
4-Chloroisocoumarins as Chlamydial Protease Inhibitors and Anti-Chlamydial Agents

Abstract
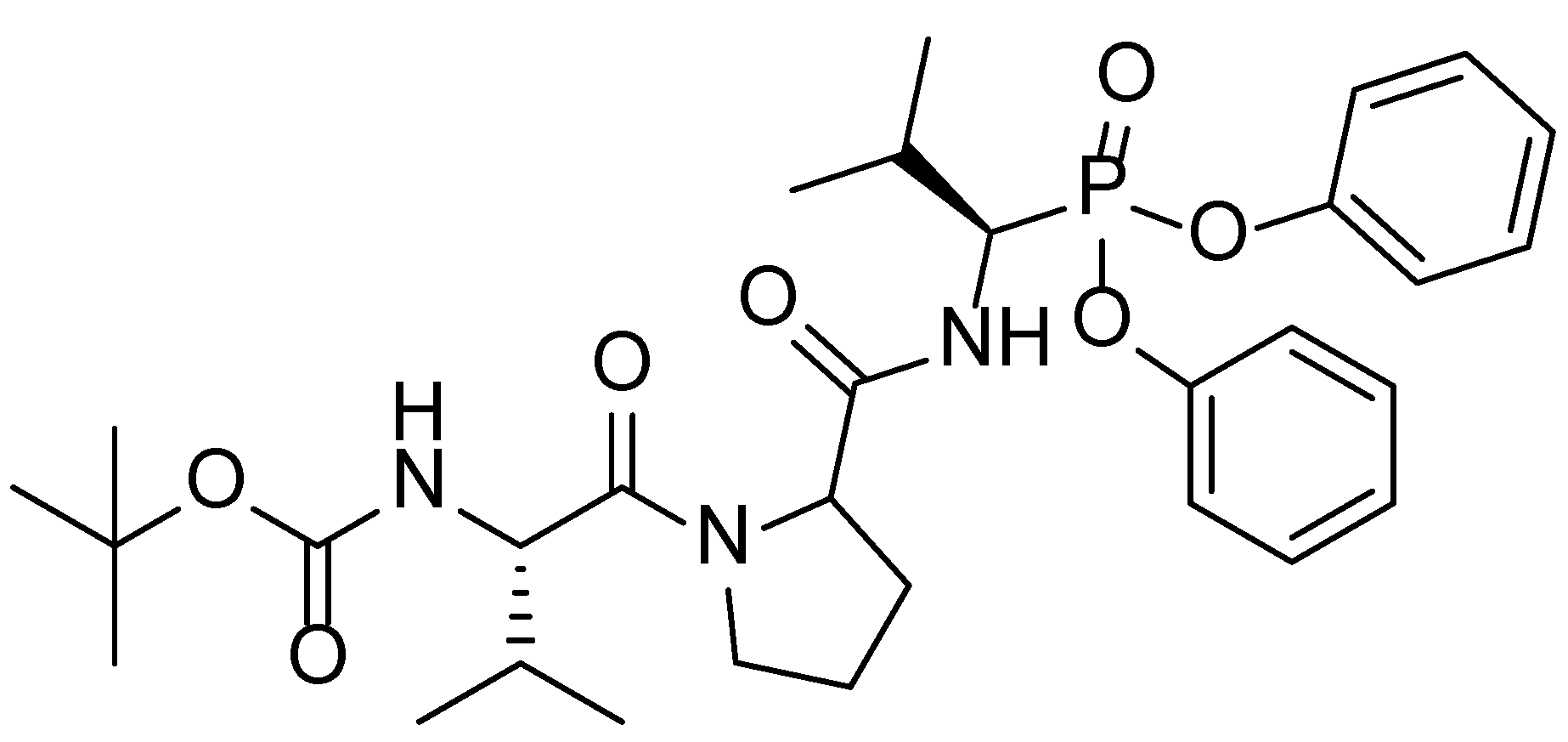
:1. Introduction
2. Results and Discussion
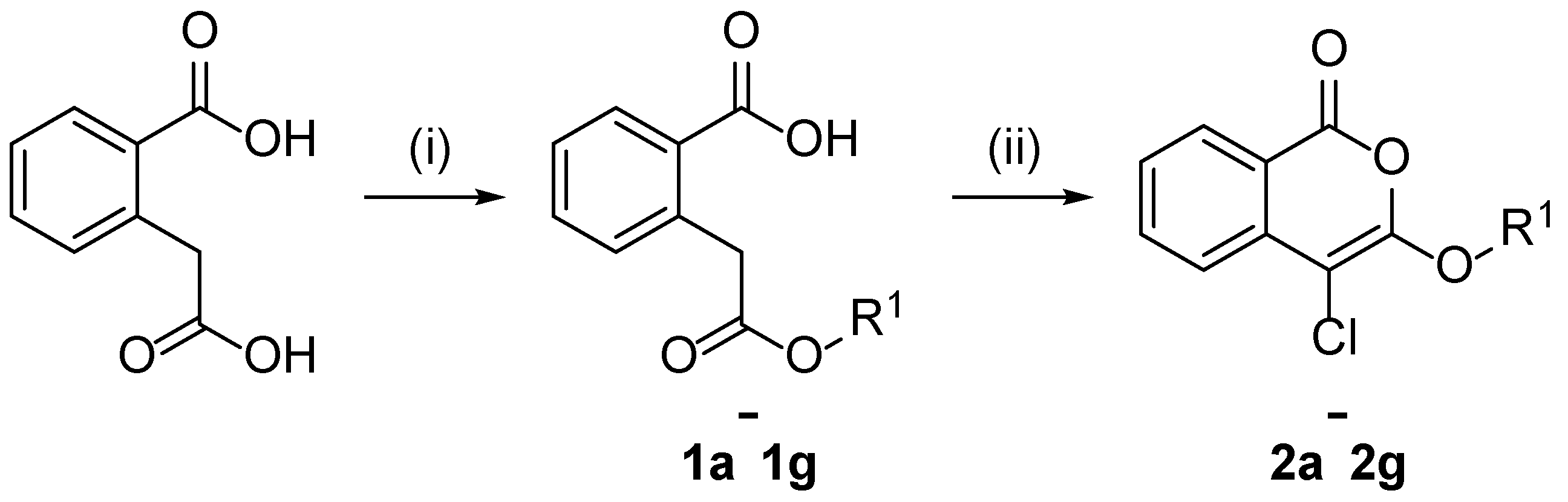
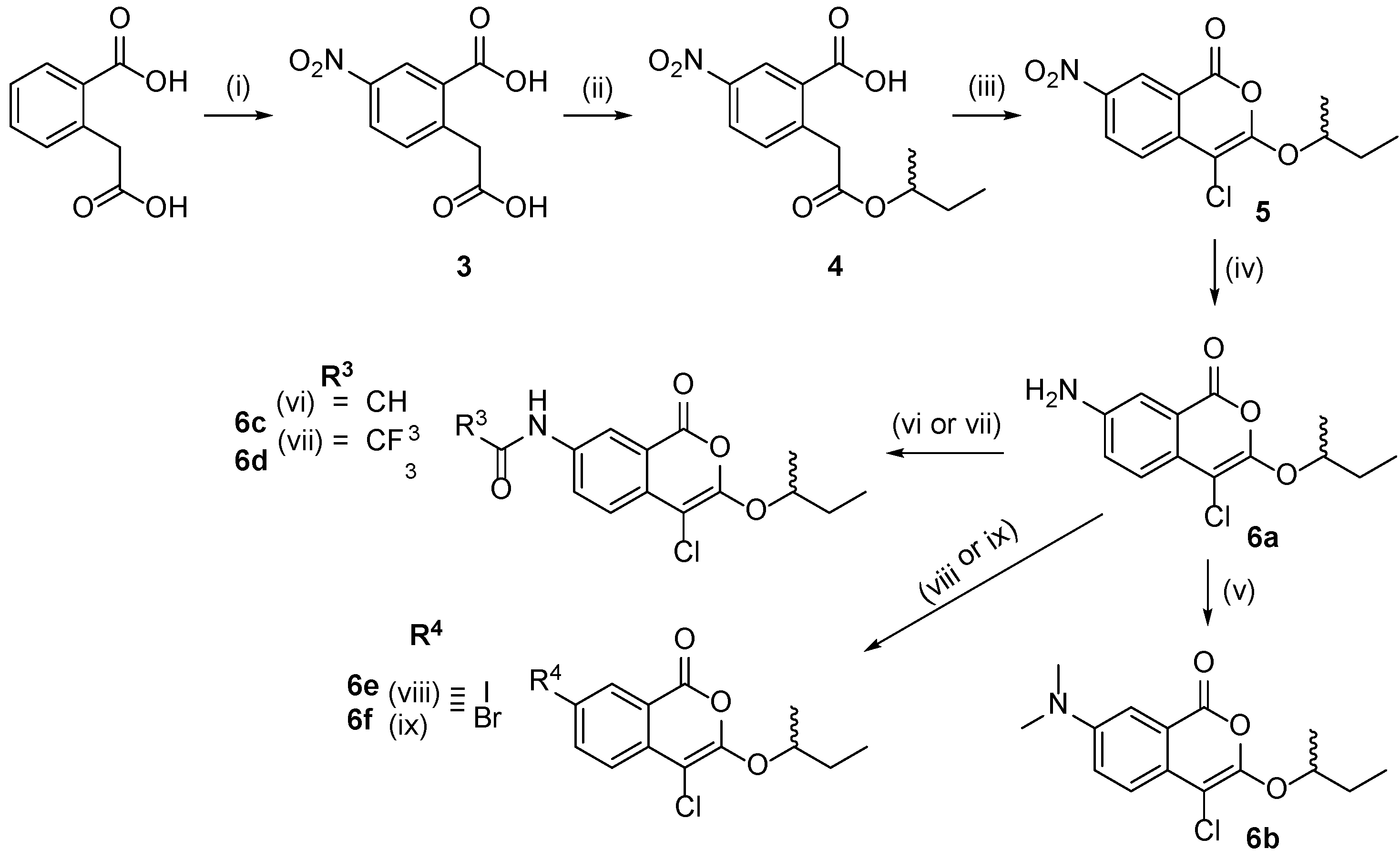
2.1. Design and Synthesis of 4-Chloroisocoumarin Compounds
2.2. Inhibitory Properties of 4-Chloroisocoumarins against CtHtrA and HLE
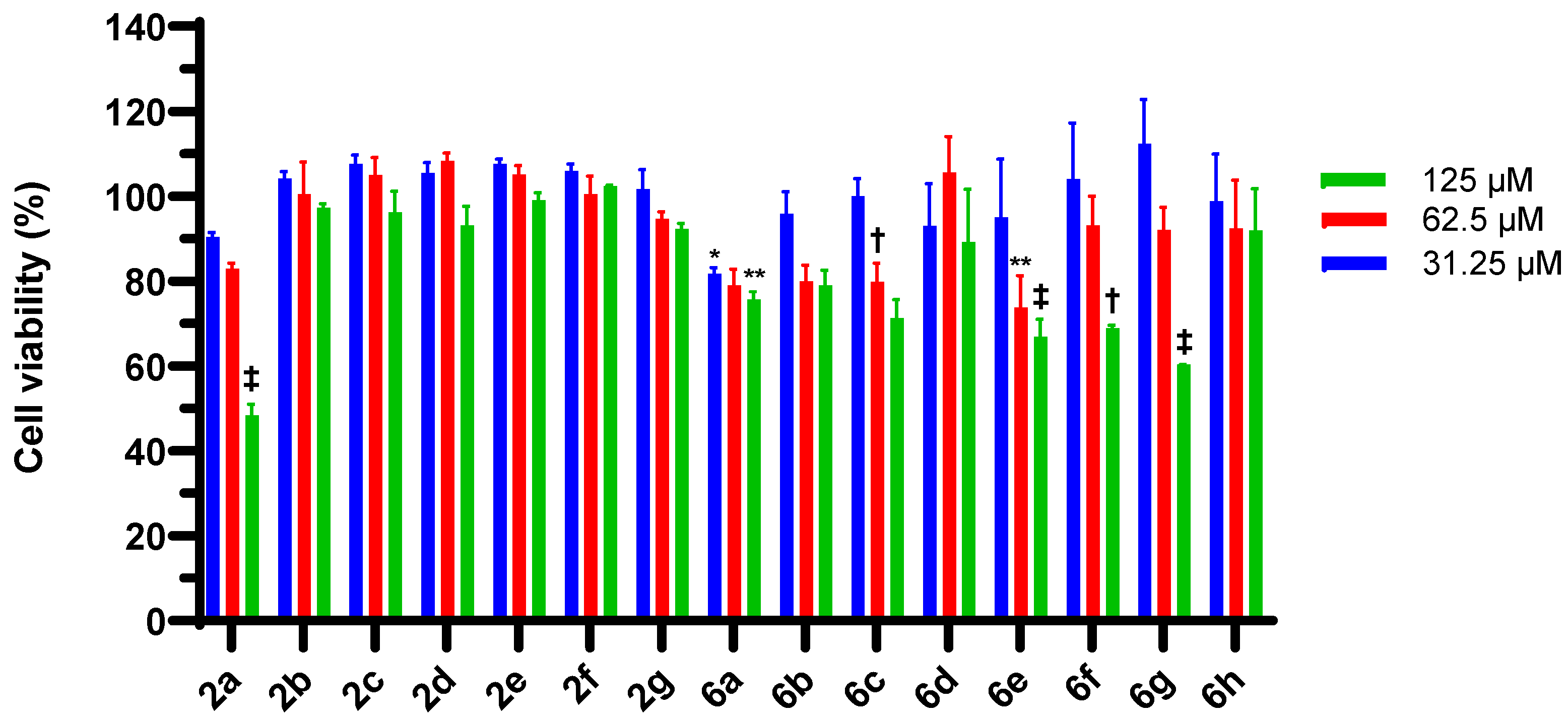
2.3. 4-Chloroisocoumarin Cytotoxic Effects in HEp-2 Cells and McCoy B Cells
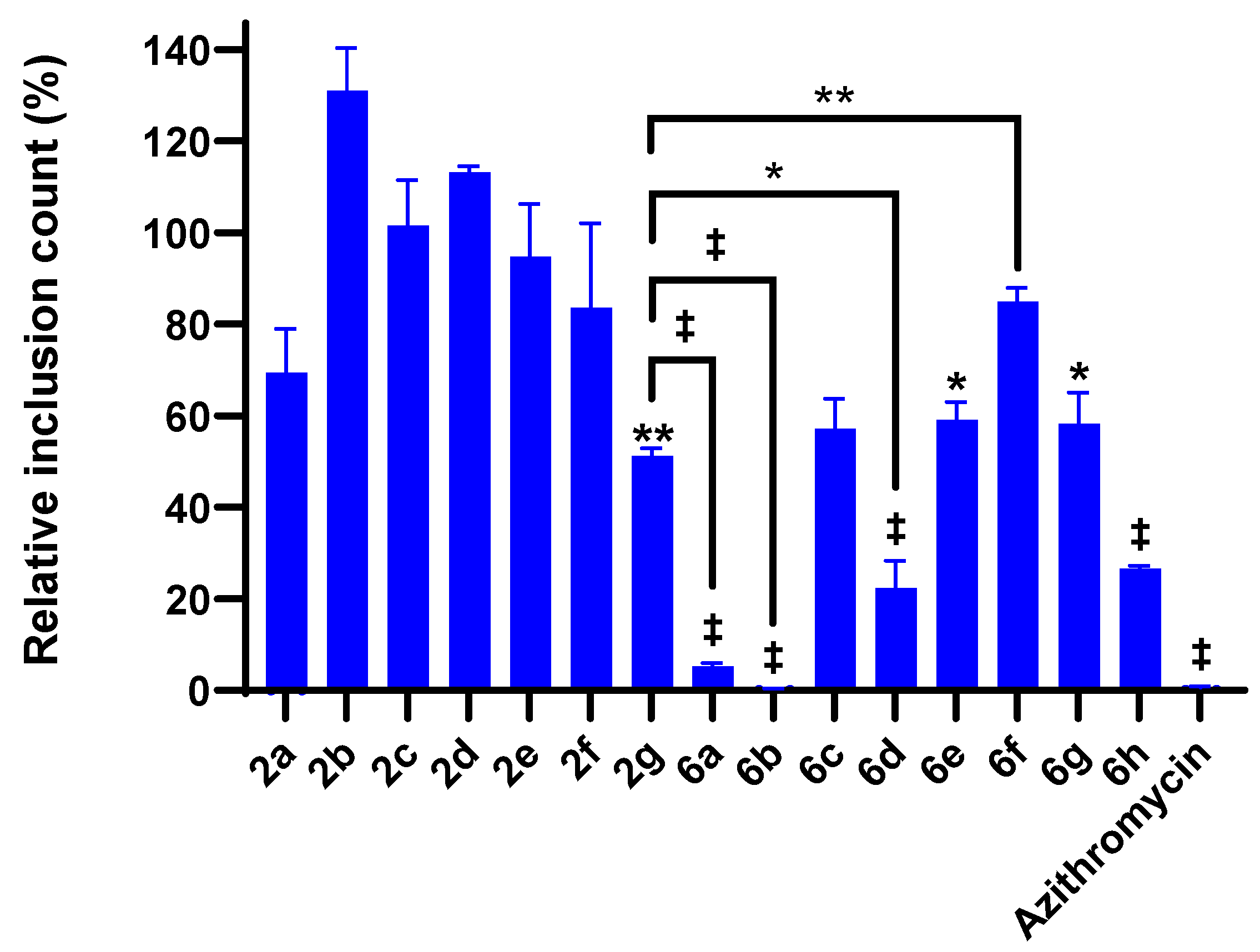
2.4. 4-Chloroisocoumarin Compound Effects against C. trachomatis
3. Materials and Methods
3.1. Chemistry
3.1.1. General Procedure for the Synthesis of Unsubstituted Homophthalate Monoesters (1a–1g)
- 2-(2-iso-butoxy-2-oxo-ethyl)benzoic acid (1f). White solid (0.800 g, 49%) 1H NMR: (400 MHz, CDCl3) δ 11.33 (br s, 1H, COOH), 8.13 (dd, J = 1.2, 8.0 Hz, 1H, H-6), 7.54 (td, J = 1.2, 7.6 Hz, 1H, H-5), 7.40 (td, J = 1.2, 7.6 Hz, 1H, H-4), 7.29 (d, J = 7.6 Hz, 1H, H-3), 4.08 (s, 2H, CH2CO2), 3.88 (d, 2H, OCH2), 1.91 (sxt, J = 6.8 Hz, 1H, CH), 0.90 (d, J = 6.8 Hz, 6H, 2 × CH3). 13C NMR: (100 MHz, CDCl3) δ 172.2, 171.5, 137.0, 133.2, 132.4, 131.9, 128.6, 127.5, 70.9, 40.7, 27.7, 19.0. HRMS (ESI/Q-TOF) m/z: [M + Na]+ Calcd for C13H16O4Na 259.0946; Found 259.0951. m.p. 120–122 °C.
- (±)-2-(2-sec-butoxy-2-oxo-ethyl)benzoic acid (1g). White solid (1.22 g, 62%), 1H NMR: (400 MHz, CDCl3) δ 8.12 (dd, J = 7.6, 1.2 Hz, 1H, H-6), 7.53, (td, J = 1.2 Hz, 7.6 Hz, 1H, H-5), 7.40 (td, J = 1.2, 7.6 Hz, 1H, H-4), 7.28 (d, J = 7.6 Hz, 1H, H-3), 4.87 (sxt, J = 6.4 Hz, 1H, C-1′), 4.03 (d, J = 2.0 Hz, 2H, CH2CO2), 1.66–1.47, m, 2H, CH2), 1.21 (d, J = 6.4 Hz, 3H, CH3), 0.88 (t, J = 7.2 Hz, 3H, CH3). 13C NMR: (100 MHz, CDCl3) δ 172.5, 171.2, 137.1, 133.2, 132.4, 131.8, 128.7, 127.4, 72.8, 41.1, 28.7, 19.3, 9.6. HRMS (ESI/Q-TOF) m/z: [M + Na]+ Calcd for C13H16O4Na 259.0946; Found 259.0942. m.p. 101–103 °C.
3.1.2. General Procedure for the Synthesis of 3-alkoxy-4-chloroisocoumarins (2a–2g) (General Procedure A)
- 3-Methoxy-4-chloroisocoumarin (2a). Pale-yellow needles (0.272 g, 20%). 1H NMR (500 MHz, CDCl3) δ 8.20 (d, J = 7.5 Hz, H-8), 7.77–7.72 (m, 2H, H-6 and H-5), 7.39 (dt, J = 8.5, 1.0 Hz, 1H, H-7), 4.46 (q, J = 7.0 Hz, 2H, CH2), 1.47 (t, J = 7.0 Hz, 3H, CH3). 13C NMR (125 MHz, CDCl3) δ 159.7, 153.1, 137.9, 135.6, 130.1, 126.2, 122.23 117.3, 91.3, 66.8, 14.9. HRMS (ESI/Q-TOF) m/z: [M + Na]+ Calcd for C11H10ClO3Na 247.0138; Found 247.0127. 1H, 13C NMR and melting point was consistent with previous reported data [31].
- 3-Ethoxy-4-chloroisocoumarin (2b). Pale-yellow needles (0.250 g, 23%). 1H NMR (500 MHz, CDCl3) δ 8.20 (d, J = 7.5 Hz, H-8), 7.77–7.72 (m, 2H, H-6 and H-5), 7.39 (dt, J = 8.5, 1.0 Hz, 1H, H-7), 4.46 (q, J = 7.0 Hz, 2H, CH2), 1.47 (t, J = 7.0 Hz, 3H, CH3). 13C NMR (125 MHz, CDCl3) δ 159.7, 153.1, 137.9, 135.6, 130.1, 126.2, 122.23 117.3, 91.3, 66.8, 14.9. HRMS (ESI/Q-TOF) m/z: [M + Na]+ Calcd for C11H10ClO3Na 247.0138; Found 247.0127. m.p. 94–96 °C. Melting point was consistent with the reported value [31].
- 3-Propoxy-4-chloroisocoumarin (2c). 1H, 13C NMR and melting point were consistent with those previously reported [19].
- 3-Butoxy-4-chloroisocoumarin (2d). 1H, 13C NMR and melting point were consistent with those previously reported [19].
- 3-iso-Propoxy-4-chloroisocoumarin (2e). Pale yellow solid (0.235 g, 34%) 1H NMR: (500 MHz, CDCl3) δ 8.21 (d, J = 8.0 Hz, 1H, H-8), 7.78–7.71 (m, 2H, H-6 and H-5), 7.40 (dt, J = 7.0, 1.5 Hz, 1H, H-7), 5.06 (sep, J = 6.0 Hz, 1H, CH), (d, J = 6.0 Hz, 6H, 2 × CH3). 13C NMR: (125 MHz, CDCl3) δ 160.0, 152.7, 137.8, 135.6, 130.1, 126.3, 122.5, 117.6, 93.0, 75.6, 22.3. HRMS (ESI/Q-TOF) m/z: [M + Na]+ Calcd for C12H11ClO3Na 261.0289; Found 261.0284. m.p. 58–60 °C.
- 3-iso-Butoxy-4-chloroisocoumarin (2f). Yellow solid (0.255 g, 34%) 1H NMR: (500 MHz, CDCl3) δ 8.20 (dd, J = 7.5, 1.5 Hz, 1H, H-8), 7.77–7.70 (m, 2H, H-6 and H-5), 7.39 (dt, J = 7.0, 1.5 Hz, 1H, H-7), 4.15 (d, J = 6.5 Hz, 2H, CH2), (sep, J = 7.0 Hz, 1H, CH), 1.05 (d, J = 7.0 Hz, 6H, 2 × CH3).13C NMR: (125 MHz, CDCl3) δ 159.7, 153.3, 138.0, 135.6, 130.1, 126.1, 122.2, 117.3, 91.0, 76.6, 28.4, 18.8. HRMS (ESI/Q-TOF) m/z: [M + Na]+ Calcd for C13H13ClO3Na 275.0451; Found 275.0444. m.p. 48–51 °C. Melting point was consistent with the reported value [16].
- (±)-3-sec-Butoxy-4-chloroisocoumarin (2g). Yellow solid (0.215 g, 28%) 1H NMR: (500 MHz, CDCl3) δ 8.21 (br d, J = 7.5 Hz, 1H, H-8), 7.77–7.71 (m, 2H, H-6 and H-5), 7.40 (dt, J = 7.5, 1.5 Hz, 1H, H-7), 4.87 (sxt, J = 6.0 Hz, 1H, CH), 1.86–1.66 (m, 2H, CH2), 1.40 (d, J = 6.5 Hz, 3H, OCHCH3), 1.03 (t, J = 7.5 Hz, 3H, CH2CH3). 13C NMR: (125 MHz, CDCl3) δ 160.0, 152.9, 137.9, 135.6, 130.1, 126.2, 122.4, 117.6, 92.6, 80.2, 29.2, 19.8, 9.5. HRMS (ESI/Q-TOF) m/z: [M + Na]+ Calcd for C13H13O3ClNa 275.0451; Found 275.0439. m.p. 57–59 °C.
3.1.3. General Procedure for the Synthesis of 5-Substituted Homophthalate Monoesters (9 and 13) (General Procedure B)
- (±)-5-Chloro-2-(2-sec-butoxy-2-oxo-ethyl)benzoic acid (9). White solid (0.300 g, 48%). 1H NMR: (400 MHz, CDCl3) δ 10.66 (br s, 1H, COOH), 8.09 (d, J = 2.4 Hz, 1H, H-6), 7.50 (dd, J = 2.4, 8.0 Hz, 1H, H-4), 7.22 (d, J = 7.6 Hz, 1H, H-3), 4.86 (sxt, J = 6.4 Hz, 1H, CH), 3.99 (d, J = 2.0 Hz, 2H, CH2CO2), 1.65–1.47 (m, 2H, CH2), 1.20 (d, J = 6.4 Hz, 3H, OCHCH3), 0.88 (t, J = 7.6 Hz, 3H, CH2CH3). 13C NMR: (100 MHz, CDCl3) δ 171.3, 170.7, 135.5, 133.7, 133.4, 133.2, 131.7, 130.1, 73.1, 40.4, 28.7, 19.3, 9.6. HRMS (ESI/Q-TOF) m/z: [M + Na]+ Calcd for C13H15O4Na 293.0557; Found 293.0548. m.p. 100–103 °C.
- (±)-5-Methoxy-2-(2-sec-butoxy-2-oxo-ethyl)benzoic acid (13). Brown solid (1.53 g, 62%) 1H NMR: (400 MHz, CDCl3) δ 7.64 (d, J = 2.8 Hz, 1H, H-6), 7.18 (d, J = 8.4 Hz, 1H, H-3), 7.07 (dd, J = 3.2 Hz, 8.4 Hz, 1H, H-4), 4.86 (sxt, J = 6.4 Hz, 1H, CH), 3.95 (d, J = 2.0 Hz, 2H, CH2CO2), 3.85 (s, 3H, OCH3), 1.65-1.43 (m, 2H, CH2), 1.20 (d, J = 6.0 Hz, 3H, OCHCH3), 0.88 (d, J = 7.6 Hz, 3H, CH2CH3). 13C NMR: (100 MHz, CDCl3) δ 172.4, 171.6, 158.5, 133.4, 129.5, 129.2, 119.3, 116.5, 72.7, 55.5, 40.2, 28.7, 19.3, 9.6. HRMS (ESI/Q-TOF) m/z: [M + Na]+ Calcd for C13H18O5Na 289.1052; Found 289.1048. m.p. 115–118 °C.
3.1.4. Synthesis of (±)-5-Nitro-2-(2-sec-butoxy-2-oxo-ethyl)benzoic acid (4)
- (±)-5-Nitro-2-(2-sec-butoxy-2-oxo-ethyl)benzoic acid (4) White solid (6.50 g, 76%) 1H NMR: (400 MHz, CDCl3) δ 8.95 (s, J = 2.4 Hz, 1H, H-6), 8.38 (dd, J = 2.4 Hz, 8.4 Hz, 1H, H-4), 7.50 (d, J = 8.4 Hz, 1H, H-3), 4.88 (sxt, J = 6.2 Hz, 1H, CH), 4.16 (d, J = 2.8 Hz, 2H, H-2′), 1.66–1.50 (m, 2H, CH2CH3), 1.22 (d, J = 6.2 Hz, 3H, OCHCH3), 0.88 (t, J = 7.4 Hz, 3H, CH2CH3). 13C NMR: (100 MHz, CDCl3) δ 170.3, 169.8, 147.1, 143.9, 133.7, 130.02, 127.4, 126.8, 73.6, 40.9, 28.7, 19.3, 9.6. HRMS (ESI/Q-TOF) m/z: [M + Na]+ Calcd for C13H15NO6Na 304.0797; Found 304.0790. m.p. 112–115 °C.
- (±)-3-sec-Butoxy-7-nitro-4-chloroisocoumarin (5). Synthesized using general procedure A with minor modifications. The reaction was performed at reflux and the product was triturated with methanol to afford yellow needles (2.49 g, 59%). 1H NMR: (400 MHz, CDCl3) δ 9.02 (d, J = 2.0 Hz, 1H, H-8), 8.51 (dd, J = 9.2, 2.4 Hz, 1H, H-6), 7.82 (d, J = 8.8 Hz, 1H, H-5), 5.02 (m, 1H, CH), 1.90–1.70 (m, 2H, CH2), 1.45 (d, J = 6.2 Hz, 3H, OCHCH3), 1.03 (t, J = 7.4 Hz, 3H, CH2CH3). 13C NMR: (100 MHz, CDCl3) δ 157.8, 155.7, 145.0, 143.2, 129.6, 126.3, 123.5, 116.8, 90.8, 81.2, 29.2, 20.0, 9.4. HRMS (ESI/Q-TOF) m/z: [M + Na]+ Calcd for C13H12NO5ClNa 320.0302; Found 320.0292. m.p. 108–111 °C.
3.1.5. Synthesis of (±)-7-Amino-3-sec-butoxy-4-chloroisocoumarin (6a)
- (±)-7-Amino-4-chloro-3-sec-butoxyisocoumarin (6a). Yellow solid (1.70 g, 95%) 1H NMR: (400 MHz, CDCl3) δ 7.53 (d, J = 8.8 Hz, 1H, H-5), 7.45 (d, J = 2.4 Hz, 1H, H-8), 7.10 (dd, J = 8.8, 2.4 Hz, 1H, H-6), 4.72 (sxt, J = 6.0 Hz, 1H, CH), 3.95 (br s, 2H, NH2), 1.84–1.63 (m, 2H, CH2), 1.35 (d, J = 6.0 Hz, 3H, OCHCH3), 1.01 (t, J = 7.6 Hz, 3H, CH2CH3). 13C NMR: (100 MHz, CDCl3) δ 160.4, 150.5, 145.4, 128.8, 123.9, 123.6, 119.2, 113.1, 94.2, 80.4, 29.2, 19.6, 9.5. HRMS (ESI/Q-TOF) m/z: [M + Na]+ Calcd for C13H14ClNO3Na 290.0560; Found 290.0566. m.p. 86–88 °C.
3.1.6. Synthesis of (±)-7-(dimethylamino)-3-sec-butoxy-4-chloroisocoumarin (6b)
- (±)-7-(Dimethylamino)-3-sec-butoxy-4-chloroisocoumarin (6b). 1H NMR: (400 MHz, CDCl3) δ 7.58 (d, J = 8.8 Hz, 1H, H-8), 7.39 (d, J = 2.8 Hz, 1H, H-5), 7.18 (dd, J = 2.8, 9.2 Hz, 1H, H-6), 4.72 (sxt, 6.0 Hz, 1H, CH), 3.03 (s, 6H, N(CH3)2), 1.85–1.63 (m, 2H, CH2), 1.36 (d, J = 6.4 Hz, 3H, OCHCH3), 1.02 (t, J = 7.6 Hz, 3H, CH2CH3). 13C NMR: (100 MHz, CDCl3) δ 160.9, 150.1, 149.1, 126.6, 123.6, 120.8, 119.1, 110.3, 94.5, 80.3, 40.5, 29.2, 19.6, 9.5. HRMS (ESI/Q-TOF) m/z: [M + Na]+ Calcd for C15H18ClNO3Na 318.0867; Found 318.0870.
3.1.7. Synthesis of (±)-N-(4-Chloro-1-oxo-3-sec-butoxy-isochromen-7-yl)acetamide (6c)
- (±)-N-(4-Chloro-1-oxo-3-sec-butoxy-isochromen-7-yl)acetamide (6c). 1H NMR: (400 MHz, CDCl3) δ 8.21 (dd, J = 2.1 Hz, 8.8 Hz, 1H, H-6), 8.11 (d, J = 2.2 Hz, 1H, H-8), 7.69 (d, J = 8.8 Hz, 1H, H-5), 7.61 (br s, 1H, NH), 4.85–4.78 (m, 1H, CH), 2.23 (s, 3H, NHCOCH3), 1.85-1.67 (m, 2H, CH2), 1.38 (d, J = 6.2 Hz, 3H, OCHCH3), 1.02 (t, J = 7.4, 3H, CH2CH3). 13C NMR: (100 MHz, CDCl3) δ 168.9, 160.0, 152.1, 136.9, 133.8, 128.0, 123.5, 119.4, 117.9, 93.2, 80.6, 29.2, 24.4, 19.8, 9.5. HRMS (ESI/Q-TOF) m/z: [M + Na]+ Calcd for C15H16ClNO4Na 332.0660; Found 332.0660,. m.p. 161 °C (decomposed with gas evolution).
3.1.8. (±)-N-(4-chloro-1-oxo-3-sec-butoxy-isochromen-7-yl)-2,2,2-trifluoroacetamide (6d)
3.1.9. General Procedure for the Synthesis of 6e and 6f
- (±)-3-sec-Butoxy-4-chloro-7-iodoisocoumarin (6e). The mixture was poured into an aqueous solution (100 mL) of 5% sodium bicarbonate containing sodium thiosulfate (7.5 g). The product was extracted into hexane (75 mL) and then washed with water (50 mL) and brine (50 mL) and then dried using anhydrous magnesium sulfate. The solvent was loaded onto a silica gel column and purified with a gradient of 0–20% dichloromethane in hexanes, affording a yellow solid (1.38 g, 65%).1H NMR: (400 MHz, CDCl3) δ 8.49 (d, J = 1.6 Hz, 1H, H-8), 7.99 (dd, J = 2.0, 8.4 Hz, 1H, H-6), 7.43 (d, J = 8.4 Hz, 1H, H-5), 4.87 (sxt, 6.0 Hz, 1H, CH), 1.86–1.59 (m, 2H, CH2), 1.39 (d, J = 6.0 Hz, 3H, CHCH3), 1.02 (t, J = 7.6 Hz, 3H, CH2CH3). 13C NMR: (100 MHz, CDCl3) δ 158.4, 153.2, 144.1, 138.4, 137.3, 124.1, 118.9, 91.9, 89.6, 80.5, 29.2, 19.9, 9.5. HRMS (ESI/Q-TOF) m/z: [M + Na]+ Calcd for C13H12ClIO3Na 400.9417; Found 400.9408. m.p. 69–72 °C
- (±)-7-Bromo-3-sec-butoxy-4-chloro-isocoumarin (6f). The solvent was removed with reduced pressure at room temperature, and the crude solid was sonicated in hexanes for 15 min. The slurry was loaded onto a silica gel column and purified with a gradient of 0–20% dichloromethane in hexanes, affording a yellow solid (0.170 g, 17%). 1H NMR: (400 MHz, CDCl3) δ 8.32 (d, J = 2.0 Hz, 1H, H-8), 7.82 (dd, J = 2.0, 8.4 Hz, 1H, H-6), 7.58 (d, J = 8.8 Hz, 1H, H-5), 4.87 (sxt, 6.0 Hz, 1H, CH), 1.87–1.66 (m, 2H, CH2), 1.39 (t, J = 6.4 Hz, 3H, CH2CH3), 1.01 (d, J = 6.4 Hz, 3H, CHCH3). 13C NMR: δ 159.6, 153.1, 138.6, 136.9, 132.3, 124.2, 119.4, 118.8, 92.0, 80.6, 29.2, 19.8, 9.5. HRMS (ESI/Q-TOF) m/z: [M + Na]+ Calcd for C13H12BrClO3Na 352.9556; Found 352.9542. m.p. 78–80 °C.
3.1.10. Synthesis of Compounds 6g and 6h
- (±)-3-sec-Butoxy-4,7-dichloro-isocoumarin (6g). Synthesized using general procedure A to afford a yellow solid (0.328 g, 63%) 1H NMR: (400 MHz, CDCl3) δ 8.16 (d, J = 1.6 Hz, 1H, H-8), 7.67–7.64 (m, 2H, H-5 and H-6) 4.87 (sxt, J = 6.4 Hz, 1H, CH), 1.87–1.66 (m, 2H, CH2), 1.40 (d, J = 6.0 Hz, 3H, OCHCH3), 1.02 (t, J = 7.6 Hz, 3H, CH2CH3). 13C NMR: (100 MHz, CDCl3) δ 158.8, 153.1, 136.5, 135.8, 132.0, 129.3, 124.1, 118.6, 92.0, 80.6, 29.2, 19.8, 9.5. HRMS (ESI/Q-TOF) m/z: [M + Na]+ Calcd for C13H12Cl2O3H+ 309.0061; Found 309.0051. m.p. 86–88 °C.
- (±)-7-Methoxy-3-sec-butoxy-4-chloroisocoumarin (6h). Synthesized using general procedure A to afford a yellow solid (0.410 g, 77%) 1H NMR: (400 MHz, CDCl3) δ 7.62 (d, J = 8.8 Hz, 1H, H-5), 7.59 (d, J = 2.8 Hz, 1H, H-8), 7.32 (dd, J = 8.8, 2.8 Hz, 1H, H-6), 4.78 (sxt, J = 6.0 Hz, 1H, CH), 3.88 (s, 3H, OCH3), 1.85-1.64 (m, 2H, CH2), 1.37 (d, J = 6.0 Hz, 3H, OCHCH3), 1.01 (t, J = 7.6 Hz, 3H, CH2CH3). 13C NMR: (100 MHz, CDCl3) δ 160.0, 158.2, 151.4, 131.4, 125.1, 124.1, 118.7, 110.4, 93.4, 80.4, 55.7, 29.2, 19.7, 9.4. HRMS (ESI/Q-TOF) m/z: [M + Na]+ Calcd for C14H15O4ClNa 305.0549; Found 305.0557. m.p. 55–56 °C.
3.2. Biological Studies
3.2.1. Recombinant Expression and Purification of CtHtrA
3.2.2. CtHtrA Protease Inhibition Assay
3.2.3. HLE Protease Inhibition Assay
3.2.4. MTS Cell Viability Assay
3.2.5. Chlamydia trachomatis Growth Inhibition Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Centre for Disease Control and Prevention. Sexually Transmitted Disease Surveillance 2018; U.S. Department of Health and Human Services: Atlanta, GA, USA, 2019.
- Menon, S.; Timms, P.; Allan, J.A.; Alexander, K.; Rombauts, L.; Horner, P.; Keltz, M.; Hocking, J.; Huston, W.M. Human and Pathogen Factors Associated with Chlamydia trachomatis-Related Infertility in Women. Clin. Microbiol. Rev. 2015, 28, 969–985. [Google Scholar] [CrossRef] [PubMed]
- Cyr, S.S.; Barbee, L.; Workowski, K.A.; Bachmann, L.; Pham, C.; Schlanger, K.; Torrone, E.; Weinstock, H.; Kersh, E.N.; Thorpe, P. Update to CDC’s Treatment Guidelines for Gonococcal Infection, 2020. Morb. Mortal. Wkly. Rep. 2020, 69, 1911–1916. [Google Scholar] [CrossRef] [PubMed]
- Cross, R.; Ling, C.; Day, N.P.J.; McGready, R.; Paris, D.H. Revisiting doxycycline in pregnancy and early childhood—Time to rebuild its reputation? Expert Opin. Drug Saf. 2016, 15, 367–382. [Google Scholar] [CrossRef] [PubMed]
- Bongard, J.; Schmitz, A.L.; Wolf, A.; Zischinsky, G.; Pieren, M.; Schellhorn, B.; Bravo-Rodriguez, K.; Schillinger, J.; Koch, U.; Nussbaumer, P.; et al. Chemical Validation of DegS As a Target for the Development of Antibiotics with a Novel Mode of Action. ChemMedChem 2019, 14, 1074–1078. [Google Scholar] [CrossRef] [PubMed]
- Hoy, B.; Löwer, M.; Weydig, C.; Carra, G.; Tegtmeyer, N.; Geppert, T.; Schröder, P.; Sewald, N.; Backert, S.; Schneider, G.; et al. Helicobacter pylori HtrA is a new secreted virulence factor that cleaves E-cadherin to disrupt intercellular adhesion. EMBO Rep. 2010, 11, 798–804. [Google Scholar] [CrossRef] [PubMed]
- Marsh, J.; Gloeckl, S.; Tyndall, J.D.A.; Huston, W.M. The role of HtrA as a chaperone and protease in bacterial pathogenesis. In Bacterial Pathogens: Virulence Mechanisms, Diagnosis and Management; Boulanger, A., Blanc, M., Eds.; Nova Science Publishers: Hauppauge, NY, USA, 2012; pp. 117–164. [Google Scholar]
- Huston, W.M.; Theodoropoulos, C.; Mathews, S.A.; Timms, P. Chlamydia trachomatis responds to heat shock, penicillin induced persistence, and IFN-gamma persistence by altering levels of the extracytoplasmic stress response protease HtrA. BMC Microbiol. 2008, 8, 190. [Google Scholar] [CrossRef] [PubMed]
- Gloeckl, S.; Ong, V.A.; Patel, P.; Tyndall, J.D.A.; Timms, P.; Beagley, K.W.; Allan, J.A.; Armitage, C.W.; Turnbull, L.; Whitchurch, C.B.; et al. Identification of a serine protease inhibitor which causes inclusion vacuole reduction and is lethal to Chlamydia trachomatis. Mol. Microbiol. 2013, 89, 676–689. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Lu, X.; Huang, D.; Lu, Y.; Zhang, H.; Zhang, L.; Yu, P.; Wang, F.; Wang, Y. A novel protease inhibitor causes inclusion vacuole reduction and disrupts the intracellular growth of Chlamydia trachomatis. Biochem. Biophys. Res. Commun. 2019, 516, 157–162. [Google Scholar] [CrossRef] [PubMed]
- Hwang, J.; Strange, N.; Phillips, M.J.A.; Krause, A.L.; Heywood, A.; Gamble, A.B.; Huston, W.M.; Tyndall, J.D.A. Optimization of peptide-based inhibitors targeting the HtrA serine protease in Chlamydia: Design, synthesis and biological evaluation of pyridone-based and N-Capping group-modified analogues. Eur. J. Med. Chem. 2021, 224, 113692. [Google Scholar] [CrossRef] [PubMed]
- Hwang, J.; Strange, N.; Mazraani, R.; Phillips, M.J.; Gamble, A.B.; Huston, W.M.; Tyndall, J.D.A. Design, synthesis and biological evaluation of P2-modified proline analogues targeting the HtrA serine protease in Chlamydia. Eur. J. Med. Chem. 2022, 230, 114064. [Google Scholar] [CrossRef] [PubMed]
- Agbowuro, A.A.; Mazraani, R.; McCaughey, L.C.; Huston, W.M.; Gamble, A.B.; Tyndall, J.D.A. Stereochemical basis for the anti-chlamydial activity of the phosphonate protease inhibitor JO146. Tetrahedron 2018, 74, 1184–1190. [Google Scholar] [CrossRef]
- Saputra, P.B.T.; Purwati, D.D.; Ulhaq, A.U.D.; Yolanda, S.; Djatioetomo, Y.C.E.D.; Rosyid, A.N.; Bakhtiar, A. Neutrophil Elastase in the Pathogenesis of Chronic Obstructive Pulmonary Disease: A Review. Curr. Respir. Med. Rev. 2023, 19, 29–35. [Google Scholar] [CrossRef]
- Harper, J.W.; Powers, J.C. 3-Alkoxy-7-amino-4-chloroisocoumarins: A New Class of Suicide Substrates for Serine Proteases. J. Am. Chem. Soc. 1984, 106, 7618–7619. [Google Scholar] [CrossRef]
- Harper, J.W.; Powers, J.C. Reaction of Serine Proteases with Substituted 3-Alkoxy-4-chloroisocoumarins and 3-Alkoxy-7-amino-4-chloroisocoumarins: New Reactive Mechanism-Based Inhibitors. Biochemistry 1985, 24, 7200–7213. [Google Scholar] [CrossRef] [PubMed]
- Powers, J.C.; Kam, C.M.; Narasimhan, L.; Oleksyszyn, J.; Hernandez, M.A.; Ueda, T. Mechanism-based isocoumarin inhibitors for serine proteases: Use of active site structure and substrate specificity in inhibitor design. J. Cell. Biochem. 1989, 39, 33–46. [Google Scholar] [CrossRef] [PubMed]
- Kerrigan, J.E.; Oleksyszyn, J.; Kam, C.M.; Selzler, J.; Powers, J.C. Mechanism-Based Isocoumarin Inhibitors for Human Leukocyte Elastase. Effect of the 7-Amino Substituent and 3-Alkoxy Group in 3-Alkoxy-7-amino-4-chloroisocoumarins on Inhibitory Potency. J. Med. Chem. 1995, 38, 544–552. [Google Scholar] [CrossRef] [PubMed]
- Heynekamp, J.J.; Hunsaker, L.A.; Vander Jagt, T.A.; Royer, R.E.; Deck, L.M.; Vander Jagt, D.L. Isocoumarin-based inhibitors of pancreatic cholesterol esterase. Bioorg. Med. Chem. 2008, 16, 5285–5294. [Google Scholar] [CrossRef] [PubMed]
- Kam, C.M.; Kerrigan, J.E.; Plaskon, R.R.; Duffy, E.J.; Lollar, P.; Suddath, F.L.; Powers, J.C. Mechanism-Based Isocoumarin Inhibitors for Blood Coagulation Serine Proteases. Effect of the 7-Substituent in 7-Amino-4-chloro-3-(isothioureidoalkoxy)isocoumarins on Inhibitory and Anticoagulant Potency. J. Med. Chem. 1994, 37, 1298–1306. [Google Scholar] [CrossRef]
- Peuchmaur, M.; Lacour, M.; Sévalle, J.; Lisowski, V.; Touati-Jallabe, Y.; Rodier, F.; Martinez, J.; Checler, F.; Hernandez, J. Further characterization of a putative serine protease contributing to the γ-secretase cleavage of β-amyloid precursor protein. Bioorg. Med. Chem. 2013, 21, 1018–1029. [Google Scholar] [CrossRef] [PubMed]
- Billamboz, M.; Bailly, F.; Barreca, M.L.; De Luca, L.; Mouscadet, J.; Calmels, C.; Andréola, M.; Witvrouw, M.; Christ, F.; Debyser, Z.; et al. Design, Synthesis, and Biological Evaluation of a Series of 2-Hydroxyisoquinoline-1,3(2H,4H)-diones as Dual Inhibitors of Human Immunodeficiency Virus Type 1 Integrase and the Reverse Transcriptase RNase H Domain. J. Med. Chem. 2008, 51, 7717–7730. [Google Scholar] [CrossRef]
- Ram, S.; Ehrenkaufer, R.E. A general procedure for mild and rapid reduction of aliphatic and aromatic nitro compounds using ammonium formate as a catalytic hydrogen transfer agent. Tetrahedron Lett. 1984, 25, 3415–3418. [Google Scholar] [CrossRef]
- Zhao, Q.; Xu, X.; Xie, Z.; Liu, X.; You, Q.; Guo, Q.; Zhong, Y.; Li, Z. Design, synthesis and biological evaluation of 3-substituted indenoisoquinoline derivatives as topoisomerase I inhibitors. Bioorg. Med. Chem. Lett. 2016, 26, 1068–1072. [Google Scholar] [CrossRef] [PubMed]
- Theodorou, V.; Skobridis, K.; Tzakos, A.G.; Ragoussis, V. A simple method for the alkaline hydrolysis of esters. Tetrahedron Lett. 2007, 48, 8230–8233. [Google Scholar] [CrossRef]
- Hansch, C.; Leo, A.; Unger, S.H.; Kim, K.H.; Nikaitani, D.; Lien, E.J. Aromatic substituent constants for structure-activity correlations. J. Med. Chem. 1973, 16, 1207–1216. [Google Scholar] [CrossRef] [PubMed]
- Suchland, R.J.; Geisler, W.M.; Stamm, W.E. Methodologies and Cell Lines Used for Antimicrobial Susceptibility Testing of Chlamydia spp. Antimicrob. Agents Chemother. 2003, 47, 636–642. [Google Scholar] [CrossRef] [PubMed]
- Bharti, S.K.; Roy, R. Quantitative 1H NMR spectroscopy. TrAC Trends Anal. Chem. 2012, 35, 5–26. [Google Scholar] [CrossRef]
- Cushman, M.; Georg, G.I.; Holzgrabe, U.; Wang, S. Absolute Quantitative 1H NMR Spectroscopy for Compound Purity Determination. J. Med. Chem. 2014, 57, 9219. [Google Scholar] [CrossRef] [PubMed]
- Gopinath, P.; Nilaya, S.; Muraleedharan, K.M. Highly Chemoselective Esterification Reactions and Boc/THP/TBDMS Discriminating Deprotections under Samarium(III) Catalysis. Org. Lett. 2011, 13, 1932–1935. [Google Scholar] [CrossRef] [PubMed]
- Tirodkar, R.B. Isocoumarins. VI. 3-Alkoxy-4-chloroisocoumarins. Indian J. Chem. 1969, 7, 1114–1116. [Google Scholar]
- Huston, W.M.; Swedberg, J.E.; Harris, J.M.; Walsh, T.P.; Mathews, S.A.; Timms, P. The temperature activated HtrA protease from pathogen Chlamydia trachomatis acts as both a chaperone and protease at 37 °C. FEBS Lett. 2007, 581, 3382–3386. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Relative Potency against CtHtrA a | Relative Potency against HLE b | Relative Selectivity c |
|---|---|---|---|
| JO146 | 1.00 | 1.00 | 1.00 |
| 2a | 2.33 | 0.0207 | 113.0 |
| 2b | 3.73 | 0.0664 | 56.4 |
| 2c | 2.23 | 0.323 | 6.92 |
| 2d | 1.57 | 0.310 | 5.08 |
| 2e | 5.32 | 0.419 | 12.7 |
| 2f | 1.50 | 0.368 | 4.08 |
| 2g | 6.58 | 0.451 | 14.6 |
| 6a | 0.326 | n.d. | n.d. |
| 6b | 0.113 | n.d. | n.d. |
| 6c | 0.158 | n.d. | n.d. |
| 6d | <0.001 | n.d. | n.d. |
| 6e | <0.001 | n.d. | n.d. |
| 6f | 0.057 | n.d. | n.d. |
| 6g | 0.390 | n.d. | n.d. |
| 6h | 0.184 | n.d. | n.d. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Phillips, M.J.A.; Huston, W.M.; McDonagh, A.M.; Rawling, T. 4-Chloroisocoumarins as Chlamydial Protease Inhibitors and Anti-Chlamydial Agents. Molecules 2024, 29, 1519. https://doi.org/10.3390/molecules29071519
Phillips MJA, Huston WM, McDonagh AM, Rawling T. 4-Chloroisocoumarins as Chlamydial Protease Inhibitors and Anti-Chlamydial Agents. Molecules. 2024; 29(7):1519. https://doi.org/10.3390/molecules29071519
Chicago/Turabian StylePhillips, Matthew J. A., Wilhelmina M. Huston, Andrew M. McDonagh, and Tristan Rawling. 2024. "4-Chloroisocoumarins as Chlamydial Protease Inhibitors and Anti-Chlamydial Agents" Molecules 29, no. 7: 1519. https://doi.org/10.3390/molecules29071519