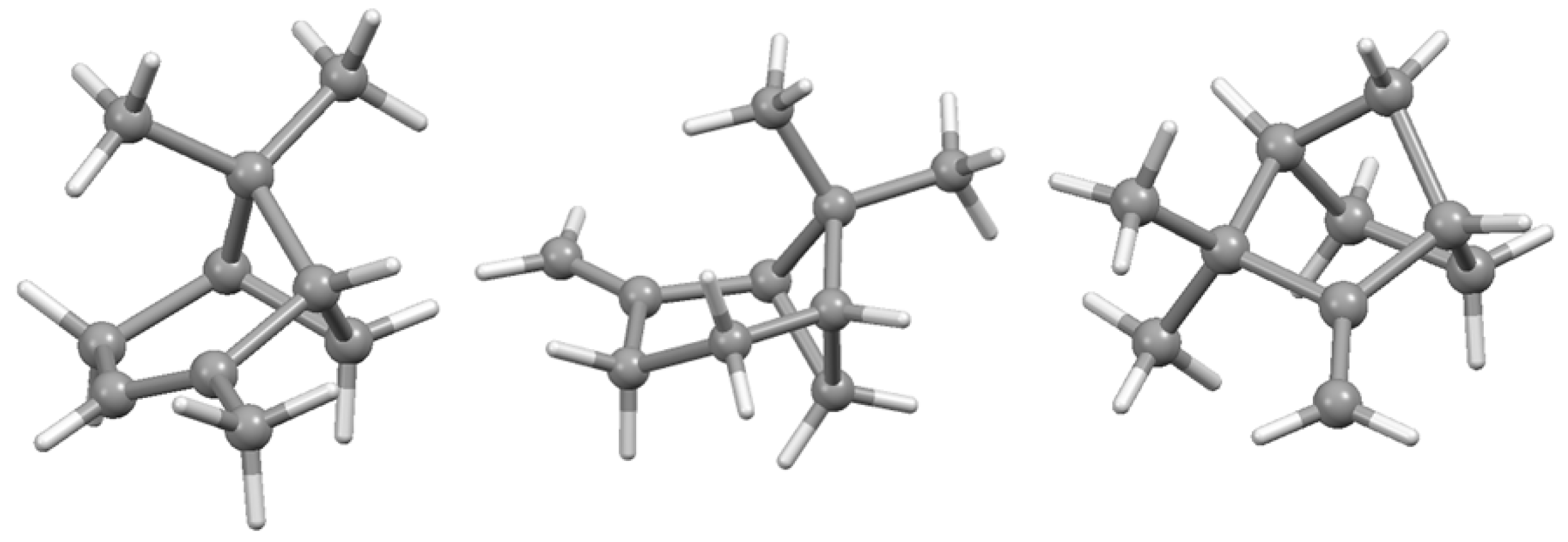
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
McGlinchey, M.J. Organotransition Metal Chemistry of Terpenes: Syntheses, Structures, Reactivity and Molecular Rearrangements. Molecules 2024, 29, 1409. https://doi.org/10.3390/molecules29061409
McGlinchey MJ. Organotransition Metal Chemistry of Terpenes: Syntheses, Structures, Reactivity and Molecular Rearrangements. Molecules. 2024; 29(6):1409. https://doi.org/10.3390/molecules29061409
Chicago/Turabian StyleMcGlinchey, Michael J. 2024. "Organotransition Metal Chemistry of Terpenes: Syntheses, Structures, Reactivity and Molecular Rearrangements" Molecules 29, no. 6: 1409. https://doi.org/10.3390/molecules29061409
APA StyleMcGlinchey, M. J. (2024). Organotransition Metal Chemistry of Terpenes: Syntheses, Structures, Reactivity and Molecular Rearrangements. Molecules, 29(6), 1409. https://doi.org/10.3390/molecules29061409