Abstract
Easy-to-handle N-hydroxyacridinecarbimidoyl chloride hydrochlorides were synthesized as convenient nitrile oxide precursors in the preparation of 3-(acridin-9/2-yl)isoxazole derivatives via 1,3-dipolar cycloaddition with terminal alkynes, 1,1-dichloroethene, and acrylonitrile. Azirines with an acridin-9/2-yl substituent attached directly or via the 1,2,3-triazole linker to the azirine C2 were also synthesized. The three-membered rings of the acridine–azirine hybrids were found to be resistant to irradiation in the UV/visible boundary region, despite their long-wave absorption at 320–420 nm, indicating that the acridine moiety cannot be used as an antenna to transfer light energy to generate nitrile ylides from azirines for photoclick cycloaddition. The acridine–isoxazole hybrids linked at the C9–C3 or C2–C3 atoms under blue light irradiation underwent the addition of such hydrogen donor solvents, such as, toluene, o-xylene, mesitylene, 4-chlorotoluene, THF, 1,4-dioxane, or methyl tert-butyl ether (MTBE), to the acridine system to give the corresponding 9-substituted acridanes in good yields. The synthesized acridine–azirine, acridine–isoxazole, and acridane–isoxazole hybrids exhibited cytotoxicity toward both all tested cancer cell lines (HCT 116, MCF7, and A704) and normal cells (WI-26 VA4).
1. Introduction
Acridine derivatives exhibit a number of biological activities, including anticancer, antimicrobial, antibiotic, antiacetylcholinesterase, antileukemic, antiprotozoal, neuroleptic, anti-dementia, telomerase inhibitory, and many others [1,2,3,4,5]. A stimulating strategy in drug discovery to maximize efficacy, minimize side effects, and combat drug resistance is the synthesis of hybrid molecules that include a combination of two biologically relevant heterocyclic moieties that act on different targets [6]. In particular, a number of molecular hybrids [7,8,9,10,11,12,13,14,15] containing an acridine moiety have been prepared because of efforts to develop new therapeutic agents. Thus, tacrine–acridine [8], cyclopentaquinoline–acridine [9], and acridine–flavone hybrids [10] are potentially useful for treating Alzheimer’s disease, quinoline–acridine and pyrrolidine–acridine–artemisinin hybrids are antimalarials [11,12,13], neocryptolepine–acridine hybrids exhibit antiproliferative activity [6,14], and thiophene–acridine hybrids demonstrate antitumor activity [3,15]. Another very important class of N-heterocyclic compound is isoxazoles because they have a wide spectrum of biological activity used as base for developing of several commercially available drugs [16,17,18,19,20,21,22,23,24,25,26]. Some biologically active hybrids of isoxazoles with various heterocycles have been synthesized and studied [20,21,22,23,24,25,26,27]. Meanwhile, not many acridines containing an isoxazole moiety have been obtained so far. Thus, 9-(4-isoxazolylanilino)acridines with antibacterial and larvicidal activity were synthesized from corresponding chalcones [28]. In an attempt to at least partially fill the gap in the synthesis of acridine–isoxazole hybrids, we report a simple approach to the synthesis of various isoxazole–acridine and azirine–acridine hybrids, in which these heterocycles are directly linked, as well as some of their photochemical reactions under irradiation in the UV/visible boundary region, allowing the easy synthesis of acridan–isoxazole hybrids. The anticancer activity of a number of synthesized acridine/acridan-substituted isoxazoles and acridine-substituted azirines is also reported.
2. Results and Discussion
The only known (acridin-9-yl)isoxazole, ethyl 3-(acridin-9-yl)-5-methylisoxazole-4-carboxylate, was prepared from acridine-9-carbaldehyde oxime 1a by converting it into unstable N-hydroxyacridine-9-carbimidoyl chloride 2a under the action of N-chlorosuccinimide, followed by immediate treatment with triethylamine and ethyl 3-(pyrrolidin-1-yl)but-2-enoate [29]. We were unsuccessful in our attempts to apply the procedure proposed in this work for generating acridinyl-substituted nitrile oxide to obtain derivatives of 9-(isoxazolyl)acridines through cycloaddition to alkenes and acetylenes. In all experiments, the only compound isolated was the hydrolysis product of N-hydroxyacridine-9-carbimidoyl chloride 2a to acridine-9-carboxylic acid. Since the synthesis of N-hydroxyacridine-9-carbimidoyl chloride 2a and its application described in the work [29] have never been applied by other authors, unlike the anthracene analogue [30], we decided to find another, more reliable approach. It was found that the use of easy-to-handle N-hydroxyacridine-9-carbimidoyl chloride hydrochlorides 2-HCl, instead of the unstable and moisture-sensitive N-hydroxyacridine-9-carbimidoyl chloride 2a, allowed for the cycloaddition of acridinyl-substituted nitrile oxides 3 to acetylenes and alkenes to be carried out at a high yield (Scheme 1). These compounds were easily prepared in good to excellent yields by the oxidation of 9-methylacridines 4a–c or 7-methylbenzo[c]acridine 4d with SeO2, the condensation of aldehydes 5a–d with hydroxylamine to give oximes 1a–d, followed by chlorination with Cl2. The reaction of nitrile oxides 3a–d, generated from compounds 2a–d-HCl in a two-phase system DCM/sat. aq. NaHCO3, with alkynes 6a–g gave 3-(acridin-9-yl)isoxazoles 7a–h in 90–98% yields. The cycloaddition of nitrile oxide 3a, generated from compounds 2a-HCl, to 1,1-dichloroethene 8 accompanied the dehydrochlorination of intermediate 5,5-dichloro-4,5-dihydroisoxazoles and gave acridinyl-substituted 5-chloroizoxazole 7i in a 62% yield (Scheme 1). The cycloaddition of nitrile oxide 3b to acrylonitrile 9 afforded 3-(2-methylacridin-9-yl)-4,5-dihydroisoxazole-5-carbonitrile 10 in an 83% yield (Scheme 1).
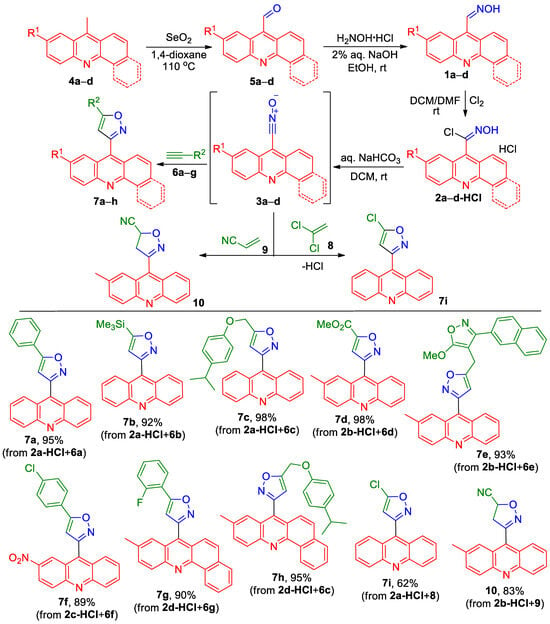
Scheme 1.
Synthesis of acridinyl-substituted isoxazole derivatives 7a–i, 10.
Analogously, by the cycloaddition of nitrile oxide 3e, generated from compound 2e-HCl, to 1,1-dichloroethene 8, 3-(acridin-2-yl)isoxazole 7j was prepared in a 64% yield (Scheme 2).
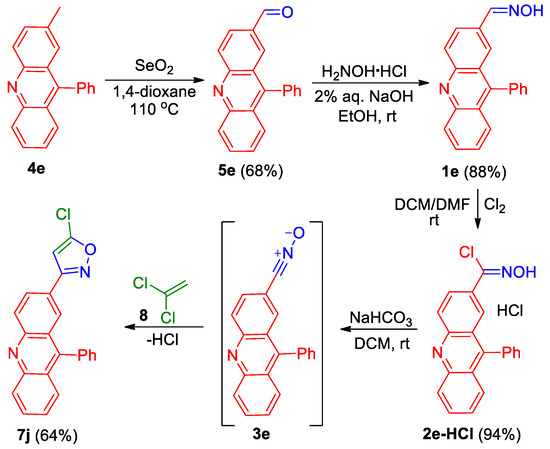
Scheme 2.
Synthesis of 3-(acridin-2-yl)isoxazole 7j.
Acridinyl-substituted 5-chloroisoxazoles 7i,j can be used for the preparation of other derivatives by the substitution of chlorine with O-nucleophiles (Scheme 3). Thus, the reaction of 5-chloroisoxazoles 7i,j with tBuOK gives 5-(tert-butoxy)isoxazoles 7k,l in a 68% and 90% yield, respectively (Scheme 3).
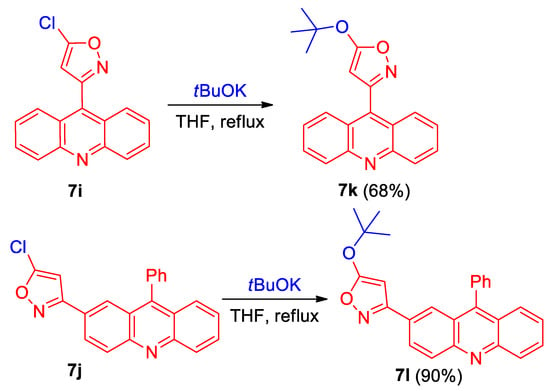
Scheme 3.
Synthesis of acridinyl-substituted isoxazoles 7k and l.
Isoxazoles with a heteroatom substituent at C5 have found wide application as convenient precursors of 2H-azirine-2-carboxylic acid derivatives, versatile building blocks for organic synthesis [31]. A very large number of structurally diverse azirines have been synthesized to date, including those which are of interest for biorthogonal chemistry [32], the chemistry of natural compounds [33], and medicine [34], but azirines with acridinyl substituents are still unknown in the literature.
Having acridinyl-substituted isoxazoles at our disposal, we decided to prepare acridinyl-substituted azirines in order to evaluate the possibility of their use in photoclick cycloaddition [35,36,37,38]. The authors of the work [35] synthesized 3-(pyren-1-yl)-2H-azirine, which absorbs light in the 400 nm region, allowing for the activation of an efficient cycloaddition reaction for azirine-based ligation, which is potentially suitable for the bioorthogonal conjugation of polymers and peptides using low-energy visible light sources. According to them, such azirine allows for avoiding handling problems during synthesis or sample preparation, and the pyrene group is not only critical for light absorption, but is also incorporated into the carbon backbone of the cycloadducts, thereby providing additional desirable functions such as an integrated fluorescent marker or an anchor for π-π stacking. We assumed that the involvement of acridine-substituted azirines in photoinitiated cycloaddition reactions, using low-energy visible light sources, cannot be ruled out, since the acridinyl substituent provides light absorbance in the UV/visible radiation boundary region (385–405 nm) [39,40]. Moreover, acridines bind to DNA and RNA by intercalation, and their fluorescence could also be of interest in the case of the implementation of the discussed cycloaddition reaction. Azirine 11a was obtained by the isoxazole–azirine isomerization [31] of isoxazole 7l catalyzed by FeCl2·4H2O at an 87% yield (Scheme 4).
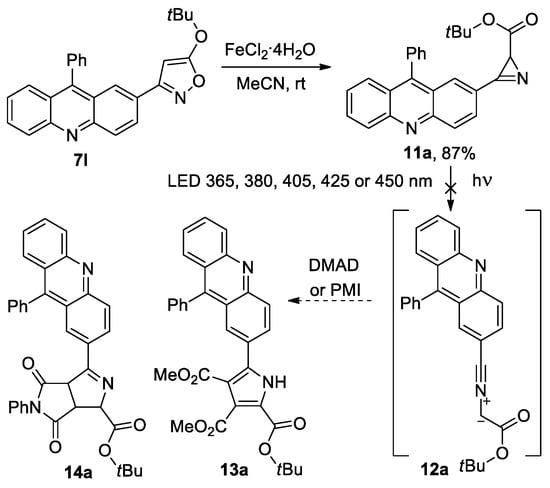
Scheme 4.
Synthesis of acridinyl-substituted azirine 11a.
In the UV spectrum of azirine 11a, there is a long-wave absorption band in the region of 320–420 nm (see Figure S3 in the Supporting Information), therefore, its photolysis in the presence of DMAD or N-phenylmaleimide (PMI) (20 equiv.) was carried out in acetonitrile using LED 365, 380, 405, 425, or 450 nm, as well with white light LED (380−760 nm). However, under all the conditions tested, nitrile ylide 12a, which was expected to be formed by photolysis [35,41,42], did not produce a cycloaddition product 13a with DMAD or 14a with PMI, and the starting azirine was isolated unchanged. The same result was also obtained during the photolysis of isoxazole 7l in the presence of DMAD or PMI, although it is known that azirines and nitrile ylides can successively be formed during the photolysis of isoxazoles [42]. Meanwhile, the photolysis of 3-(pyren-1-yl)-2H-azirine 15, which has a long-wave absorption band in the region of 360–400 nm, using LED (410–420 nm) in acetonitrile in the presence of DMAD gave, via nitrile ylide 16, cycloaddition product 17 with a yield of 75% [35] (Scheme 5). In an attempt to find the reason for this difference in the reactivity of azirines 15 and 11a, we performed DFT calculations (the B3LYP-D3/6-311+G(d,p) level of theory with SMD model for MeCN, see Supporting Information for details) of the cycloaddition of the nitrile ylides formed from azirine 15 and model azirines 19 and 23 to DMAD (Scheme 5).
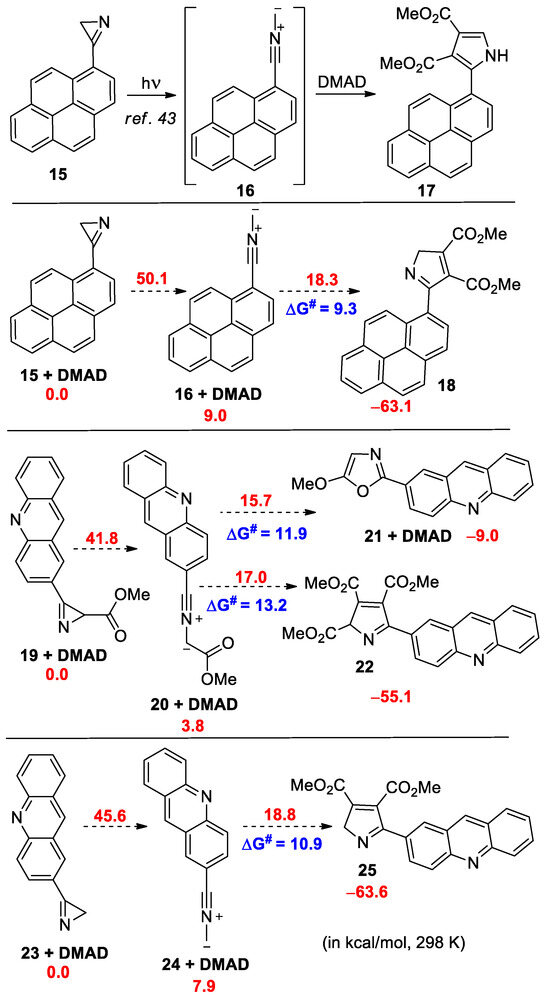
Scheme 5.
Relative Gibbs free energies for the transformations of pyrenyl- and acridinyl-substituted nitrile ylides (in kcal/mol, 298 K, DFT B3LYP-D3/6-311+G(d,p) level with SMD model for MeCN).
From the calculation results, it follows that, although the barrier for the cycloaddition of nitrile ylide 20 to DMAD is higher than the barrier for the cycloaddition of nitrile ylide 16, it is still low enough to be easily overcome at room temperature (ΔG# 13.2 vs. 9.3 kcal/mol). At the same time, nitrile ylide 24, without a methoxycarbonyl substituent, has a very similar barrier to its cycloaddition to DMAD (ΔG# 10.9 vs. 9.3 kcal/mol). A significant difference between the chemical behavior of nitrile ylides 16 and 20 is that, in the case of nitrile ylide 20, there is an additional possibility of its transformation into oxazole 21 through a lower energy barrier than that for cycloaddition. Although the formation of oxazole type 21 was not detected during the photolysis of azirine 11a, we decided to synthesize azirine 28 to ensure that the presence of an additional conversion channel in the case of nitrile ylide 12a was critical.
Azirine 28 was prepared the usual way from aldehyde 5e (Scheme 6). However, the photolysis of azirine 28 in acetonitrile, in the presence of DMAD or PMI (20 equiv.) using LED 365, 380, or 405 nm, led to the same result as the photolysis of azirine 11a: the starting azirine was isolated unchanged.
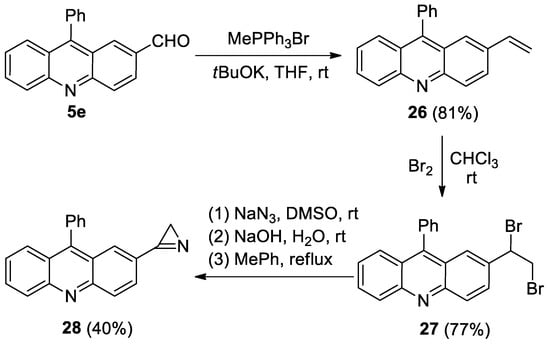
Scheme 6.
Synthesis of acridinyl-substituted azirine 7j.
Azirine 32, linked to acridine by a triazole bridge, was also synthesized by the reaction of 1-(3-phenyl-2H-azirin-2-yl)-2-(triphenylphosphoranylidene)ethanone 30 with 9-azidoacridine 31, according to the procedure developed in the work [43], at a 77% yield (Scheme 7). However, the photolysis of azirine 32 in acetonitrile, in the presence of DMAD or PMI (20 equiv.) using LED 365, 380, 405, or 425 nm, led to the same result as the photolysis of azirine 11a: the starting azirine was isolated unchanged.
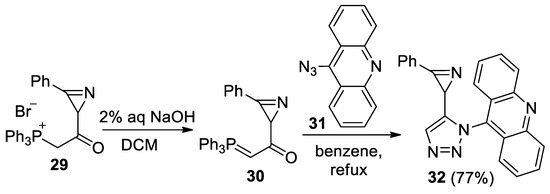
Scheme 7.
Synthesis of acridinyl-substituted azirine 32.
One of the possible reasons for the different behavior of pyrenyl-substituted azirine 15 and acridinyl-substituted azirines 11a,28,32 during photolysis in the presence of DMAD may be the fast relaxation of the singlet excited state of acridinyl-substituted azirines through intersystem crossing into a triplet excited state, characteristic of acridine systems in acetonitrile [44]. This may prevent the energy required to break the azirine C2–C3 bond from being transferred from the excited acridine moiety to the azirine moiety and block, therefore, the formation of nitrile ylide from azirine.
It was previously found that the photolysis of 9-phenylacridine in toluene using a high-pressure mercury lamp leads to 9-benzyl-9-phenyl-9,10-dihydroacridine (49%) [45] through the probable formation and coupling of 9-phenyl-9,10-dihydroacridine-9-yl and benzyl radicals [45,46,47]. To expand the range of isoxazole-substituted acridine derivatives, we decided to carry out the photolysis of isoxazolyl- and azirinyl-substiuted acridines in the presence of hydrogen donor solvents, but by using LED light in the UV/visible radiation boundary region (385–405 nm) instead of with a high-pressure mercury lamp. The irradiation of azirine 28 in toluene under these conditions gave a complex mixture of products, probably because free radicals can open the azirine ring [48]. Fortunately, the irradiation of isoxazolylacridines 7 using LED 385 or 405 nm in the presence of toluene, mesitylene, o-xylene, 4-chlorotoluene, THF, MTBE, and 1,4-dioxane gave 9,9-disubstituted acridanes 33a–n, in good yields in most cases, while the reaction with tetrahydrothiophene led to unstable products (Scheme 8).
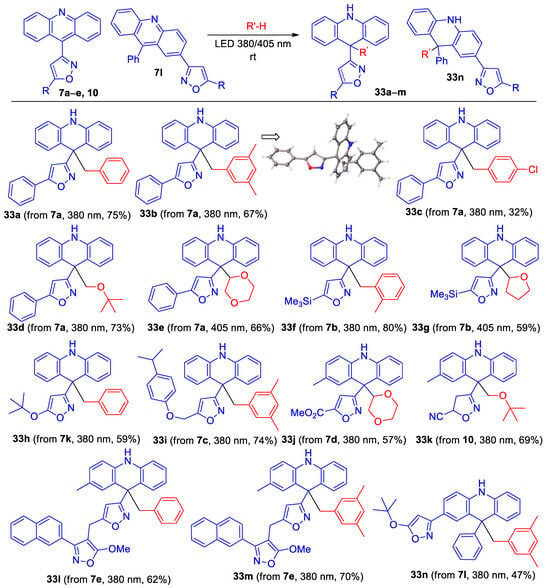
Scheme 8.
Synthesis of 9,9-disubstituted acridanes 33a–n.
Note that nitro-substituted derivative 7f reacts very slowly, and long-term irradiation leads to its decomposition. All new compounds were characterized by 1H, 13C NMR, and HRMS methods. The structure of acridane 33b was also confirmed by single-crystal X-ray diffraction analysis.
The evaluation of the antiproliferative activity of the synthesized compounds, including acridine–azirine hybrids, acridine–isoxazoles, and acridane–isoxazole hybrids, was conducted using the MTT assay with a compound concentration of 30 μM over a 72 h period. This comprehensive assessment was performed against a diverse set of cancer cell lines such as HCT 116 (colon cancer), MCF7 (breast cancer), and A704 (kidney cancer), as well as against normal fibroblast cells (WI-26 VA4) to determine the compounds’ selectivity. The findings revealed that the majority of the compounds demonstrated antiproliferative activity, impacting both cancerous and normal cell lines. Notably, derivatives containing the isoxazole moiety, especially compounds 7g,i and 7l, were identified as having significant cytotoxic activity (Figure 1). However, the lack of pronounced selectivity between the cancerous and normal cells highlights an area for future refinement to improve the possible therapeutic efficacy of these compounds.
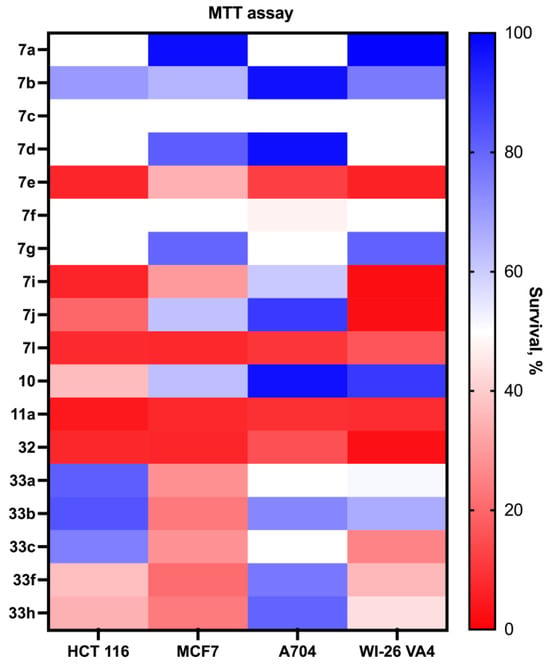
Figure 1.
The cytotoxicity of acridine–azirine, acridine–isoxazole, and acridane–isoxazole hybrids.
3. Materials and Methods
3.1. General Instrumentation
Melting points were determined on a melting point apparatus. 1H (400 MHz) and 13C (100 MHz) spectra were recorded on a Bruker AVANCE 400 spectrometer (Billerica, MA, USA) in CDCl3 or DMSO-d6. Chemical shifts (δ) are reported in parts per million downfield from tetramethylsilane (TMS, δ = 0.00). 1H NMR spectra were calibrated according to the residual peak of H-analogues of CDCl3 (7.26 ppm), DMSO-d6 (2.50 ppm), and C6D6 (7.16 ppm). For all new compounds, 13C{1H} and 13C DEPT-135 spectra were recorded and calibrated according to the peak of CDCl3 (77.00 ppm), DMSO-d6 (39.51 ppm), and C6D6 (128.06 ppm). Electrospray ionization (ESI) mass spectra were recorded on a Bruker MaXis mass spectrometer, HRMS-ESI-QTOF. Single-crystal X-ray data were collected by the means of “XtaLAB Synergy” diffractometer (Rigaku Oxford Diffraction, Akishima, Japan). Crystallographic data for the structure 33b (CCDC 2328401) were deposited with the Cambridge Crystallographic Data Centre. Thin-layer chromatography (TLC) was conducted on aluminum sheets with 0.2 mm silica gel and a fluorescent indicator. The physical and spectral data of 7,9-dimethylbenz[c]acridine 4b [49], 9-methyl-2-nitroacridine 4c [50], 2-methylacridine-9-carbaldehyde 5a [51], acridine-9-carbaldehyde oxime 1a [29], (2-oxo-2-(3-phenyl-2H-azirin-2-yl)ethyl)triphenylphosphonium bromide 29 [52], and 9-azidoacridine 31 [53], prepared according to the published procedures, were in agreement with previously reported values.
3.2. General Experimental Procedures
3.2.1. General Procedure A (GP-A) for the Preparation of Chlorooxime Hydrochlorides 2-HCl
A suspension of oxime 1 in DCM/DMF 20:1 (v/v) was bubbled with chlorine gas at rt for 0.5–1.5 h (the suspension changed from a brick (orange) color to bright yellow or orange). The reaction mixture was stirred at rt for 1 d in a closed reaction vessel and diluted with DCM, and the product was filtered, washed with DCM, and dried in air.
3.2.2. General Procedure B (GP-B) for the Preparation of Isoxazoles 7
Chlorooxime hydrochloride 2-HCl (1 equiv.), excess of appropriate acetylene or alkene, and DCM/sat. aq. NaHCO3 2:1 (v/v) were placed in round-bottom flask with a plastic stopper. The suspension was shaken well until the reaction mixture became lighter in color and the chloroxime hydrochloride began to dissolve. Then, the reaction mixture was vigorously stirred at rt overnight. The layers were separated and the water layer was extracted with DCM. The combined organic layers were washed with water and brine, dried over Na2SO4, and after the evaporation of the solvent, the product was purified by chromatography on silica gel.
3.2.3. General Procedure C (GP-C) for the Reaction of Isoxazoles 7a–e,l and 10 with Hydrogen Donor Solvents
Isoxazoles in an appropriate hydrogen donor solvent (when reacting with 4-chlorotoluene, α,α,α-trifluorotoluene was added as a co-solvent) were irradiated using LED 385 or 405 nm at rt (TLC control). The solvent was evaporated and the residue was purified by chromatography on silica gel.
3.2.4. Specific Procedures and Characterization
- 2-Methyl-9-phenylacridine (4e). Compound 4e was prepared according to the published procedure [54] from 4-methyl-N-phenylaniline (1.83 g, 10 mmol), benzoic acid (2.44 g, 20 mmol), and anhydrous ZnCl2 (2.72 g, 20 mmol) at 220 °C overnight to give a pure product of 1.62 g (60% yield), after column chromatography on silica (light petroleum/ethyl acetate, 5:1–1:1, (v/v)) as a light yellow solid: mp 110–111 °C (ethyl acetate); 1H NMR (400 MHz, CDCl3) δ 8.26 (d, 1H, J = 8.8 Hz), 8.18 (d, 1H, J = 8.9 Hz), 7.73 (ddd, 1H, J = 8.5, 6.5, 1.3 Hz), 7.66 (dd, 1H, J = 8.8, 1.3 Hz), 7.67–7.58 (m, 4H), 7.45–7.38 (m, 4H), 2.46 (s, 3H); 13C{1H} NMR (100 MHz, CDCl3) δ 148.3 (C), 147.8 (C), 146.0 (C), 136.2 (C), 135.5 (C), 132.9 (CH), 130.5 (CH), 129.6 (CH), 129.5 (CH), 129.4 (CH), 128.5 (CH), 128.2 (CH), 126.8 (CH), 125.5 (CH), 125.3 (C), 125.2 (C), 124.8 (CH), 22.0 (CH3); HRMS (ESI) m/z [M + H]+ calcd for C20H16N+ 270.1277, found 270.1280.
- 2-Nitroacridine-9-carbaldehyde (5c). Compound 5c was prepared according to the published procedure [55] from 9-methyl-2-nitroacridine 4c (325 mg, 1.35 mmol) and SeO2 (160 mg, 1.43 mmol) in 1,4-dioxane (5 mL) at 115 °C (bath temperature) to give a pure product of 214 mg (62% yield), after column chromatography on silica (light petroleum/ethyl acetate, 1:1–0:1, (v/v)) as a brown-orange solid: mp 182–184 °C (ethyl acetate); 1H NMR (400 MHz, DMSO-d6) δ 11.52 (s, 1H), 9.78 (d, 1H, J = 2.6 Hz), 8.94 (d, 1H, J = 8.8 Hz), 8.55 (dd, 1H, J = 9.4, 2.7 Hz), 8.44 (d, 1H, J = 9.6 Hz), 8.34 (d, 1H, J = 8.7 Hz), 8.09–8.06 (m, 1H), 7.92–7.88 (m, 1H); 13C{1H} NMR (100 MHz, DMSO-d6) δ 195.1 (CH), 150.9 (C), 148.9 (C), 146.3 (C), 136.0 (C), 132.6 (CH), 131.8 (CH), 130.1 (CH), 129.7 (CH), 124.4 (C), 124.1 (CH), 123.2 (CH), 122.9 (CH), 120.4 (C); HRMS (ESI) m/z [M + H]+ calcd for C14H9N2O3+ 253.0608, found 253.0599.
- 9-Methylbenzo[c]acridine-7-carbaldehyde (5d). Compound 5d was prepared according to the published procedure [55] from 7,9-dimethylbenzo[c]acridine 4d (1.5 g, 7.24 mmol) and SeO2 (843 mg, 7.6 mmol) in 1,4-dioxane (50 mL) at 115 °C (bath temperature) to give a pure product of 1.32 g (82% yield), after column chromatography on silica (light petroleum/ethyl acetate, 1:1–0:1, (v/v)) as an orange solid: mp 165–166 °C (ethyl acetate); 1H NMR (400 MHz, CDCl3) δ 11.44 (d, 1H, J = 6.7 Hz), 9.50–9.47 (m, 1H), 8.49 (d, 1H, J = 4.5 Hz), 8.45–8.41 (m, 1H), 8.33 (dd, 1H, J = 8.7, 5.0 Hz), 7.88–7.73 (m, 4H), 7.69 (dt, 1H, J = 9.3, 2.2 Hz), 2.64 (s, 3H); 13C{1H} NMR (100 MHz, CDCl3) δ 193.9 (CH), 146.9 (C), 146.4 (C), 138.8 (C), 132.7 (C), 132.2 (CH), 131.4 (C), 131.0 (C), 130.8 (CH), 130.3 (CH), 129.2 (CH), 127.79 (CH), 127.75 (CH), 125.2 (CH), 123.6 (C), 123.1 (C), 121.8 (CH), 120.3 (CH), 22.4 (CH3); HRMS (ESI) m/z [M + H]+ calcd for C19H14NO+ 272.1070, found 272.1065.
- 9-Phenylacridine-2-carbaldehyde (5e). Compound 5e was prepared according to the published procedure [55] from 2-methyl-9-phenylacridine 4e (1.0 g, 3.7 mmol) and SeO2 (4.3 g, 38.7 mmol, 6 portions every 12 h) 1,4-dioxane (35 mL) at 115 °C (bath temperature) to give a pure product of 714 mg (68% yield), after column chromatography on silica (light petroleum/ethyl acetate, 1:1–0:1, (v/v)) as a light yellow solid: mp 185–186 °C (ethyl acetate); 1H NMR (400 MHz, CDCl3) δ 10.02 (d, 1H, J = 0.8 Hz), 8.32 (td, 2H, J = 9.6, 0.8 Hz), 8.23–8.20 (m, 2H), 7.77–7.75 (m, 1H), 7.68–7.64 (m, 3H), 7.52–7.47 (, 3H); 13C{1H} NMR (CDCl3, 100 MHz) δ 191.5 (CH), 150.5 (C), 150.4 (C), 149.9 (C), 135.4 (CH), 134.9 (C), 133.7 (C), 131.5 (CH), 131.1 (CH), 130.4 (CH), 129.8 (CH), 129.0 (CH), 128.7 (CH), 127.2 (CH), 126.5 (CH), 125.7 (CH), 125.5 (C), 124.3 (C); HRMS (ESI) m/z [M + H]+ calcd for C20H14NO+ 284.1070, found 284.1062.
- 2-Methylacridine-9-carbaldehyde oxime (1b). Compound 1b was prepared according to the published procedure [29] from acridinecarbaldehyde 5b (1.0 g, 4.5 mmol) and H2NOH·HCl (628 mg, 9 mmol) in EtOH (20 mL) and 2% aq. NaOH (2 mL) to give a pure product of 885 mg (83% yield), after filtration as a brick-yellow solid: mp 196–197 °C (EtOH/H2O); 1H NMR (400 MHz, DMSO-d6) δ 12.35 (s, 1H), 9.28 (s, 1H), 8.55 (d, 1H, J = 8.8 Hz), 8.31–8.30 (m, 1H), 8.19 (d, 1H, J = 8.6 Hz), 8.12 (d, 1H, J = 8.9 Hz), 7.89 (ddd, 1H, J = 8.4, 6.5, 1.4 Hz), 7.77 (dd, 1H, J = 8.9, 1.9 Hz), 7.68 (ddd, 1H, J = 8.2, 6.6, 1.3 Hz), 2.56 (s, 1H); 13C{1H} NMR (100 MHz, DMSO-d6) δ 146.3 (br. s, C), 145.8 (br. s, C), 145.1 (CH), 136.8 (C), 135.1 (br. s, C), 133.9 (CH), 130.7 (CH), 128.1 (br. s, CH), 127.9 (br. s, CH), 126.9 (CH), 125.8 (CH), 123.64 (CH), 123.61 (C), 123.56 (C), 21.7 (CH3); HRMS (ESI) m/z [M + H]+ calcd for C15H13N2O+ 237.1022, found 237.1017.
- 2-Nitroacridine-9-carbaldehyde oxime (1c). Compound 1c was prepared according to the published procedure [29] from acridinecarbaldehyde 5c (200 mg, 0.8 mmol) and H2NOH·HCl (138 mg, 2 mmol) in EtOH (5 mL) and 2% aq. NaOH (0.5 mL) to give a pure product of 162 mg (76% yield), after filtration as a yellow-green solid: mp 202–214 °C (dec., EtOH/H2O); 1H NMR (400 MHz, DMSO-d6) δ 12.67 (s, 1H), 9.65 (d, 1H, J = 2.5 Hz), 9.44 (s, 1H), 8.56 (d, 1H, J = 8.8 Hz), 8.49 (dd, 1H, J = 9.5, 2.7 Hz), 8.34 (d, 1H, J = 9.5 Hz), 8.24 (d, 1H, J = 8.6 Hz), 8.01 (t, 1H, J = 7.7 Hz), 7.77 (t, 1H, J = 7.7 Hz); 13C{1H} NMR (100 MHz, DMSO-d6) δ 150.0 (C), 148.7 (C), 145.0 (C), 144.5 (CH), 141.7 (CH), 137.7 (C), 132.0 (CH), 131.4 (CH), 129.5 (CH), 127.5 (CH), 124.9 (CH), 124.0 (CH), 122.3 (CH), 120.9 (C); HRMS (ESI) m/z [M + H]+ calcd for C14H10N3O3+ 268.0717, found 268.0719.
- 9-Methylbenzo[c]acridine-7-carbaldehyde oxime (1d). Compound 1d was prepared according to the published procedure [29] from acridinecarbaldehyde 5d (935 mg, 3.5mmol) and H2NOH·HCl (600 mg, 8.6 mmol) in EtOH (20 mL) and 2% aq. NaOH (2 mL) to give a pure product of 984 mg (99% yield), after filtration as a brick-orange solid: mp 237–238 °C (EtOH/H2O); 1H NMR (400 MHz, DMSO-d6) δ 12.02 (br. s, 1H), 9.42–9.40 (m, 1H), 9.23 (s, 1H), 8.31–8.27 (m, 3H), 8.02–8.00 (m, 1H), 7.90 (d, 1H, J = 9.5 Hz), 7.83–7.78 (m, 3H), 2.61 (s, 3H); 13C{1H} NMR (100 MHz, DMSO-d6) δ 145.5 (C), 145.2 (CH), 145.0 (C), 136.9 (C), 133.9 (C), 133.0 (C), 132.9 (CH), 130.3 (C), 129.5 (CH), 129.0 (CH), 128.3 (CH), 128.0 (CH), 127.6 (CH), 124.8 (CH), 124.2 (C), 123.8 (CH), 122.9 (CH), 122.3 (C), 21.7 (CH3); HRMS (ESI) m/z [M + H]+ calcd for C19H15N2O+ 287.1179, found 287.1170.
- 9-Phenylacridine-2-carbaldehyde oxime (1e). Compound 1e was prepared according to the published procedure [29] from acridinecarbaldehyde 5e (693 mg, 2.5 mmol) and H2NOH·HCl (425 mg, 6.1 mmol) in EtOH (20 mL) and 2% aq. NaOH (2 mL) to give a pure product of 639 mg (88% yield), after filtration as a bright yellow solid: mp 212–215 °C (dec., EtOH/H2O); 1H NMR (400 MHz, DMSO-d6) δ 11.71 (br. s, 1H), 8.62–8.42 (m, 3H0, 8.34 (s, 1H), 8.24–8.16 (m, 1H), 7.92–7.90 (m,1H), 7.81–7.73 (m, 5H), 7.61–7.57 (m, 2H); 13C{1H} NMR (100 MHz, DMSO-d6) δ 155.8 (br. s, C), 147.2 (CH), 142.1 (br. s, C), 141.4 (br. s, C), 135.2 (br. s, CH), 133.3 (C), 132.4 (br. s, C), 132.3 (CH), 130.0 (CH), 129.8 (CH), 128.9 (CH), 128.0 (CH), 127.6 (CH), 125.5 (CH), 125.2 (C), 124.9 (C), 123.1 (br. s, CH), 122.3 (br. s, CH); HRMS (ESI) m/z [M + H]+ calcd for C20H15N2O+ 299.1179, found 299.1183.
- N-Hydroxyacridine-9-carbimidoyl chloride hydrochloride (2a-HCl). Compound 2a-HCl was prepared according to the general procedure GP-A from oxime 1a (2.0 g, 9 mmol) in DCM (30 mL) and DMF (3 mL) to give a pure product of 1.84 g (70% yield), after filtration as a bright yellow solid: mp 226–230 °C (dec., DCM); 1H NMR (400 MHz, DMSO-d6) δ 13.29 (s, 1H), 8.42 (d, 2H, J = 8.7 Hz), 8.14–8.07 (m, 4H), 7.89–7.85 (m, 2H), 4.46 (br. s); 13C{1H} NMR (100 MHz, DMSO-d6) δ 144.9 (C), 140.4 (C), 133.6 (CH), 128.7 (CH), 127.4 (C), 125.8 (CH), 125.2 (CH), 123.6 (C); HRMS (ESI) m/z [M − Cl]+ calcd for C14H10ClN2O+ 257.0476, found 257.0482.
- N-Hydroxy-2-methylacridine-9-carbimidoyl chloride hydrochloride (2b-HCl). Compound 2b-HCl was prepared according to the general procedure GP-A from oxime 1b (842 mg, 2.3 mmol) in DCM (20 mL) and DMF (2 mL) to give a pure product of 838 mg (83% yield), after filtration as a bright yellow-orange solid: mp 231–232 °C (dec., DCM); 1H NMR (400 MHz, DMSO-d6) δ 13.33 (s, 1H), 8.46–8.44 (m, 2H), 8.39 (d, 1H, J = 9.0 Hz), 8.12–8.08 (m, 2H), 7.99 (dd, 1H, J = 8.9, 1.9 Hz), 7.90–7.86 (m, 2H), 7.61 (s, 3H), 5.81 (br. s); 13C{1H} NMR (100 MHz, DMSO-d6) δ 143.2 (C), 142.8 (C), 140.0 (C), 138.8 (C), 136.6 (CH), 133.3 (CH), 128.4 (CH), 127.0 (C), 124.9 (CH), 124.6 (CH), 124.4 (CH), 123.6 (C), 123.6 (C), 122.8 (CH), 21.4 (CH3); HRMS (ESI) m/z [M − Cl]+ calcd for C14H20ClN2O+ 257.0476, found 257.0482.
- N-Hydroxy-2-nitroacridine-9-carbimidoyl chloride hydrochloride (2c-HCl). Compound 2c-HCl was prepared according to the general procedure GP-A from oxime 1c (155 mg, 0.6 mmol) in DCM (3 mL) and DMF (0.3 mL) to give a pure product of 136 mg (69% yield), after filtration as an orange-brown solid: mp 229–230 °C (dec., DCM); 1H NMR (400 MHz, DMSO-d6) δ 13.47 (s, 1H), 8.86 (t, 1H, J = 2.0 Hz), 8.55 (dt, 1H, J = 9.4, 2.4 Hz), 8.45 (dd, 1H, J = 9.4, 2.2 Hz), 8.32 (d, 1H, J = 8.7 Hz), 8.12 (d, 1H, J = 8.8 Hz), 8.10–8.06 (m, 1H), 7.88 (dd, 1H, J = 8.7, 6.7 Hz), 7.51–6.61 (br. s); 13C{1H} NMR (100 MHz, DMSO-d6) δ 150.2 (C), 148.4 (C), 145.6 (C), 139.9 (C), 133.3 (CH), 131.7 (CH), 129.6 (CH), 129.4 (CH), 127.4 (C), 125.1 (CH), 123.9 (C), 123.7 (CH), 122.0 (CH), 121.5 (C); HRMS (ESI) m/z [M − Cl]+ calcd for C14H9ClN3O3+ 302.0327, found 302.0320.
- N-Hydroxy-9-methylbenzo[c]acridine-7-carbimidoyl chloride hydrochloride (2d-HCl). Compound 2d-HCl was prepared according to the general procedure GP-A from oxime 1d (855 mg, 3 mmol) in DCM (30 mL) and DMF (3 mL) to give a pure product of 908 mg (85% yield), after filtration as a bright yellow solid: mp 267–272 °C (dec., DCM); 1H NMR (400 MHz, DMSO-d6) δ 13.07 (s, 1H), 9.39–9.37 (m, 1H), 8.31 (d, 1H, J = 8.7 Hz), 8.07–8.04 (m, 1H), 8.01 (d, 1H, J = 9.4 Hz), 7.86–7.76 (m, 5H), 5.61–5.16 (br. s), 2.61 (s, 3H); 13C{1H} NMR (100 MHz, DMSO-d6) δ 146.0 (C), 145.6 (C), 138.0 (C), 135.1 (C), 133.3 (CH), 133.0 (C), 130.5 (C), 129.78 (CH), 129.75 (CH), 129.5 (CH), 128.8 (C), 128.4 (CH), 128.1 (CH), 124.6 (CH), 123.8 (C), 122.6 (CH), 122.2 (C), 121.8 (CH), 21.7 (CH3); HRMS (ESI) m/z [M − Cl]+ calcd for C19H14ClN2O+ 321.0789, found 321.0801.
- N-Hydroxy-9-phenylacridine-2-carbimidoyl chloride hydrochloride (2e-HCl). Compound 2e-HCl was prepared according to the general procedure GP-A from oxime 1e (623 mg, 2.1 mmol) in DCM (20 mL) and DMF (2 mL) to give a pure product of 721 mg (94% yield), after filtration as a bright yellow solid: mp 228–229 °C (dec., DCM); 1H NMR (400 MHz, DMSO-d6) δ 12.94 (s, 1H), 8.63–8.56 (m, 3H), 8.27–8.23 (m, 1H), 8.16 (d, 1H, J = 1.9 Hz), 7.83 (d, 1H, J = 3.9 Hz), 7.79–7.75 (m, 3H), 7.64–7.62 (m, 2H); 13C{1H} NMR (100 MHz, DMSO-d6) δ 156.7 (C), 142.0 (C), 141.8 (C), 135.9 (CH), 134.4 (C), 133.1 (C), 132.1 (CH), 131.1 (C), 130.0 (CH), 130.0 (CH), 128.9 (CH), 128.3 (CH), 127.7 (CH), 125.8 (CH), 125.3 (C), 124.4 (C), 123.0 (CH), 122.3 (CH); HRMS (ESI) m/z [M + H]+ calcd for C20H14ClN2O+ 333.0789, found 333.0777.
- 3-(Acridin-9-yl)-5-phenylisoxazole (7a). Compound 7a was prepared according to the general procedure GP-B from chlorooxime 2a-HCl (171 mg, 0.58 mmol) and phenylacetylene 6a (298 mg, 2.92 mmol, 5 eq.) in DCM (10 mL) and sat. aq. NaHCO3 (5 mL) to give a pure product of 178 mg (95% yield), after column chromatography on silica (light petroleum/ethyl acetate, 6:1, (v/v)) as a bright yellow solid: mp 258–260 °C (light petroleum/ethyl acetate); 1H NMR (400 MHz, CDCl3): δ 8.32 (d, 2H, J = 8.7 Hz), 8.01 (d, 2H, J = 8.7 Hz), 7.96–7.94 (m, 2H), 7.83 (ddd, 2H, J = 8.5, 6.6, 1.4 Hz), 7.58–7.53 (m, 5H), 6.85 (s, 1H); 13C{1H} NMR (100 MHz, CDCl3) δ 170.8 (C), 159.4 (C), 148.7 (C), 133.7 (C), 130.8 (CH), 130.3 (CH), 129.9 (CH), 129.2 (CH), 127.0 (C), 126.8 (CH), 126.1 (CH), 125.8 (CH), 125.0 (C), 102.6 (CH); HRMS (ESI) m/z [M + H]+ calcd for C29H23N2O+ 415.1805, found 415.1799.
- 3-(Acridin-9-yl)-5-(trimethylsilyl)isoxazole (7b). Compound 7b was prepared according to the general procedure GP-B from chlorooxime 2a-HCl (255 mg, 0.87 mmol) and trimethylsilylacetylene 6b (1.94 g, 17.4 mmol, 20 eq.) in DCM (15 mL) and sat. NaHCO3 aq. (8 mL) to give a pure product of 256 mg (92% yield), after column chromatography on silica (light petroleum/ethyl acetate, 6:1, (v/v)) as a bright yellow solid: mp 151–152 °C (light petroleum/ethyl acetate); 1H NMR (400 MHz, CDCl3) δ 8.29 (dt, 2H, J = 8.8, 1.0 Hz), 7.87 (ddd, 2H, J = 8.7, 1.4, 0.7 Hz), 7.80 (ddd, 2H, J = 8.9, 6.6, 1.4 Hz), 7.52 (ddd, 2H, J = 8.7, 6.6, 1.2 Hz), 6.74 (s, 1H), 0.49 (s, 9H); 13C{1H} NMR (100 MHz, CDCl3) δ 179.1 (C), 156.9 (C), 148.6 (C), 134.1 (C), 130.2 (CH), 129.8 (CH), 126.5 (CH), 125.9 (CH), 125.1 (C), 115.4 (CH), −1.8 (CH3); HRMS (ESI) m/z [M + H]+ calcd for C19H19N2OSi+ 319.1261, found 319.1258.
- 3-(Acridin-9-yl)-5-((4-isopropylphenoxy)methyl)isoxazole (7c). Compound 7c was prepared according to the general procedure GP-B from chlorooxime 2a-HCl (155 mg, 0.51 mmol) and 1-isopropyl-4-(prop-2-yn-1-yloxy)benzene 6c (446 mg, 2.56 mmol, 5 eq.) in DCM (10 mL) and sat. NaHCO3 aq. (5 mL) to give a pure product of 198 mg (98% yield), after column chromatography on silica (light petroleum/ethyl acetate, 10:1, (v/v)) as a bright yellow solid: mp 94–95 °C (light petroleum/ethyl acetate); 1H NMR (400 MHz, CDCl3) δ 8.30 (d, 2H, J = 8.8 Hz), 7.91 (d, 2H, J = 8.7 Hz), 7.83–7.79 (m, 2H), 7.56–7.52 (m, 2H), 7.21 (d, 2H, J = 8.2 Hz), 6.99 (d, 2H, J = 8.2 Hz), 6.67 (s, 1H), 5.37 (s, 2H), 2.90 (hept, 1H, J = 7.0 Hz), 1.25 (d, 6H, J = 7.0 Hz); 13C{1H} NMR (100 MHz, CDCl3) δ 169.2 (C), 158.9 (C), 155.8 (C), 148.6 (C), 142.7 (C), 133.3 (C), 130.3 (CH), 129.9 (CH), 127.6 (CH), 126.8 (CH), 125.7 (CH), 124.9 (C), 114.8 (CH), 106.4 (CH), 61.8 (CH2), 33.3 (CH), 24.1 (CH3); HRMS (ESI) m/z [M + H]+ calcd for C26H23N2O2+ 395.1754, found 395.1752.
- Methyl 3-(2-methylacridin-9-yl)isoxazole-5-carboxylate (7d). Compound 7d was prepared according to the general procedure GP-B from chlorooxime 2b-HCl (330 mg, 1.1 mmol) and methyl propiolate 6d (452 mg, 5.4 mmol, 5 eq.) in DCM (20 mL) and sat. NaHCO3 aq. (10 mL) to give a pure product of 179 mg (52% yield), after column chromatography on silica (light petroleum/ethyl acetate, 6:1, (v/v)) as a yellow solid: mp 169–170 °C (light petroleum/ethyl acetate); 1H NMR (400 MHz, CDCl3) δ 8.28 (dd, 1H, J = 9.1, 1.2 Hz), 8.20 (d, 1H, J = 8.9 Hz), 7.80–776 (m, 2H), 7.65 (dd, 1H, J = 8.9, 1.6 Hz), 7.55–7.51 (m, 2H), 7.25 (s, 1H), 4.08 (s, 3H), 2.52 (s, 3H); 13C{1H} NMR (100 MHz, CDCl3) δ 160.9 (C), 159.6 (C), 157.0 (C), 148.0 (C), 147.6 (C), 137.3 (C), 133.3 (CH), 130.5 (C), 130.0 (CH), 129.8 (CH), 129.7 (CH), 127.0 (CH), 125.0 (CH), 124.8 (C), 124.8 (C), 123.1 (CH), 112.3 (CH), 53.1 (CH3), 22.1 (CH3); HRMS (ESI) m/z [M + H]+ calcd for C19H15N2O3+ 319.1077, found 319.1076.
- 5-Methoxy-4-((3-(2-methylacridin-9-yl)isoxazol-5-yl)methyl)-3-(naphthalen-2-yl)isoxazole (7e). Compound 7e was prepared according to the general procedure GP-B from chlorooxime 2b-HCl (150 mg, 0.49 mmol) and 5-methoxy-3-(naphthalen-2-yl)-4-(prop-2-yn-1-yl)isoxazole 6e (296 mg, 1.12 mmol, 2.3 eq.) in DCM (20 mL) and sat. NaHCO3 aq. (10 mL) to give a pure product of 227 mg (93% yield), after column chromatography on silica (light petroleum/ethyl acetate, 20:1–6:1, (v/v)) as a bright yellow solid: mp 157–158 °C (light petroleum/ethyl acetate); 1H NMR (400 MHz, CDCl3) δ 8.21 (d, 1H, J = 8.7 Hz), 8.14 (d, 1H, J = 8.7 Hz), 8.13 (s, 1H), 7.96 (d, 1H, J = 8.5 Hz), 7.92–7.89 (m, 2H), 7.78 (dd, 1H, J = 8.5, 1.8 Hz), 7.70 (ddd, 1H, J = 8.1, 6.7, 1.3 Hz), 7.61–7.52 (m, 5H), 7.24–7.20 (m, 1H), 6.23 (s, 1H), 4.25 (s, 3H), 4.18 (s, 2H), 2.40 (s, 3H); 13C{1H} NMR (100 MHz, CDCl3) δ 171.3 (C), 170.4 (C), 164.3 (C), 159.0 (C), 147.8 (C), 147.4 (C), 136.7 (C), 133.8 (C), 133.1 (CH), 133.0 (C), 132.2 (C), 129.6 (CH), 129.6 (CH), 129.4 (CH), 128.8 (CH), 128.5 (CH), 127.8 (CH), 127.6 (CH), 127.2 (CH), 126.8 (CH), 126.7 (C), 126.4 (CH), 125.3 (CH), 124.9 (C), 124.8 (C), 124.6 (CH), 123.5 (CH), 105.2 (CH), 85.9 (CH), 58.1 (CH3), 22.0 (CH3), 20.1 (CH2); HRMS (ESI) m/z [M + H]+ calcd for C32H24N3O3+ 498.1812, found 498.1807.
- 5-(4-Chlorophenyl)-3-(2-nitroacridin-9-yl)isoxazole (7f). Compound 7f was prepared according to the general procedure GP-B from chlorooxime 2c-HCl (150 mg, 0.44 mmol) and 1-chloro-4-ethynylbenzene 6f (305 mg, 2.2 mmol, 5 eq.) in DCM (20 mL) and sat. aq. NaHCO3 (10 mL) to give a pure product of 159 mg (89% yield), after column chromatography on silica (light petroleum/ethyl acetate, 6:1–0:1, (v/v)) as a bright yellow solid: mp 232–233 °C (ethyl acetate); 1H NMR (400 MHz, DMSO-d6) δ 8.90 (d, 1H, J = 2.5 Hz), 8.55 (dd, 1H, J = 9.5, 2.5 Hz), 8.48 (d, 1H, J = 9.5 Hz), 8.36 (d, 1H, J = 8.8 Hz), 8.11–8.05 (m, 5H), 7.79 (dd, 1H, J = 8.7, 6.6 Hz), 7.71–7.69 (m, 2H); 13C{1H} NMR (CDCl3, 100 MHz) δ 169.3 (C), 158.0 (C), 149.9 (C), 148.4 (C), 145.1 (C), 136.4 (C), 135.5 (C), 132.4 (CH), 131.5 (CH), 129.4 (CH), 129.1 (CH), 128.2 (CH), 127.5 (CH), 125.6 (CH), 124.9 (C), 124.4 (C), 122.9 (CH), 122.8 (CH), 122.0 (C), 103.9 (CH); HRMS (ESI) m/z [M + H]+ calcd for C22H13ClN3O3+ 402.0640, found 402.0635.
- 5-(2-Fluorophenyl)-3-(9-methylbenzo[c]acridin-7-yl)isoxazole (7g). Compound 7g was prepared according to the general procedure GP-B from chlorooxime 2d-HCl (170 mg, 0.48 mmol) and 1-ethynyl-2-fluorobenzene 6g (230 mg, 1.9 mmol, 4 eq.) in DCM (10 mL) and sat. NaHCO3 aq. (5 mL) to give a pure product of 174 mg (90% yield), after column chromatography on silica (light petroleum/ethyl acetate, 6:1, (v/v)) as a brick-yellow solid: mp 234–235 °C (ethyl acetate); 1H NMR (CDCl3, 400 MHz) δ 9.56 (d, 1H, J = 7.9 Hz), 8.36–8.34 (m, 1H), 8.21 (td, 1H, J = 7.6, 1.8 Hz), 7.86 (dd, 1H, J = 7.6, 1.5 Hz), 7.82–7.78 (m, 1H), 7.76–7.68 (m, 5H), 7.52 (tdd, 1H, J = 7.5, 5.1, 1.8 Hz), 7.40 (td, 1H, J = 7.5, 1.2 Hz), 7.29–7.25 (m, 1H), 7.06 (d, 1H, J = 3.8 Hz), 2.55 (s, 3H); 13C{1H} NMR (CDCl3, 100 MHz) δ 164.5 (d, C, J = 2.5 Hz), 160.1 (C), 159.3 (d, C, J = 253.6 Hz), 146.6 (C), 146.0 (C), 136.9 (C), 133.3 (C), 132.4 (CH), 132.1 (d, CH, J = 8.6 Hz), 131.55 (C), 131.48 (C), 129.9 (CH), 129.0 (CH), 128.6 (CH), 127.84 (CH), 127.82 (CH), 127.5 (CH), 125.4 (C), 125.3 (CH), 124.9 (d, CH, J = 3.6 Hz), 123.8 (CH), 123.6 (C), 122.9 (CH), 116.4 (d, CH, J = 21.0 Hz), 115.6 (d, C, J = 12.3 Hz), 106.7 (d, CH, J = 11.3 Hz), 22.3 (CH3); HRMS (ESI) m/z [M + H]+ calcd for C27H18FN2O+ 405.1398, found 405.1392.
- 5-((4-Isopropylphenoxy)methyl)-3-(9-methylbenzo[c]acridin-7-yl)isoxazole (7h). Compound 7h was prepared according to the general procedure GP-B from chlorooxime 2d-HCl (150 mg, 0.58 mmol) and 1-isopropyl-4-(prop-2-yn-1-yloxy)benzene 6c (370 mg, 2.1 mmol, 5 eq.) in DCM (10 mL) and sat. NaHCO3 aq. (5 mL) to give a pure product of 157 mg (95% yield), after column chromatography on silica (light petroleum/ethyl acetate, 10:1, (v/v)) as a bright yellow solid: mp 169–170 °C (light petroleum/ethyl acetate); 1H NMR (400 MHz, CDCl3) δ 9.58 (br. s, 1H), 8.38 (br. s, 1H), 7.87–7.85 (m, 1H), 7.82–7.72 (m, 2H), 7.69 (d, 2H, J = 9.2 Hz), 7.63–7.61 (m, 2H), 7.24–7.20 (m, 2H), 7.02–6.98 (m, 2H), 6.66 (s, 1H), 5.39 (s, 2H), 2.91 (hept, 1H, J = 7.0 Hz), 2.55 (s, 3H), 1.25 (d, 6H, J = 7.0 Hz); 13C{1H} NMR (100 MHz, CDCl3) δ 169.1 (C), 159.3 (C), 155.9 (C), 146.6 (C), 146.0 (C), 142.6 (C), 136.9 (C), 133.3 (C), 132.4 (CH), 131.6 (C), 131.3 (C), 129.9 (CH), 129.1 (CH), 128.6 (CH), 127.8 (CH), 127.6 (CH), 127.5 (CH), 125.4 (C), 125.3 (CH), 123.7 (CH), 123.5 (C), 122.9 (CH), 114.8 (CH), 106.4 (CH), 61.9 (CH2), 33.3 (CH), 24.2 (CH3), 22.1 (CH3); HRMS (ESI) m/z [M + H]+ calcd for C31H27N2O2+ 459.2068, found 459.2061.
- 3-(Acridin-9-yl)-5-chloroisoxazole (7i). Compound 7i was prepared according to the general procedure GP-B from chlorooxime 2a-HCl (365 mg, 0.58 mmol) and 1,1-dichloroethylene 8 (2 mL, 24.9 mmol, 20 eq.) in DCM (20 mL) and sat. NaHCO3 aq. (10 mL) to give a pure product of 218 mg (62% yield), after column chromatography on silica (light petroleum/ethyl acetate, 6:1, (v/v)) as a bright yellow solid: mp 181–182 °C (light petroleum/ethyl acetate); 1H NMR (400 MHz, CDCl3) δ 8.31 (dt, 2H, J = 8.8, 1.0 Hz), 7.91 (dt, 2H, J = 8.8, 1.0 Hz), 7.83 (ddd, 2H, J = 8.8, 6.6, 1.4 Hz), 7.52 (ddd, 2H, J = 8.7, 6.6, 1.2 Hz), 6.52 (s, 1H); 13C{1H} NMR (100 MHz, CDCl3) δ 160.9 (C), 155.9 (C), 148.6 (C), 132.1 (C), 130.3 (CH), 130.0 (CH), 127.1 (CH), 125.3 (CH), 124.7 (C), 104.5 (CH); HRMS (ESI) m/z [M + H]+ calcd for C16H10ClN2O+ 291.0477, found 291.0470.
- 5-Chloro-3-(9-phenylacridin-2-yl)isoxazole (7j). Compound 7j was prepared according to the general procedure GP-B from chlorooxime 2d-HCl (620 mg, 1.7 mmol) and 1,1-dichloroethylene 8 (2.7 mL, 33.6 mmol, 20 eq.) in DCM (30 mL) and sat. aq. NaHCO3 (15 mL) to give a pure product of 381 mg (64% yield), after column chromatography on silica (light petroleum/ethyl acetate, 6:1, (v/v)) as a bright yellow solid: mp 159–160 °C (light petroleum/ethyl acetate); 1H NMR (400 MHz, CDCl3) δ 8.36 (d, 1H, J = 9.0 Hz), 8.29 (d, 1H, J = 8.8 Hz), 8.23–8.20 (m, 1H), 8.01 (d, 1H, J = 1.9 Hz), 7.82 (ddd, 1H, J = 8.6, 6.6, 1.4 Hz), 7.72 (dd, 1H, J = 8.8, 1.3 Hz), 7.68–7.61 (m, 3H), 7.49–7.45 (m, 3H), 6.41 (s, 1H); 13C{1H} NMR (CDCl3, 100 MHz) δ 163.7 (C), 155.3 (C), 149.5 (C), 149.0 (C), 148.2 (C), 135.2 (C), 130.8 (CH), 130.7 (CH), 130.4 (CH), 129.7 (CH), 128.8 (CH), 128.7 (CH), 127.3 (CH), 127.0 (CH), 126.2 (CH), 125.8 (CH), 125.5 (C), 125.4 (C), 124.6 (C), 99.6 (CH); HRMS (ESI) m/z [M + H]+ calcd for C22H14ClN2O+ 357.0789, found 357.0785.
- 3-(Acridin-9-yl)-5-(tert-butoxy)isoxazole (7k). Compound 7k was prepared according to the published procedure [56] from chloroisoxazole 7i (165 mg, 0.59 mmol) and tBuOK (100 mg, 0.88 mmol) in THF (6 mL) to give a pure product of 128 mg (68% yield), after column chromatography on silica (light petroleum/ethyl acetate, 8:1, (v/v)) as a colorless solid: mp 166–167 °C (light petroleum/ethyl acetate); 1H NMR (400 MHz, CDCl3) δ 8.28 (dt, 2H, J = 8.9, 0.9 Hz), 8.02 (dt, 2H, J = 8.7, 1.1 Hz), 7.80 (ddd, 2H, J = 8.7, 6.6, 1.4 Hz), 7.55 (ddd, 2H, J = 8.7, 6.6, 1.2 Hz), 1.64 (s, 9H); 13C{1H} NMR (CDCl3, 100 MHz) δ 172.2 (C), 160.6 (C), 148.7 (C), 134.3 (C), 130.2 (CH), 129.8 (CH), 126.6 (CH), 125.8 (CH), 124.8 (C), 87.3 (CH), 85.7 (C), 28.4 (CH3); HRMS (ESI) m/z [M + H]+ calcd for C20H19N2O2+ 319.1441, found 319.1438.
- 5-(tert-Butoxy)-3-(9-phenylacridin-2-yl)isoxazole (7l). Compound 7l was prepared according to the published procedure [56] from chloroisoxazole 7j (320 mg, 0.9 mmol) and tBuOK (152 mg, 1.35 mmol) in THF (10 mL) to give a pure product of 318 mg (90% yield), after column chromatography on silica (light petroleum/ethyl acetate, 10:1, (v/v)) as a light yellow solid: mp 114–115 °C (light petroleum/ethyl acetate); 1H NMR (400 MHz, CDCl3) δ 8.34 (d, J = 9.1 Hz, 1H), 8.29 (d, 1H, J = 8.6 Hz), 8.21 (dd, 1H, J = 9.1, 1.9 Hz), 8.04 (dd, 1H, J = 1.9, 0.7 Hz), 7.79 (ddd, 1H, J = 8.7, 6.5, 1.4 Hz), 7.71 (ddd, 1H, J = 8.8, 1.4, 0.7 Hz), 7.66–7.60 (m, 3H), 7.48–7.43 (m, 3H), 5.58 (s, 1H), 1.52 (s, 9H); 13C{1H} NMR (CDCl3, 100 MHz) δ 172.2 (C), 163.4 (C), 149.2 (C), 149.0 (C), 148.0 (C), 135.4 (C), 130.5 (CH), 130.4 (CH), 129.7 (CH), 128.63 (CH), 128.57 (CH), 127.7 (CH), 127.0 (C), 126.9 (CH), 126.0 (CH), 125.5 (C), 125.0 (CH), 124.7 (C), 85.3 (C), 82.6 (CH), 28.4 (CH3); HRMS (ESI) m/z [M + H]+ calcd for C26H23N2O2+ 395.1754, found 395.1753.
- 3-(2-Methylacridin-9-yl)-4,5-dihydroisoxazole-5-carbonitrile (10). Compound 10 was prepared according to the general procedure GP-B from chlorooxime 2b-HCl (150 mg, 0.49 mmol) and acrylonitrile 9 (518 mg, 9.8 mmol, 20 eq.) in DCM (20 mL) and sat. NaHCO3 aq. (10 mL) to give a pure product of 116 mg (83% yield), after column chromatography on silica (light petroleum/ethyl acetate, 6:1–0:1, (v/v)) as a bright yellow solid: mp 217–218 °C (ethyl acetate); 1H NMR (CDCl3, 400 MHz) δ 8.28 (d, 1H, J = 8.8 Hz), 8.21–8.19 (m, 1H), 7.92 (d, 1H, J = 8.8 Hz), 7.82 (ddd, 1H, J = 8.4, 6.7, 1.4 Hz), 7.70–7.67 (m, 2H), 7.64 (dd, 1H, J = 8.2, 6.7, 1.2 Hz), 5.65 (dd, 1H, J = 10.7, 4.8 Hz), 3.93 (dd, 1H, J = 17.6, 10.7 Hz), 3.81 (dd, 1H, J = 17.6, 4.8 Hz), 2.61 (s, 3H); 13C{1H} NMR (CDCl3, 100 MHz) δ 154.6 (C), 147.8 (C), 147.5 (C), 138.0 (C), 133.6 (CH), 130.2 (CH), 130.1 (CH), 129.9 (CH), 129.5 (C), 127.6 (CH), 124.2 (C), 124.2 (C), 124.0 (CH), 122.1 (CH), 117.0 (C), 66.8 (CH), 46.2 (CH2), 22.2 (CH3); HRMS (ESI) m/z [M + H]+ calcd for C18H14N3O+ 288.1131, found 288.1125.
- tert-Butyl 3-(9-phenylacridin-2-yl)-2H-azirine-2-carboxylate (11a). A mixture of isoxazole 7l (123 mg, 0.31 mmol) and FeCl2·4H2O (6.2 mg, 0.03 mmol) was stirred in MeCN (10 mL) at rt overnight. After the evaporation of the solvent, the product was filtered through a pad of silica (light petroleum/ethyl acetate, 10:1, (v/v)) to give pure product 11a at 107 mg (87% yield) as a yellow solid: mp 155–156 °C (light petroleum/ethyl acetate); 1H NMR (400 MHz, CDCl3) δ 8.42 (d, 1H, J = 8.9 Hz), 8.31 (d, 1H, J = 8.7 Hz), 8.21–8.17 (m, 2H), 7.89–7.85 (m,1H), 7.77–7.75 (m, 1H), 7.67–7.58 (m, 3H), 7.57–7.46 (m, 2H), 7.44–7.41 (m, 1H), 2.77 (s, 1H), 1.43 (s, 9H); 13C{1H} NMR (CDCl3, 100 MHz) δ 170.7 (C), 159.0 (C), 150.4 (C), 149.7 (C), 149.6 (C), 134.8 (C), 132.6 (CH), 131.6 (CH), 131.4 (CH), 130.4 (CH), 130.3 (CH), 129.9 (CH), 129.0 (CH), 128.8 (CH), 128.6 (CH), 128.1 (CH), 127.2 (CH), 126.6 (CH), 125.6 (C), 124.5 (C), 119.8 (C), 81.7 (C), 30.9 (CH), 28.0 (CH3); HRMS (ESI) m/z [M + H]+ calcd for C26H23N2O2+ 395.1754, found 395.1753.
- 9-Phenyl-2-vinylacridine (26). Compound 26 was prepared according to the published procedure [57] from aldehyde 5e (400 mg, 1.4 mmol), methyltriphenylphosphonium bromide (1.51 g, 4.2 mmol) and tBuOK (475 mg, 4.2 mmol) in THF (25 mL) at rt for 48 h to give a pure product pf 397 mg (81% yield), after column chromatography on silica (light petroleum/ethyl acetate, 10:1, (v/v)) as a bright yellow solid: mp 108–109 °C (light petroleum/ethyl acetate); 1H NMR (400 MHz, CDCl3) δ 8.24 (dd, 2H, J = 12.2, 8.9 Hz), 7.98 (dd, 1H, J = 9.2, 2.0 Hz), 7.75 (ddd, 1H, J = 8.5, 6.6, 11.5 Hz), 7.67 (dd, 1H, J = 8.8, 1.3 Hz), 7.65–7.58 (m, 3H), 7.52 (d, 1H, J = 2.0 Hz), 7.46–7.40 (m, 3H), 6.78 (dd, 1H, J = 17.6, 10.9 Hz), 5.83 (d, 1H, J = 17.6 Hz), 5.32 (d, 1H, J = 10.9 Hz); 13C{1H} NMR (CDCl3, 100 MHz) δ 148.8 (C), 148.6 (C), 147.0 (C), 136.5 (CH), 135.8 (C), 134.6 (C), 130.5 (CH), 129.94 (CH), 129.89 (CH), 129.6 (CH), 128.5 (CH), 128.4 (CH), 127.1 (CH), 126.8 (CH), 125.7 (CH), 125.5 (C), 125.3 (CH), 125.2 (C), 115.1 (CH2); HRMS (ESI) m/z [M + H]+ calcd for C21H16N+ 282.1277, found 282.1290.
- 2-(1,2-Dibromoethyl)-9-phenylacridine (27). Compound 27 was prepared according to the published procedure [58] from styrene 26 (320 mg, 1.14 mmol) and Br2 (0.7 mL, 1.4 mmol0) in CHCl3 (5 mL) at 0 °C for 30 min to give a pure product of 387 mg (77% yield), after column chromatography on silica (light petroleum/ethyl acetate, 10:1, (v/v)) as a bright yellow solid: mp 154–155 °C (light petroleum/ethyl acetate); 1H NMR (400 MHz, CDCl3) δ 8.34 (d, 1H, J = 9.1 Hz), 8.29 (d, 1H, J = 8.8 Hz), 7.84–7.79 (m, 2H), 7.71 (d, 1H, J = 8.8 Hz), 7.72–7.60 (m, 4H), 7.47–7.43 (m, 3H), 5.22 (dd, 1H, J = 10.6, 5.5 Hz), 4.11–4.01 (m, 2H); 13C{1H} NMR (CDCl3, 100 MHz) δ 149.0 (br. s, C), 148.3 (br. s, C), 135.3 (C), 130.9 (br. s, C), 130.6 (br. s, CH), 130.5 (CH), 130.4 (CH), 129.4 (br. s, CH), 128.7 (CH), 128.6 (C), 128.6 (CH), 128.3 (br. s, CH), 126.9 (CH), 126.3 (CH), 126.1 (CH), 125.5 (C), 124.3 (C), 51.1 (CH), 34.3 (CH2); HRMS (ESI) m/z [M + H]+ calcd for C21H16Br2N+439.9644, found 439.9648.
- 2-(2H-Azirin-3-yl)-9-phenylacridine (28). Compound 28 was prepared according to the published procedure [59] from dibromide 27 (380 mg, 0.86 mmol), NaN3 (84 mg, 1.3 mmol), and NaOH (40 mg, 1 mmol) in DMSO (2 mL) and toluene (5 mL) to give a pure product of 102 mg (40% yield), after column chromatography on silica (light petroleum/ethyl acetate, 10:1, (v/v)) as a yellow-brown solid: mp 192–193 °C (light petroleum/ethyl acetate); 1H NMR (400 MHz, CDCl3) δ 8.41 (d, 1H, J = 9.0 Hz), 8.32 (d, 1H, J = 8.8 Hz), 8.26 (dd, 1H, J = 8.9, 1.8 Hz), 8.20 (d, 1H, J = 1.6 Hz), 7.86 (ddd, 1H, J = 8.5, 6.5, 1.4 Hz), 7.75 (d, 1H, J = 8.6 Hz), 7.68–7.62 (m, 3H), 7.52–7.47 (m, 3H), 1.82 (s, 2H); 13C{1H} NMR (CDCl3, 100 MHz) δ 165.7 (C), 150.0 (C), 149.7 (C), 149.4 (C), 135.0 (C), 131.6 (CH), 131.3 (CH), 131.0 (CH), 130.4 (CH), 129.7 (CH), 128.9 (CH), 128.7 (CH), 127.9 (CH), 127.2 (CH), 126.5 (CH), 125.6 (C), 124.6 (C), 122.8 (C), 20.5 (CH2); HRMS (ESI) m/z [M + H]+ calcd for C21H15N2+ 295.1230, found 295.1238.
- 9-(5-(3-Phenyl-2H-azirin-2-yl)-1H-1,2,3-triazol-1-yl)acridine (32). Compound 32 was prepared according to the published procedure [55] from phosphonium salt 29 (250 mg, 0.5 mmol) and azide 31 (165 mg, 0.75 mmol) in benzene (10 mL) for 4 h to give a pure product of 139 mg (77% yield), after column chromatography on silica (light petroleum/ethyl acetate, 8:1–3:1, (v/v)) as a beige solid: mp 194–196 °C (dec., light petroleum/ethyl acetate); 1H NMR (400 MHz, CDCl3) δ 8.30 (d, 1H, J = 8.8 Hz), 8.23 (d, 1H, J = 8.8 Hz), 7.89–7.85 (m, 1H), 7.84–7.80 (m, 1H), 7.82 (s, 1H), 7.66–7.58 (m, 2H), 7.51–7.47 (m, 1H), 7.38 (d, 2H, J = 9.2 Hz), 7.32–7.28 (m, 4H), 2.86 (s, 1H); 13C{1H} NMR (CDCl3, 100 MHz) δ 161.4 (C), 149.2 (C), 149.1 (C), 141.4 (C), 136.1 (C), 133.7 (CH), 132.3 (CH), 130.8 (CH), 130.7 (CH), 129.76 (CH), 129.74 (CH), 129.1 (CH), 129.0 (CH), 128.4 (CH), 128.2 (CH), 123.2 (C), 123.2 (C), 122.6 (CH), 122.4 (CH), 122.0 (C), 23.1 (CH); HRMS (ESI) m/z [M + H]+ calcd for C23H16N5+ 362.1400, found 362.1399.
- 3-(9-Benzyl-9,10-dihydroacridin-9-yl)-5-phenylisoxazole (33a). Compound 33a was prepared according to the general procedure GP-C from isoxazole 7a (34 mg, 0.105 mmol) in toluene (4 mL) at 380 nm for 50 min to give a pure product of 33 mg (75% yield), after column chromatography on silica (light petroleum/ethyl acetate, 15:1, (v/v)) as a colorles solid: mp 143–144 °C (light petroleum/ethyl acetate); 1H NMR (400 MHz, CDCl3) δ 7.76–7.73 (m, 2H), 7.467.40 (m, 3H), 7.12–7.04 (m, 5H), 6.94–6.90 (m, 1H), 6.88–6.84 (m, 1H), 6.45–6.43 (m, 2H), 6.32–6.30 (m, 2H), 6.23 (s, 1H), 5.65 (s, 1H), 3.60 (s, 2H); 13C{1H} NMR (CDCl3, 100 MHz) δ 170.7 (C), 169.4 (C), 138.5 (C), 137.0 (C), 130.7 (CH), 129.9 (CH), 129.3 (CH), 128.8 (CH), 128.0 (CH), 127.5 (C), 126.9 (CH), 125.83 (CH), 125.77 (CH), 122.0 (C), 120.4 (CH), 113.2 (CH), 101.0 (CH), 50.6 (CH2), 48.2 (C); HRMS (ESI) m/z [M + H]+ calcd for C9H23N2O+ 415.1805, found 415.1810.
- 3-(9-(3,5-Dimethylbenzyl)-9,10-dihydroacridin-9-yl)-5-phenylisoxazole (33b). Compound 33b was prepared according to the general procedure GP-C from isoxazole 7a (52 mg, 0.16 mmol) in mesitylene (10 mL) at 380 nm for 90 min to give a pure product of 48 mg (67% yield), after column chromatography on silica (light petroleum/ethyl acetate, 15:1, (v/v)) as a light yellow solid: mp 187–188 °C (light petroleum/ethyl acetate); 1H NMR (400 MHz, CDCl3) δ 7.73 (dd, 2H, J = 7.7, 2.0 Hz), 7.44–7.38 (m, 3H), 7.10–7.06 (m, 4H), 6.84 (t, 2H, J = 7.5 Hz), 6.68 (br. s, 1H), 6.41 (dd, 2H, J = 8.3, 1.2 Hz), 6.23 (s, 1H), 5.86 (s, 2H), 5.64 (br. s, 1H), 3.47 (s, 2H), 1.98 (s, 6H); 13C{1H} NMR (CDCl3, 100 MHz) δ 170.6 (C), 169.3 (C), 138.7 (C), 136.7 (C), 136.0 (C), 129.9 (CH), 129.3 (CH), 128.8 (CH), 128.6 (CH), 127.8 (CH), 127.6 (C), 127.2 (CH), 125.8 (CH), 122.2 (C), 120.3 (CH), 112.9 (CH), 101.1 (CH), 50.7 (CH2), 48.3 (C), 21.0 (CH3); HRMS (ESI) m/z [M + H]+ calcd for C31H27N2O+ 443.2118, found 443.2124.
- 3-(9-(4-Chlorobenzyl)-9,10-dihydroacridin-9-yl)-5-phenylisoxazole (33c). Compound 33c was prepared according to the general procedure GP-C from isoxazole 7a (60 mg, 0.19 mmol) and 4-chlorotoluene (2.36 g, 18.6 mmol, 100 eq.) in α,α,α-trifluorotoluene (12 mL) at 380 nm for 40 h to give a pure product of 27 mg (32% yield), after column chromatography on silica (light petroleum/ethyl acetate, 25:1, (v/v)) as a light yellow solid: mp 183–184 °C (light petroleum/ethyl acetate); 1H NMR (400 MHz, C6D6) δ 7.36–7.34 (m, 2H), 7.10–7.09 (m, 2H), 6.97–6.92 (m, 5H), 6.84–6.81 (m, 2H), 6.770–6.66 (m, 2H), 6.21–6.19 (m, 2H), 6.03–6.00 (m, 2H), 5.97 (s, 1H), 4.92 (br. s, 1H), 3.71 (s, 2H); 13C{1H} NMR (C6D6, 100 MHz) δ 170.8 (C), 169.9 (C), 138.9 (C), 136.3 (C), 132.5 (CH), 132.4 (C), 129.9 (CH), 129.7 (CH), 128.9 (CH), 128.4 (CH), 128.2 (CH), 127.5 (CH), 126.1 (CH), 122.4 (C), 120.9 (CH), 113.6 (CH), 101.4 (CH), 50.6 (CH2), 48.6 (C); HRMS (ESI) m/z [M + Na]+ calcd for C29H21ClN2NaO+ 471.1235, found 471.1236.
- 3-(9-(tert-Butoxymethyl)-9,10-dihydroacridin-9-yl)-5-phenylisoxazole (33d). Compound 33d was prepared according to the general procedure GP-C from isoxazole 7a (60 mg, 0.19 mmol) in MTBE (7 mL) at 380 nm for 2.5 h to give a pure product of 56 mg (73% yield), after column chromatography on silica (light petroleum/ethyl acetate, 20:1, (v/v)) as a colorless solid: mp 180–181 °C (light petroleum/ethyl acetate); 1H NMR (400 MHz, C6D6): δ 7.36–7.33 (m, 2H), 7.24–7.22 (m, 2H), 7.05–7.01 (m, 2H), 6.96–6.93 (m, 3H), 6.77–6.73 (m, 2H), 6.37–6.34 (m, 2H), 6.03 (s,1H), 5.62–5.60 (br. s, 1H), 4.32 (s, 2H), 0.87 (s, 9H); 13C NMR (100 MHz, C6D6): δ 169.5 (C), 169.2 (C), 139.4 (C), 130.0 (CH), 129.7 (CH), 128.9 (CH), 128.4 (CH), 128.0 (CH), 126.1 (CH), 122.7 (C), 120.6 (CH), 113.6 (CH), 101.4 (CH), 73.0 (C), 71.2 (CH2), 48.4 (C), 27.3 (CH3); HRMS (ESI) m/z [M + H]+ calcd for C27H27N2O2+ 411.2064, found 411.2067.
- 3-(9-(1,4-Dioxan-2-yl)-9,10-dihydroacridin-9-yl)-5-phenylisoxazole (33e). Compound 33e was prepared according to the general procedure C from isoxazole 7a (50 mg, 0.16 mmol) in 1,4-dioxane (5 mL) at 405 nm for 90 min to give a pure product pf 52 mg (66% yield), after column chromatography on silica (light petroleum/ethyl acetate, 20:1, (v/v)) as a light brown semi-solid; NMR spectra indicate that the product is a mixture of two diastereomers in a ~1:1 ratio; 1H NMR (400 MHz, CDCl3): δ 7.72–7.68 (m, 2H), 7.42–7.36 (m, 3H), 7.22–7.18 (m, 1H), 7.14–7.11 (m, 2H), 7.04–7.02 (m, 1H), 6.87–6.71 (m, 4H), 6.27 (br. S, 1H), 6.12 (s, 1H), 4.46–4.43 (m, 1H), 3.84–3.79 (m, 2H), 3.64–3.54 (m, 2H), 3.37–3.31 (m, 1H), 3.24–3.19 (m, 1H); 13C NMR (100 MHz, CDCl3): δ 168.9 ©, 168.3 (br. s, C), 138.4 (br. s, C), 138.3 (C), 132.2 (br. s, CH), 129.9 (CH), 129.6 (br. s, CH), 128.8 (CH), 128.5 (br. s, CH), 128.3 (br. s, CH), 127.5 (C), 125.7 (CH), 120.7 (br. s, CH), 120.1 (br. s, CH), 119.6 (br. s, C), 119.1 (br. s, C), 113.7 (br. s, CH), 113.3 (br. s, CH), 101.2 (CH), 81.2 (CH), 67.7 (CH2), 67.3 (CH2), 66.2 (CH2), 48.7 (br. s, C); HRMS (ESI) m/z [M + H]+ calcd for C26H23N2O3+ 411.1703, found 411.1700.
- 3-(9-(2-Methylbenzyl)-9,10-dihydroacridin-9-yl)-5-(trimethylsilyl)isoxazole (33f). Compound 33f was prepared according to the general procedure GP-C from isoxazole 7b (57 mg, 0.18 mmol) in o-xylene (8 mL) at 380 nm for 60 min to give a pure product of 61 mg (80% yield), after column chromatography on silica (light petroleum/ethyl acetate, 10:1, (v/v)) as a light yellow solid: mp 187–188 °C (light petroleum/ethyl acetate); 1H NMR (400 MHz, CDCl3) δ 7.09 (td, 2H, J = 7.7, 1.5 Hz), 6.96–6.92 (m, 3H), 6.85–6.81 (m, 3H), 6.67 (td, 1H, J = 7.5, 1.5 Hz), 6.42 (d, 2H, J = 7.9 Hz), 6.12 (s, 1H), 6.08 (dd, 1H, J = 7.7, 1.4 Hz), 5.65 (br. s, 1H), 3.62 (s, 2H), 1.48 (s, 3H), 0.32 (s, 9H); 13C NMR (100 MHz, CDCl3): δ 177.5 (C), 168.3 (C), 139.0 (C), 138.0 (C), 135.3 (C), 131.7 (CH), 129.6 (CH), 129.4 (CH), 127.8 (CH), 125.9 (CH), 124.2 (CH), 122.6 (C), 120.4 (CH), 114.1 (CH), 113.2 (CH), 48.1 (C), 47.2 (CH2), 18.8 (CH3), −1.9 (CH3); HRMS (ESI) m/z [M + Na]+ calcd for C27H28N2NaOSi+ 447.1863, found 447.1868.
- 3-(9-(Tetrahydrofuran-2-yl)-9,10-dihydroacridin-9-yl)-5-(trimethylsilyl)isoxazole (33g). Compound 33g was prepared according to the general procedure GP-C from isoxazole 7b (100 mg, 0.31 mmol) in THF (10 mL) at 405 nm for 3 h to give a pure product of 72 mg (59% yield), after column chromatography on silica (light petroleum/ethyl acetate, 20:1, (v/v)) as a colorless solid: mp 79–80 °C (light petroleum/ethyl acetate); NMR spectra indicate that the product is a mixture of two diastereomers in a ~1:1 ratio; 1H NMR (400 MHz, CDCl3): δ 7.16–7.05 (m, 3H), 6.95–6.92 (m, 1H), 6.82–6.75 (m, 2H), 6.2–6.68 (m, 2H), 6.23 (br. s, 1H), 6.05 (s, 1H), 4.86 (t, 1H, J = 7.2 Hz), 3.75 (td, 1H, J = 7.4, 4.8 Hz), 3.36 (q, 1H, 7.4 Hz), 1.73–1.69 (m, 2H), 1.33–1.23 (m, 2H); 13C NMR (100 MHz, CDCl3): δ 176.9 (C), 167.0 (C), 138.8 (C), 138.2 (C), 132.1 (CH), 129.7 (CH), 128.1 (CH), 127.9 (CH), 125.1 (C), 121.2 (C), 120.2 (CH), 120.0 (CH), 119.9 (C), 115.4 (C), 114.1 (CH), 113.4 (CH), 112.9 (CH), 86.2 (CH), 69.2 (CH2), 49.8 (C), 27.7 (CH2), 25.7 (CH2), −1.9 (CH3); HRMS (ESI) m/z [M + Na]+ calcd for C23H26N2NaO2Si+ 413.1656, found 413.1658.
- 3-(9-Benzyl-9,10-dihydroacridin-9-yl)-5-(tert-butoxy)isoxazole (33h). Compound 33h was prepared according to the general procedure GP-C from isoxazole 7k (41 mg, 0.13 mmol) in toluene (5 mL) at 380 nm for 90 min to give a pure product of 31 mg (59% yield), after column chromatography on silica (light petroleum/ethyl acetate, 20:1, (v/v)) as a colorless solid: mp 161–162 °C (light petroleum/ethyl acetate); 1H NMR (400 MHz, CDCl3) δ 7.11–7.00 (m, 5H), 6.90–6.82 (m, 4H), 6.38 (dd, 2H, J = 7.9, 1.2 Hz), 6.26–6.23 (m, 2H), 5.58 (s, 1H), 5.00 (s, 1H), 3.45 (s, 2H), 1.43 (s, 9H); 13C NMR (100 MHz, C6D6) δ 172.0 (C), 171.8 (C), 138.9 (C), 137.8 (C), 131.3 (CH), 129.8 (CH), 128.4 (CH), 128.4 (CH), 127.9 (CH), 127.3 (CH), 126.2 (CH), 122.9 (C), 120.7 (CH), 113.5 (CH), 85.7 (CH), 83.7 (C), 50.7 (CH2), 49.2 (C), 27.9 (CH3); HRMS (ESI) m/z [M + H]+ calcd for C27H27N2O2+ 411.2067, found 411.2070.
- 3-(9-(3,5-Dimethylbenzyl)-9,10-dihydroacridin-9-yl)-5-((4-isopropylphenoxy)methyl)isoxazole (33i). Compound 33i was prepared according to the general procedure GP-C from isoxazole 7k (55 mg, 0.14 mmol) in mesitylene (10 mL) at 380 nm for 90 min to give a pure product of 53 mg (74% yield), after column chromatography on silica (light petroleum/ethyl acetate, 20:1, (v/v)) as a colorless solid: mp 167–168 °C (light petroleum/ethyl acetate); 1H NMR (400 MHz, CDCl3) δ 7.15–7.12 (m, 2H), 7.08–7.04 (m, 2H), 6.98 (d, 2H, J = 7.6 Hz), 6.87–6.81 (m, 4H), 6.67 (s, 1H), 6.40–6.38 (m, 2H), 6.04 (s, 1H), 5.83 (s, 2H), 5.60 (s, 1H), 5.08 (s, 2H), 3.42 (s, 2H), 2.85 (hept, 1H, J = 6.9 Hz), 1.97 (s, 6H), 1.22 (d, 6H, J = 6.9 Hz); 13C NMR (100 MHz, CDCl3) δ 169.9 (C), 167.6 (C), 156.1 (C), 142.3 (C), 138.7 (C), 136.7 (C), 136.0 (C), 129.2 (CH), 128.7 (CH), 127.8 (CH), 127.4 (CH), 127.2 (CH), 122.2 (C), 120.3 (CH), 114.9 (CH), 113.0 (CH), 104.8 (CH), 61.9 (CH2), 50.7 (CH2), 48.3 (C), 33.3 (CH), 24.1 (CH3), 20.9 (CH3); HRMS (ESI) m/z [M + H]+ calcd for C35H35N2O2+ 515.2693, found 515.2692.
- Methyl 3-(9-(1,4-dioxan-2-yl)-2-methyl-9,10-dihydroacridin-9-yl)isoxazole-5-carboxylate (33j). Compound 33j was prepared according to the general procedure GP-C from isoxazole 7d (90 mg, 0.28 mmol) in 1,4-dioxane (20 mL) at 380 nm for 2 h to givea pure product of 65 mg (57% yield), after column chromatography on silica (light petroleum/MTBE, 3:1, (v/v)) as a light yellow solid: mp 79–81 °C (light petroleum/MTBE); NMR spectra indicate that the product is a mixture of two diastereomers in a ~1:1 ratio; 1H NMR (400 MHz, C6D6) δ 7.31–7.29 (m, 0.5 H), 7.05–7.00 (m, 0.5 H), 6.99–6.96 (m, 1H), 6.89–6.86 (m, 0.5 H), 6.78–6.74 (m, 1H), 6.64–6.59 (m, 0.5 H), 6.49 (s, 0.5 H), 6.48 (s, 0.5 H), 6.30–6.24 (m, 1H), 6.21–6.14 (m, 1H), 5.44 (s, 0.5 H), 5.43 (s, 0.5 H), 4.78 (dd, 0.5 H, J = 10.2, 2.3 Hz), 4.72 (dd, 0.5 H, J = 10.2, 2.2 Hz), 3.91–3.86 (m, 1H), 3.70–3.60 (m, 1H), 3.44–3.37 (m, 2H), 3.14 (s, 1.5 H), 3.14 (s, 1.5 H), 3.16–3.10 (m, 1H), 3.06–3.00 (m, 1H), 2.04 (s, 1.5 H), 1.92 (s, 1.5 H); 13C NMR (100 MHz, C6D6) δ 168.99 (C), 168.96 (C), 160.1 (C), 157.04 (C), 157.03 (C), 139.1 (C), 136.7 (C), 136.5 (C), 132.8 (CH), 132.5 (CH), 130.3 (C), 129.78 (CH), 129.75 (CH), 129.6 (CH), 129.5 (CH), 128.66 (CH), 128.62 (CH), 128.4 (CH), 120.8 (CH), 120.3 (CH), 119.6 (C), 119.5 (C), 119.3 (C), 119.1 (C), 114.3 (CH), 114.1 (CH), 113.9 (CH), 113.7 (CH), 111.48 (CH), 111.46 (CH), 82.1 (CH), 81.9 (CH), 67.9 (CH2), 67.72 (CH2), 67.66 (CH2), 66.16 (CH2), 66.12 (CH2), 51.8 (CH3), 49.33 (C), 49.25 (C), 30.2 (C), 20.9 (CH3), 20.7 (CH3); HRMS (ESI) m/z [M + H]+ calcd for C23H25N2O5+ 407.1601, found 407.1601.
- 3-(9-(tert-Butoxymethyl)-2-methyl-9,10-dihydroacridin-9-yl)-4,5-dihydroisoxazole-5-carbonitrile (33k). Compound 33k was prepared according to the general procedure GP-C from isoxazole 10 (60 mg, 0.18 mmol) in MTBE (20 mL) at 380 nm for 2 h to give a pure product of 47 mg (69% yield), after column chromatography on silica (light petroleum/MTBE, 1:1, (v/v)) as a light brow oil; NMR spectra indicate that the product is a mixture of two diastereomers in a ~1:1 ratio; 1H NMR (400 MHz, C6D6) δ 7.23–7.19 (m, 1 H), 7.03–6.91 (m, 2 H), 6.84–6.78 (m, 1.5 H), 6.73–6.69 (m, 0.5 H), 6.26–6.19 (m, 2H), 5.38 (s, 0.5 H), 5.37 (s, 0.5 H), 4.07–3.94 (m, 3H), 2.51–2.27 (m, 2H), 2.21 (s, 1.5 H), 2.08 (s, 1.5 H), 0.77 (s, 4.5 H), 0.77 (s, 4.5 H); 13C NMR (100 MHz, C6D6) δ 161.1 (C), 160.9 (C), 139.9 (C), 139.4 (C), 137.3 (C), 136.9 (C), 129.6 (C), 129.4 (CH), 129.3 (CH), 128.63 (CH), 128.56 (CH), 128.45 (CH), 128.42 (CH), 128.39 (CH), 121.1 (CH), 120.3 (CH), 120.0 (C), 119.8 (C), 119.2 (C), 119.1 (C), 117.9 (C), 117.7 (C), 114.1 (CH), 114.0 (CH), 113.94 (CH), 113.90 (CH), 73.0, 71.68 (CH2), 71.63 (CH2), 65.9 (CH), 65.8 (CH), 48.6, 48.5, 42.0 (CH2), 27.1 (CH3); HRMS (ESI) m/z [M + H]+ calcd for C23H26N3O2+ 376.2020, found 376.2013.
- 4-((3-(9-Benzyl-2-methyl-9,10-dihydroacridin-9-yl)isoxazol-5-yl)methyl)-5-methoxy-3-(naphthalen-2-yl)isoxazole (33l). Compound 33l was prepared according to the general procedure GP-C from isoxazole 7e (50 mg, 0.10 mmol) in toluene (10 mL) at 380 nm for 5h to give a pure product of 37 mg (62% yield), after column chromatography on silica (light petroleum/ethyl acetate, 20:1, (v/v)) as a colorless solid: mp 157–158 °C (light petroleum/ethyl acetate); 1H NMR (400 MHz, CDCl3) δ 7.91 (s, 1H), 7.86–7.84 (m, 2H), 7.71 (d, 1H, J = 8.0 Hz), 7.61 (d, 1H, J = 8.6 Hz), 7.54 (t, 1H, J = 7.3 Hz), 7.49 (t, 1H, J = 7.4 Hz), 7.02 (d, 1H, J = 7.8 Hz), 6.98 (d, 1H, J = 8.4 Hz), 6.89–6.78 (m, 5H), 6.622 (t, 1H, J = 7.7 Hz), 6.34 (d, 1H, J = 7.9 Hz), 6.29 (d, 1H, J = 8.0 Hz), 6.24 (d, 2H, J = 7.6 Hz), 5.65 (s, 1H), 5.50 (s, 1H), 4.12 (s, 3H), 3.85 (s, 2H), 3.51–3.43 (m, 2H), 2.13 (s, 3H); 1H NMR (400 MHz, C6D6) δ 8.03 (s, 1H), 7.83–7.81 (m, 1H), 7.58–7.56 (m, 2H), 7.53–7.51 (m, 1H), 7.28–7.20 (m, 2H), 7.03–6.98 (m, 2H), 6.92–6.84 (m, 4H), 6.75–6.73 (m, 1H), 6.57–6.53 (m, 1H), 6.50–6.47 (m, 2H), 5.98–5.94 (m, 2H), 5.55 (s, 1H), 4.87 (s, 1H), 4.87 (s, 1H), 3.89–3.79 (m, 2H), 3.48 (s, 2H), 3.33 (s, 3H), 1.93 (s, 3H); 13C NMR (100 MHz, C6D6) δ 170.7 ©, 170.6 (C), 170.4 (C), 164.5 (C), 139.0 (C), 137.8 (C), 136.6 (C), 134.2 (C), 133.6 (C), 131.2 (CH), 129.8 (CH), 129.68 (CH), 129.66 (C), 129.02 (CH), 128.9 (CH), 128.8 (CH), 128.4 (CH), 128.2 (CH), 128.0 (CH), 127.9 (CH), 127.8 (C), 127.3 (CH), 127.1 (CH), 126.7 (CH), 126.3 (CH), 125.5 (CH), 122.5 (C), 120.3 (CH), 113.6 (CH), 113.4 (C), 103.6 (CH), 86.8 (C), 57.4 (CH3), 51.2 (CH2), 48.7 (C), 20.7 (CH3), 20.1 (CH2); HRMS (ESI) m/z [M + H]+ calcd for C39H32N3O3+ 590.2438, found 590.2424.
- 4-((3-(9-(3,5-Dimethylbenzyl)-2-methyl-9,10-dihydroacridin-9-yl)isoxazol-5-yl)methyl)-5-methoxy-3-(naphthalen-2-yl)isoxazole (33m). Compound 33m was prepared according to the general procedure GP-C from isoxazole 7e (50 mg, 0.10 mmol) in mesytilene (10 mL) at 380 nm for 5h to give a pure product of 43 mg (70% yield), after column chromatography on silica (light petroleum/ethyl acetate, 20:1, (v/v)) as a beige solid: mp 162–163 °C (light petroleum/ethyl acetate); 1H NMR (400 MHz, C6D6): δ 8.03 (s, 1H), 7.84–7.81 (m, 1H), 7.58–7.56 (m, 2H), 7.54–7.51 (m, 1H), 7.27–7.20 (m, 2H), 7.03–7.00 (m, 2H), 6.92–6.88 (m, 1H), 6.77–6.74 (m, 1H), 6.61 (s, 1H), 6.57–6.53 (m, 1H), 6.10 (s, 2H), 6.01–5.97 (m, 2H), 5.58 (s, 1H), 4.91 (s, 1H), 3.84–3.75 (m, 2H), 3.49 (s, 2H), 3.33 (s, 3H), 1.99 (s, 6H), 1.94 (s, 3H); 13C NMR (100 MHz, C6D6) δ 170.7 (C), 170.5 (C), 170.4 (C), 164.5 (C), 139.2 (C), 137.4 (C), 136.8 (C), 136.0 (C), 134.2 (C), 133.6 (C), 129.8 (CH), 129.7 (CH), 129.6 (C), 129.4 (CH), 129.0 (CH), 128.9 (CH), 128.6 (CH), 128.4 (CH), 128.4 (CH), 128.2 (CH), 128.0 (CH), 127.9 (CH), 127.7 (CH), 127.1 (CH), 126.7 (CH), 125.5 (CH), 122.8 (C), 120.3 (CH), 113.3 (CH), 113.2 (CH), 103.7 (CH), 86.8 (C), 57.4 (CH3), 51.2 (CH2), 48.8 (C), 21.3 (CH3), 20.7 (CH3), 20.1 (CH2); HRMS (ESI) m/z [M + H]+ calcd for C41H36N3O3+ 618.2751, found 618.2738.
- 5-(tert-Butoxy)-3-(9-(3,5-dimethylbenzyl)-9-phenyl-9,10-dihydroacridin-2-yl)isoxazole (33n). Compound 33n was prepared according to the general procedure GP-C from isoxazole 7l (56 mg, 0.14 mmol) in mesytilene (15 mL) at 380 nm for 8h to give a pure product of 34 mg (47% yield), after column chromatography on silica (light petroleum/MTBE, 20:1, (v/v)) as a colorless solid: mp 138–139 °C (light petroleum/ethyl acetate); 1H NMR (400 MHz, C6D6) δ 7.68–7.66 (m, 1H), 7.53–7.47 (m, 3H), 7.12–7.10 (m, 2H), 7.04–7.01 (m, 1H), 6.92–6.88 (m, 1H), 6.82–6.80 (m, 1H), 6.68–6.64 (m, 2H), 6.02 (s, 2H), 5.99–5.94 (m, 2H), 5.37 (s, 1H), 4.92 (s, 1H), 3.40–3.30 (m, 2H), 2.00 (s, 6H), 1.11 (s, 9H); 13C NMR (100 MHz, C6D6) δ 172.3 (C), 163.9 (C), 151.3 (C), 140.4 (C), 138.6 (C), 137.6 (C), 136.1 (C), 130.8 (CH), 130.0 (CH), 129.1 (CH), 129.0 (CH), 128.4 (CH), 128.3 (CH), 127.8 (CH), 127.3 (C), 127.1 (C), 127.0 (CH), 126.4 (CH), 125.5 (CH), 122.4 (C), 120.8 (CH), 113.3 (CH), 113.0 (CH), 84.1 (C), 82.6 (CH), 52.5 (CH2), 51.7 (C), 28.2 (CH3), 21.3 (CH3); HRMS (ESI) m/z [M + H]+ calcd for C35H35N2O2+ 515.2693, found 515.2690.
3.2.5. Cell Culture
The MCF7 breast cancer cell line, HCT 116 colorectal carcinoma cell line, A-704 kidney adenocarcinoma cell line, and WI-26 VA4 lung epithelial-like cells were purchased from the ATCC. The MCF7 cells were maintained in MEM (Gibco, Paisley, UK) supplemented with 10% fetal bovine serum (FBS, Gibco, UK), human recombinant insulin (0.01 mg/mL), penicillin (100 UI mL−1), streptomycin (100 µg mL−1), and GlutaMax (2 mM, Gibco, UK). The HCT 116 cells were maintained in McCoy’s 5A (Gibco, UK) supplemented with 10% fetal bovine serum (FBS, Gibco, UK), penicillin (100 UI mL−1), streptomycin (100 µg mL−1), and GlutaMax (1.5 mM, Gibco, UK). A-704 and WI-26 VA4 cells were maintained in MEM (Gibco, UK) supplemented with 10% fetal bovine serum (FBS, Gibco, UK), penicillin (100 UI mL−1), streptomycin (100 µg mL−1), and GlutaMax (1.87 mM, Gibco, UK). All cell lines’ cultivation was performed under a humidified atmosphere of 95% air/5% CO2 at 37 °C. Subconfluent monolayers, in the log growth phase, were harvested by a brief treatment with TrypLE Express solution (Gibco, UK) in phosphate-buffered saline (PBS, Capricorn Scientific, Ebsdorfergrund, Germany) and washed three times in serum-free PBS. The number of viable cells was determined by trypan blue exclusion.
3.2.6. Antiproliferative Assay
The effects of the synthesized compounds on cell viability were determined using the MTT colorimetric test. All examined cells were diluted with the growth medium to 3.5 × 104 cells per mL and the aliquots (7 × 103 cells per 200 μL) were placed in individual wells in 96-multiplates (Eppendorf, Germany) and incubated for 24 h. The next day, the cells were then treated with synthesized compounds separately at a final concentration of 30 μM and incubated for 72 h at 37 °C in a 5% CO2 atmosphere. After incubation, the cells were then treated with 40 μL of MTT solution (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide, 5 mg mL−1 in PBS) and incubated fir 4 h. After an additional 4h incubation, the medium with MTT was removed and DMSO (150 μL) was added to dissolve the crystals formazan. The plates were shaken for 10 min. The optical density of each well was determined at 560 nm using a microplate reader GloMax Multi+ (Promega, Madison, WI, USA). Each of the tested compounds was evaluated for cytotoxicity in three separate experiments.
4. Conclusions
Easy-to-handle N-hydroxyacridinecarbimidoyl chloride hydrochlorides were prepared from oximes in 69–94% by chlorination using Cl2. The cycloaddition of nitrile oxides, which were generated from them, to terminal alkynes gave 3-(acridin-9-yl)isoxazoles in a 80–98% yield. The reaction with 1,1-dichloroethene, accompanied by the dehydrochlorination of intermediate 5,5-dichloro-4,5-dihydroisoxazoles, gave acridinyl-substituted 5-chloroizoxazoles in a 62–64% yield, and the reaction with acrylonitrile gave 4,5-dihydroisoxazole-5-carbonitrile in an 83% yield. Acridinyl-containing azirines with a long-wave absorption of 320–420 nm were prepared from acridinyl isoxazoles or by traditional methods, in order to evaluate the possibility of their use in photoclick cycloaddition in the UV/visible radiation boundary region. However, their photolysis in the presence of DMAD or PMI in acetonitrile using 365, 380, 405, 425, or 450 nm LEDs, as well as white light LED (380–760 nm), did not produce the expected photolysis cycloaddition products of the nitrile ylide. A comparison of the results of DFT calculations of the cycloaddition of nitrile ylides, derived from acridinylazirines and 3-(pyren-1-yl)-2H-azirine, for which such cycloaddition was previously implemented, did not reveal any obstacles in terms of energy barriers to the cycloaddition of acridinyl-substituted nitrile ylides. This means that the acridine fragment, unlike the pyrene fragment, cannot be an antenna for transmitting light energy for the formation of nitrile ylides from azirines. One of the reasons for this may be the fast relaxation of the singlet excited state of acridinyl-substituted azirines through intersystem crossing into a triplet excited state, characteristic of acridine heterocyclic systems. This may prevent the energy required to break the azirine C2-C3 bond from being transferred from the excited acridine moiety to the azirine moiety and block, therefore, the formation of nitrile ylide from azirine. It was found that irradiation of the synthesized isoxazolylacridines with LED light already in the UV/visible radiation boundary region (385–405 nm) in the presence of toluene, o-xylene, mesitylene, 4-chlorotoluene, THF, 1,4-dioxane, and MTBE gave 9-alkylated acridane derivatives, usually in good yields. A series of synthesized acridine/acridane-substituted isoxazoles and acridine-substituted azirines were tested for their cytotoxicity. Azirines were toxic to all cell lines, including normal cells (WI-26 VA4). 5-Methoxy-4-((3-(2-methylacridin-9-yl)isoxazol-5-yl)methyl)-3-(naphthalen-2-yl)isoxazole, 3-(acridin-9-yl)-5-chloroisoxazole, and 5-(tert-butoxy)-3-(9-phenylacridin-2-yl)isoxazole are among the compounds that were active, but not particularly selective.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/molecules29071538/s1, X-ray diffraction experiment; UV-VIS absorption spectra of compounds 7; 10; 11a; 28; NMR spectra of compounds 4e; 5; 1; 2; 7; 10; 11a; 26; 27; 28; 32; 33; Computational details. References [60,61,62,63,64,65,66,67,68,69] are cited in the Supplementary Materials.
Author Contributions
Conceptualization, A.F.K. and M.S.N.; methodology, A.F.K. and E.E.G.; synthesis, E.E.G.; writing, A.F.K., E.E.G., M.S.N. and A.S.B.; antiproliferative assay, A.S.B.; funding acquisition, A.F.K. All authors have read and agreed to the published version of the manuscript.
Funding
This research was supported by the Russian Science Foundation, grant number 22-13-00011.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data are contained within the article.
Acknowledgments
In commemoration of the 300th anniversary of St Petersburg State University’s founding. This research was carried out using resources of the Centre for Magnetic Resonance, the Research Centre for X-ray Diffraction Studies, the Centre for Chemical Analysis and Materials, and the Computer Centre of the Science Park of St. Petersburg State University.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Gensicka-Kowalewska, M.; Cholewiński, G.; Dzierzbicka, K. Recent developments in the synthesis and biological activity of acridine/acridone analogues. RSC Adv. 2017, 7, 15776–15804. [Google Scholar] [CrossRef]
- Ježek, J.; Hlaváček, J.; Šebestík, J. Biomedical Applications of Acridines; Progress in Drug Research; Rainsford, R.D., Ed.; Springer: Cham, Switzerland, 2017; Volume 72, pp. 1–243. [Google Scholar]
- Varakumar, P.; Rajagopal, K.; Aparna, B.; Raman, K.; Byran, G.; Gonçalves Lima, C.M.; Rashid, S.; Nafady, M.H.; Emran, T.B.; Wybraniec, S. Acridine as an Anti-Tumour Agent: A Critical Review. Molecules 2023, 28, 193. [Google Scholar] [CrossRef]
- Piorecka, K.; Kurjata, J.; Stanczyk, W.A. Acriflavine, an acridine derivative for biomedical application: Current state of the art. J. Med. Chem. 2022, 65, 11415–11432. [Google Scholar] [CrossRef] [PubMed]
- Prasher, P.; Sharma, M. Medicinal chemistry of acridine and its analogues. MedChemComm 2018, 9, 1589–1618. [Google Scholar] [CrossRef] [PubMed]
- Viegas, C.J.; Danuello, A.; Bolzani, V.S.; Barreiro, E.J.; Fraga, C.M. Molecular Hybridization: A Useful Tool in the Design of New Drug Prototypes. Curr. Med. Chem. 2007, 14, 1829–1852. [Google Scholar]
- Zhang, Q.; Yu, X. Current Scenario of Acridine Hybrids with Anticancer Potential. Curr. Top. Med. Chem. 2021, 21, 1773–1786. [Google Scholar] [CrossRef] [PubMed]
- Chufarova, N.; Czarnecka, K.; Skibiński, R.; Cuchra, M.; Majsterek, I.; Szymański, P. New tacrine–acridine hybrids as promising multifunctional drugs for potential treatment of Alzheimer’s disease. Arch. Pharm. 2018, 351, e1800050. [Google Scholar] [CrossRef]
- Maciejewska, K.; Czarnecka, K.; Kręcisz, P.; Niedziałek, D.; Wieczorek, G.; Skibiński, R.; Szymański, P. Novel Cyclopentaquinoline and Acridine Analogs as Multifunctional, Potent Drug Candidates in Alzheimer’s Disease. Int. J. Mol. Sci. 2022, 23, 5876. [Google Scholar] [CrossRef]
- Remya, R.S.; Ramalakshmi, N.; Nalini, C.N.; Amuthalakshmi, S. Design, synthesis, and in vitro evaluation of novel acridine derivatives as monoamine oxidase inhibitors. Rasayan J. Chem. 2022, 15, 2318–2325. [Google Scholar] [CrossRef]
- Fonte, M.; Tassi, N.; Gomes, P.; Teixeira, C. Acridine-Based Antimalarials—From the Very First Synthetic Antimalarial to Recent Developments. Molecules 2021, 26, 600. [Google Scholar] [CrossRef]
- Kumar, A.; Srivastava, K.; Kumar, S.R.; Puri, S.K.; Chauhan, P.M. Synthesis of new 4-aminoquinolines and quinoline–acridine hybrids as antimalarial agents. Bioorganic Med. Chem. Lett. 2010, 20, 7059–7063. [Google Scholar] [CrossRef]
- Pandey, S.K.; Biswas, S.; Gunjan, S.; Chauhan, B.S.; Singh, S.; Srivastava, K.; Singh, S.; Batra, S.; Tripathi, R. Pyrrolidine-Acridine hybrid in Artemisinin-based combination: A pharmacodynamic study. Parasitology 2016, 143, 1421–1432. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, A.A.; Awad, H.M.; El-Sayed, I.E.T.; El Gokha, A.A. Synthesis and antiproliferative activity of new hybrids bearing neocryptolepine, acridine and α-aminophosphonate scaffolds. J. Iran. Chem. Soc. 2020, 17, 1211–1221. [Google Scholar] [CrossRef]
- Lisboa, T.; Silva, D.; Duarte, S.; Ferreira, R.; Andrade, C.; Lopes, A.L.; Ribeiro, J.; Farias, D.; Moura, R.; Reis, M.; et al. Toxicity and antitumor activity of a thiophene–acridine hybrid. Molecules 2019, 25, 64. [Google Scholar] [CrossRef] [PubMed]
- Pandhurnekar, C.P.; Pandhurnekar, H.C.; Mungole, A.J.; Butoliya, S.S.; Yadao, B.G. A review of recent synthetic strategies and biological activities of isoxazole. J. Heterocycl. Chem. 2022, 60, 537–565. [Google Scholar] [CrossRef]
- Agrawal, N.; Mishra, P. The synthetic and therapeutic expedition of isoxazole and its Analogs. Med. Chem. Res. 2018, 27, 1309–1344. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Mo, J.; Lin, H.Z.; Chen, Y.; Sun, H.P. The Recent Progress of Isoxazole in Medicinal Chemistry. Bioorg. Med. Chem. 2018, 26, 3065–3075. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Hu, Q.; Tang, H.; Pan, X. Isoxazole/Isoxazoline Skeleton in the Structural Modification of Natural Products: A Review. Pharmaceuticals 2023, 16, 228. [Google Scholar] [CrossRef] [PubMed]
- Saeedi, M.; Mohtadi-Haghighi, D.; Mirfazli, S.S.; Mahdavi, M.; Hariri, R.; Lotfian, H.; Edraki, N.; Iraji, A.; Firuzi, O.; Akbarzadeh, T. Design and synthesis of selective acetylcholinesterase inhibitors: Arylisoxazole-phenylpiperazine derivatives. Chem. Biodivers. 2019, 16, e180043. [Google Scholar] [CrossRef]
- Rastegari, A.; Safavi, M.; Vafadarnejad, F.; Najafi, Z.; Hariri, R.; Bukhari, S.N.A.; Iraji, A.; Edraki, N.; Firuzi, O.; Saeedi, M.; et al. Synthesis and evaluation of novel arylisoxazoles linked to tacrine moiety: In vitro and in vivo biological activities against Alzheimer’s disease. Mol. Divers. 2022, 26, 409–428. [Google Scholar] [CrossRef]
- Wang, M.; Li, L.; Yang, S.; Guo, F.; Zhu, G.; Zhu, B.; Chang, J. Synthesis of novel oxazol-5-one derivatives containing chiral trifluoromethyl and isoxazole moieties as potent antitumor agents and the mechanism investigation. Bioorg. Chem. 2023, 135, 106505. [Google Scholar] [CrossRef] [PubMed]
- Lingala, A.K.; Murahari, K.K.; Desireddi, J.R.; Mothe, T.; Mait, B.; Manchal, R. Design, synthesis and biological evaluation of isoxazole bearing 1,3-oxazole-1,3,4-oxadiazole derivatives as anticancer agents. Chem. Data Collect. 2023, 43, 100959. [Google Scholar] [CrossRef]
- Hawash, M.; Kahraman, D.C.; Ergun, S.G.; Cetin-Atalay, R.; Baytas, S.N. Synthesis of novel indole-isoxazole hybrids and evaluation of their cytotoxic activities on hepatocellular carcinoma cell lines. BMC Chem. 2021, 15, 66. [Google Scholar] [CrossRef] [PubMed]
- Güiza, F.M.; Duarte, Y.B.; Mendez-Sanchez, S.C.; Bohórquez, A.R.R. Synthesis and in vitro evaluation of substituted tetrahydroquinoline-isoxazole hybrids as anticancer agents. Med. Chem. Res. 2019, 28, 1182–1196. [Google Scholar] [CrossRef]
- Xie, X.; Xiong, S.S.; Li, X.; Huang, H.; Wu, F.B.; Shen, P.F.; Peng, C.; He, G.; Han, B. Design and organocatalytic synthesis of spirooxindole–cyclopentene–isoxazole hybrids as novel MDM2–p53 inhibitors. Org. Chem. Front. 2021, 8, 1836–1843. [Google Scholar] [CrossRef]
- Kushwaha, P.K.; Srivastava, K.S.; Kumari, N.; Kumar, R.; Mitra, D.; Sharon, A. Synthesis and anti-HIV activity of a new isoxazole containing disubstituted 1,2,4-oxadiazoles analogs. Bioorg. Med. Chem. 2022, 56, 116612. [Google Scholar] [CrossRef] [PubMed]
- Kalirajan, R.; Rafick, M.H.; Sankar, S.; Gowramma, B. Green synthesis of some novel chalcone and isoxazole substituted 9-anilinoacridine derivatives and evaluation of their antimicrobial and larvicidal activities. Indian J. Chem. 2018, 57B, 583–590. [Google Scholar]
- Mosher, M.D.; Natale, N.R. The preparation of intercalating isoxazoles via a nitrile oxide cycloaddition. J. Heterocycl. Chem. 1995, 32, 779–781. [Google Scholar] [CrossRef]
- Ahmed, S.M.; Hussain, F.H.; Quadrelli, P. 9-Anthraldehyde oxime: A synthetic tool for variable applications. Monatshefte Für Chem.-Chem. Mon. 2020, 151, 1643–1658. [Google Scholar] [CrossRef]
- Galenko, E.E.; Khlebnikov, A.F.; Novikov, M.S. Isoxazole-azirine isomerization as a reactivity switch in the synthesis of heterocycles. Chem. Heterocycl. Compd. 2016, 52, 637–650. [Google Scholar] [CrossRef]
- Lim, R.K.; Lin, Q. Azirine ligation: Fast and selective protein conjugation via photoinduced azirine–alkene cycloaddition. Chem. Commun. 2010, 46, 7993–7995. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; He, Z.; Ma, W.; Bao, G.; Li, Y.; Yu, C.; Li, J.; E, R.; Xu, Z.; Wang, R.; et al. Copper (I)-Catalyzed Late-Stage Introduction of Oxime Ethers into Peptides at the Carboxylic Acid Site. Org. Lett. 2022, 24, 9248–9253. [Google Scholar] [CrossRef]
- Sakharov, P.A.; Novikov, M.S.; Rostovskii, N.V. 2H-Azirines in medicinal chemistry. Chem. Heterocycl. Compd. 2021, 57, 512–521. [Google Scholar] [CrossRef]
- Mueller, J.O.; Schmidt, F.G.; Blinco, J.P.; Barner-Kowollik, C. Visible-Light-Induced Click Chemistry. Angew. Chem. Int. Ed. 2015, 54, 10284–10288. [Google Scholar] [CrossRef] [PubMed]
- Kumar, G.S.; Lin, Q. Light-Triggered Click Chemistry. Chem. Rev. 2021, 121, 6991–7031. [Google Scholar] [CrossRef] [PubMed]
- Fairbanks, B.D.; Macdougall, L.J.; Mavila, S.; Sinha, J.; Kirkpatrick, B.E.; Anseth, K.S.; Bowman, C.N. Photoclick Chemistry: A Bright Idea. Chem. Rev. 2021, 121, 6915–6990. [Google Scholar] [CrossRef] [PubMed]
- Luque, A.; Paternoga, J.; Opatz, T. Strain Release Chemistry of Photogenerated Small-Ring Intermediates. Chem. Eur. J. 2021, 27, 4500–4516. [Google Scholar] [CrossRef]
- 9-Phenyl Acridine. Available online: https://www.hampfordresearch.com/wp-content/uploads/2022/11/FP_5400_9_PA.pdf (accessed on 11 July 2014).
- Hansda, S.; Ghosh, G.; Ghosh, R. 9-phenyl acridine photosensitizes A375 cells to UVA radiation. Helion 2020, 6, e04733. [Google Scholar] [CrossRef]
- Padwa, A. Cycloaddition and cyclization chemistry of 2H-azirines. In Advances in Heterocyclic Chemistry; Academic Press: Cambridge, MA, USA, 2010; Volume 99, pp. 1–31. [Google Scholar]
- Khlebnikov, A.F.; Novikov, M.S. Ring Expansions of Azirines and Azetines. Top. Heterocycl. Chem. 2015, 41, 143–232. [Google Scholar]
- Krivolapova, Y.V.; Tomashenko, O.A.; Funt, L.D.; Spiridonova, D.V.; Novikov, M.S.; Khlebnikov, A.F. Azirine-triazole hybrids: Selective synthesis of 5-(2H-azirin-2-yl)-, 5-(1H-pyrrol-2-yl)-1H-1,2,3-triazoles and 2-(5-(2H-azirin-2-yl)-1H-1,2,3-triazol-1-yl)pyridines. Org. Biomol. Chem. 2022, 20, 5434–5443. [Google Scholar] [CrossRef]
- Periasamy, N. Nanosecond laser flash photolysis of acridine in organic solvents. Proc. Indian Acad. Sci. (Chem. Sci.) 1984, 93, 1361–1375. [Google Scholar] [CrossRef]
- Braun, D.; Sfudenroth, R. Synthese von 9-Phenyl-9-benzylacridan. Liebigs Ann. Chem. 1981, 1981, 999–1002. [Google Scholar] [CrossRef]
- Castellano, A.; Catteau, J.-P.; Lablache-Combier, A.; Allan, G. Photochemical Studies. XVI. Second Moment Analysis of the Electron Spin Resonance Spectra of Radicals Formed by Ultraviolet Irradiation of 9-Phenylacridine and of Acridine. Can. J. Chem. 1973, 51, 3508–3513. [Google Scholar] [CrossRef]
- Niizuma, S.; Koizumi, M. Radicals Produced by UV Irradiation of Acridane and Acridine in Glassy and Crystalline Medium at 77°K. Bull. Chem. Soc. Jpn. 1968, 41, 1090–1096. [Google Scholar] [CrossRef]
- Agafonova, A.V.; Sakharov, P.A.; Smetanin, I.A.; Rostovskii, N.V.; Khlebnikov, A.F.; Novikov, M.S. Stannyl radical-mediated synthesis of 6H-1,3-oxazin-6-ones from 2-acyloxyazirines or whether free radicals can open the azirine ring? Org. Chem. Front. 2022, 9, 4118–4127. [Google Scholar] [CrossRef]
- Ye, Y.; Holder, G.M.; Duke, C.C. The preparation of 2H and 3H-labelled 7, 9-, and 7, 10-dimethylbenz[c]acridine by catalytic dehalogenation. J. Label. Compd. Radiopharm. 1993, 33, 1–10. [Google Scholar] [CrossRef]
- Rosewear, J.; Wilshire, J.F. The preparation of some 2-nitroacridines and related compounds. Aust. J. Chem. 1981, 34, 839–853. [Google Scholar]
- Tsuge, O.; Torii, A. Compounds Related to Acridine. X. The Reaction of 9-Ethynylacridine with Active Methylene Compounds. Bull. Chem. Soc. Jpn. 1973, 46, 283–285. [Google Scholar] [CrossRef]
- Sakharov, P.A.; Novikov, M.S.; Khlebnikov, A.F. 2-Diazoacetyl-2H-azirines: Source of a Variety of 2H-Azirine Building Blocks with Orthogonal and Domino Reactivity. J. Org. Chem. 2018, 83, 8304–8314. [Google Scholar] [CrossRef]
- Singer, B.; Maas, G. Dikationether, 3 [1]. Substitutionen an Bis (acridinium) ethern und 9-Trifloxyacridinium-Salzen mit Halogeniden, Pseudohalogeniden und Schwefel-Nucleophilen. Z. Für Naturforschung B 1984, 39, 1399–1408. [Google Scholar] [CrossRef]
- Suzuki, H.; Tanaka, Y. An unusually acidic methyl group directly bound to acridinium cation. J. Org. Chem. 2001, 66, 2227–2231. [Google Scholar] [CrossRef] [PubMed]
- Eberhard, J.; Peuntinger, K.; Fröhlich, R.; Guldi, D.M.; Mattay, J. Synthesis and properties of acridine and acridinium dye functionalized bis (terpyridine) ruthenium (II) complexes. Eur. J. Org. Chem. 2018, 2018, 2682–2700. [Google Scholar] [CrossRef]
- Rostovskii, N.V.; Ruvinskaya, J.O.; Novikov, M.S.; Khlebnikov, A.F.; Smetanin, I.A.; Agafonova, A.V. Switchable Synthesis of Pyrroles and Pyrazines via Rh(II)-Catalyzed Reaction of 1,2,3-Triazoles with Isoxazoles: Experimental and DFT Evidence for the 1,4-Diazahexatriene Intermediate. J. Org. Chem. 2017, 82, 256–268. [Google Scholar] [CrossRef] [PubMed]
- Baś, S.; Yamashita, Y.; Kobayashi, S. Development of Brønsted Base–Photocatalyst Hybrid Systems for Highly Efficient C–C Bond Formation Reactions of Malonates with Styrenes. ACS Catal. 2020, 10, 10546–10550. [Google Scholar] [CrossRef]
- Molloy, J.J.; Seath, C.P.; West, M.J.; McLaughlin, C.; Fazakerley, N.J.; Kennedy, A.R.; Nelson, D.J.; Watson, A.J. Interrogating Pd (II) anion metathesis using a bifunctional chemical probe: A transmetalation switch. J. Am. Chem. Soc. 2018, 140, 126–130. [Google Scholar] [CrossRef] [PubMed]
- Mosiagin, I.P.; Tomashenko, O.A.; Spiridonova, D.V.; Novikov, M.S.; Tunik, S.P.; Khlebnikov, A.F. Free-radical cyclization approach to polyheterocycles containing pyrrole and pyridine rings. Beilstein J. Org. Chem. 2021, 17, 1490–1498. [Google Scholar] [CrossRef] [PubMed]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Cryst. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Cryst. 2015, A71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. Crystal Structure Refinement with SHELXL. Acta Cryst. 2015, C71, 3. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision A.03; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A Consistent and Accurate ab Initio Parametrization of Density Functional Dispersion Correction (DFT-D) for the 94 Elements H-Pu. J. Chem. Phys. 2010, 132, 1054104. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions. J. Phys. Chem. B 2009, 11, 6378–6396. [Google Scholar] [CrossRef]
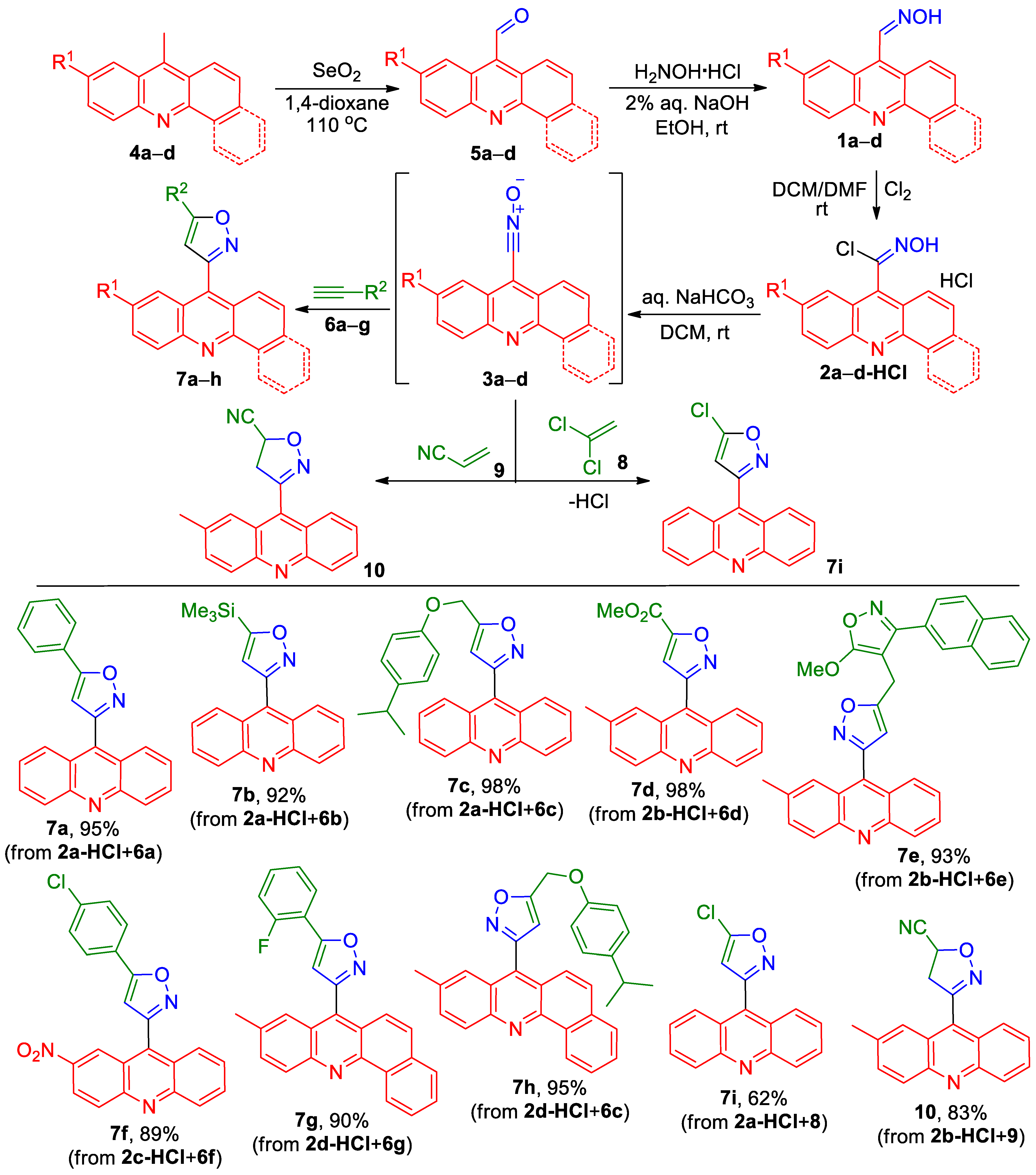
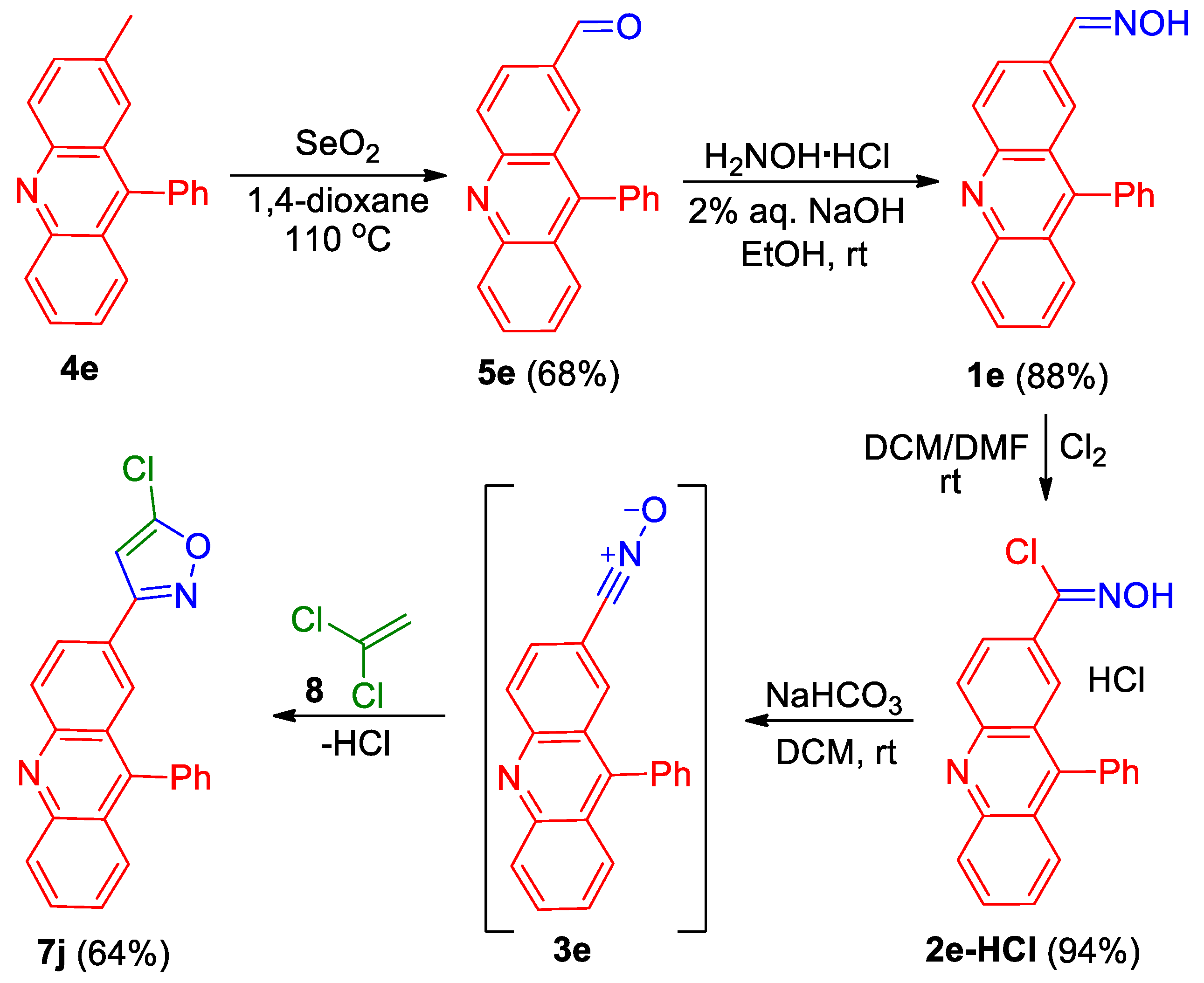
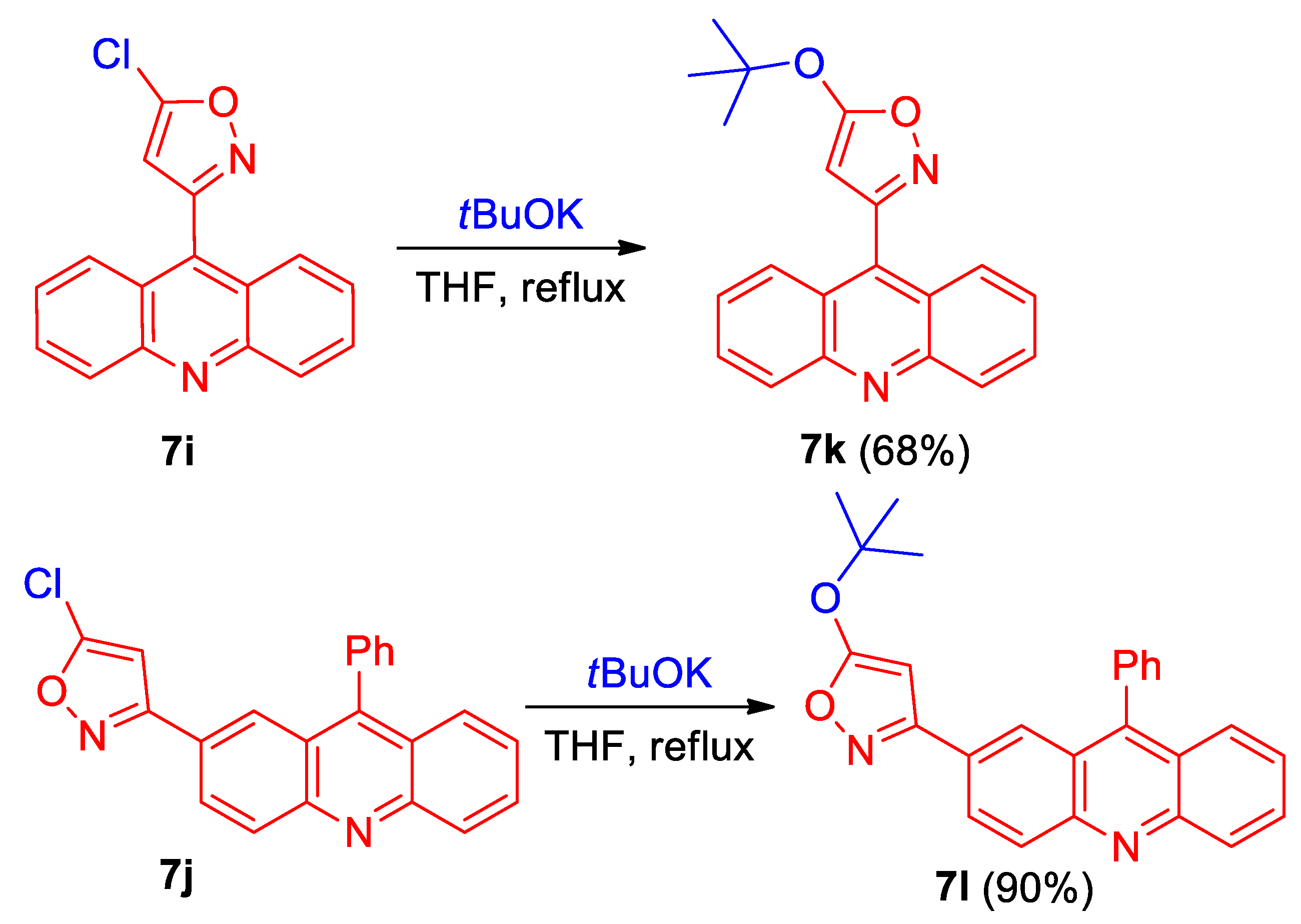
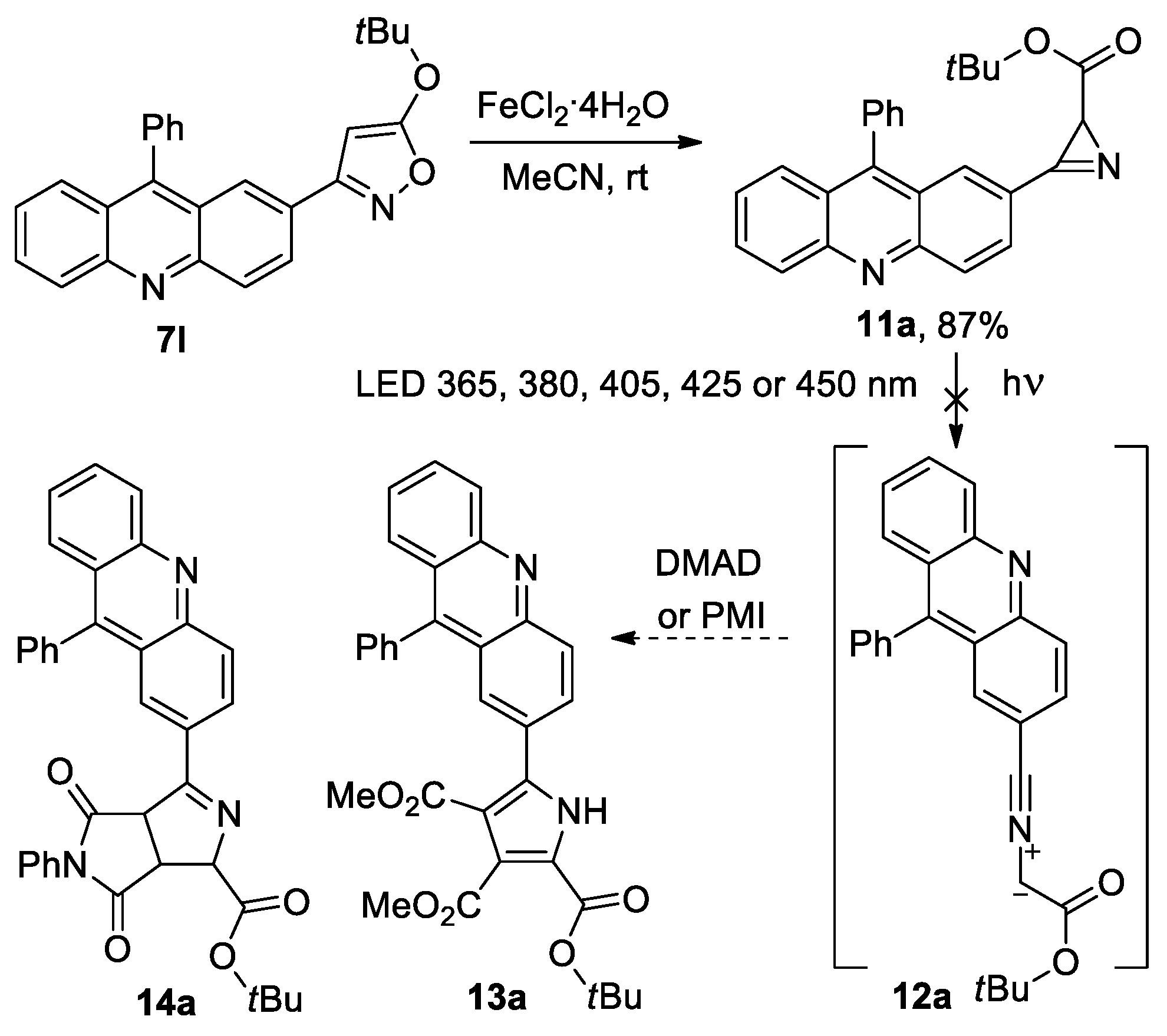
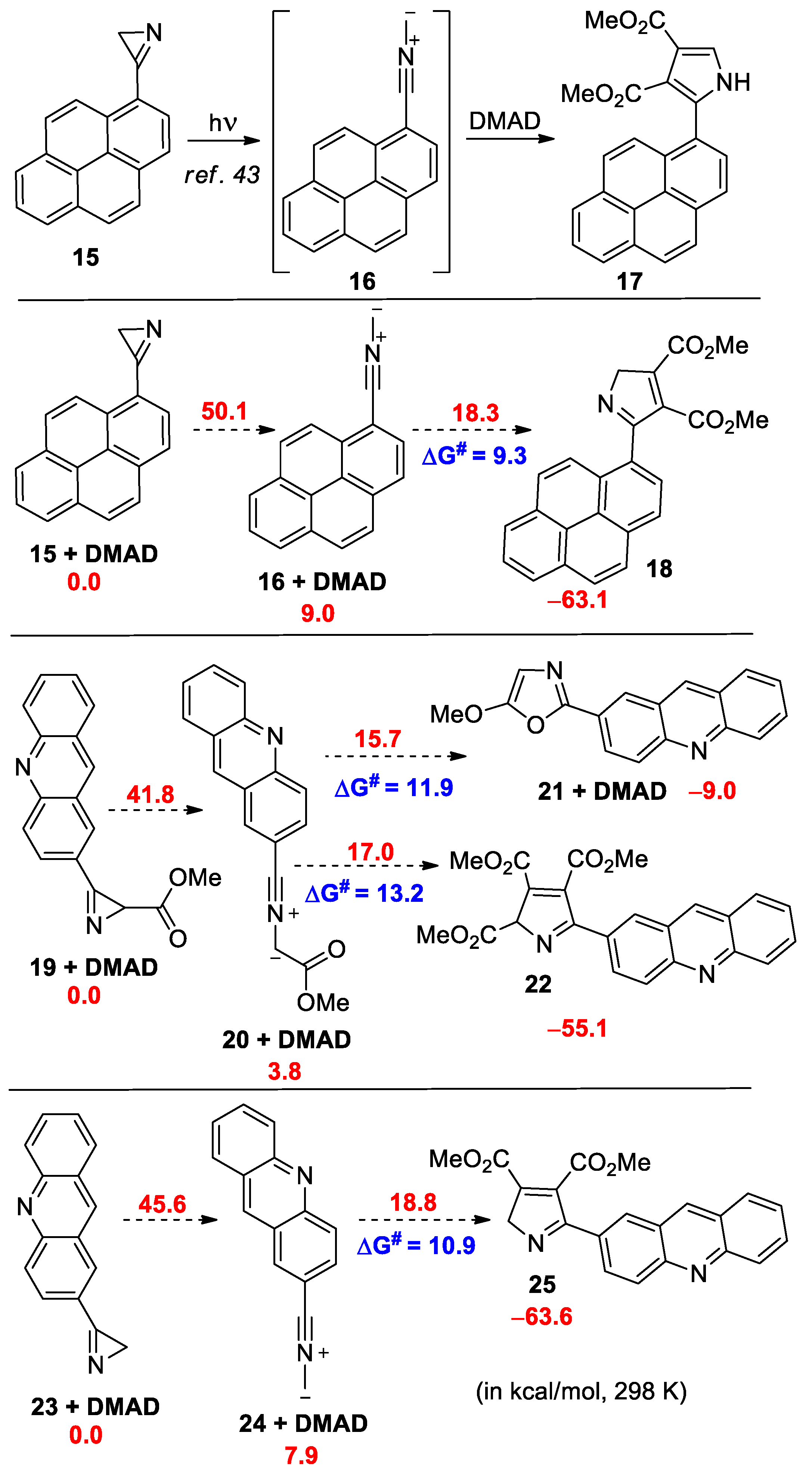
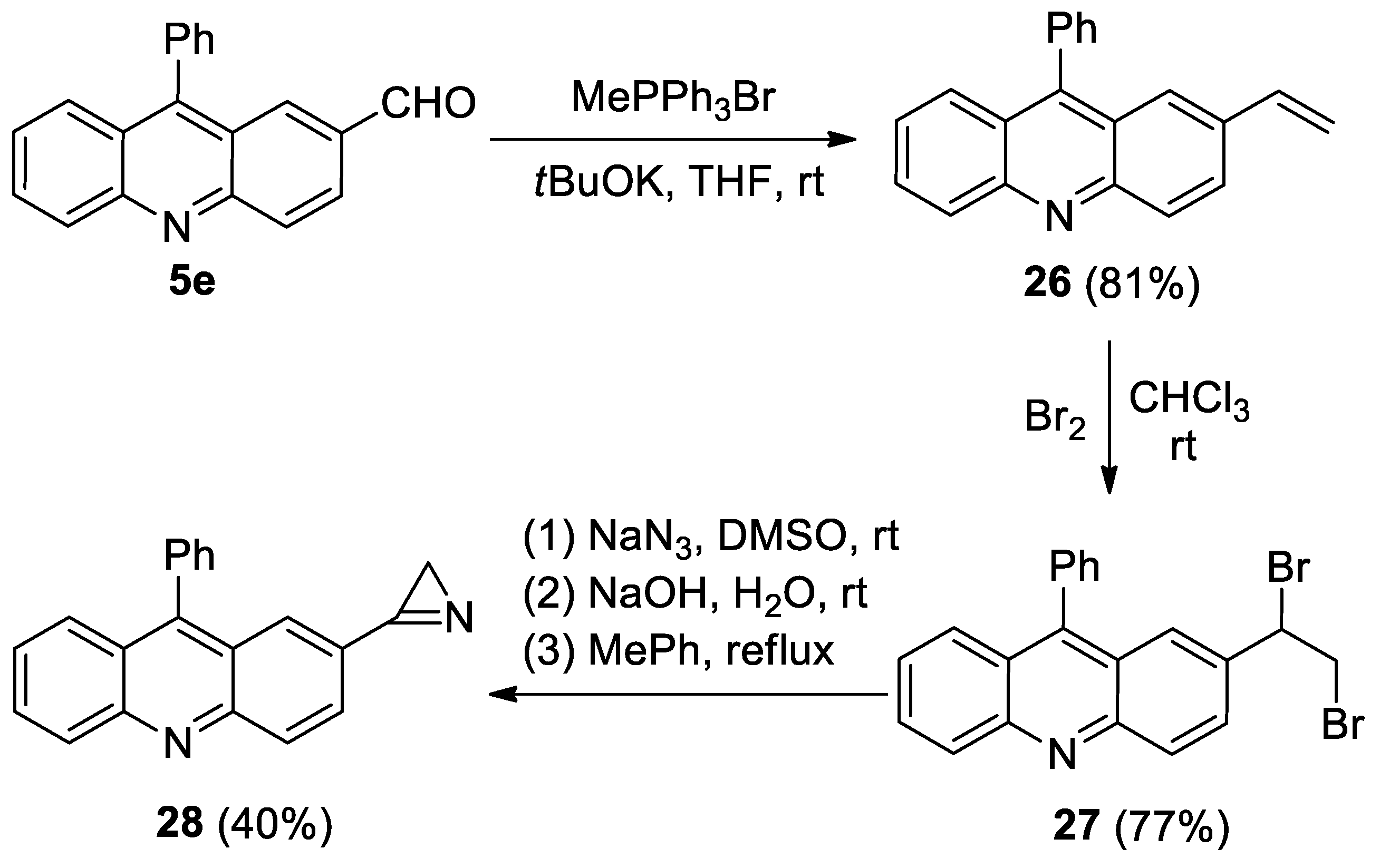
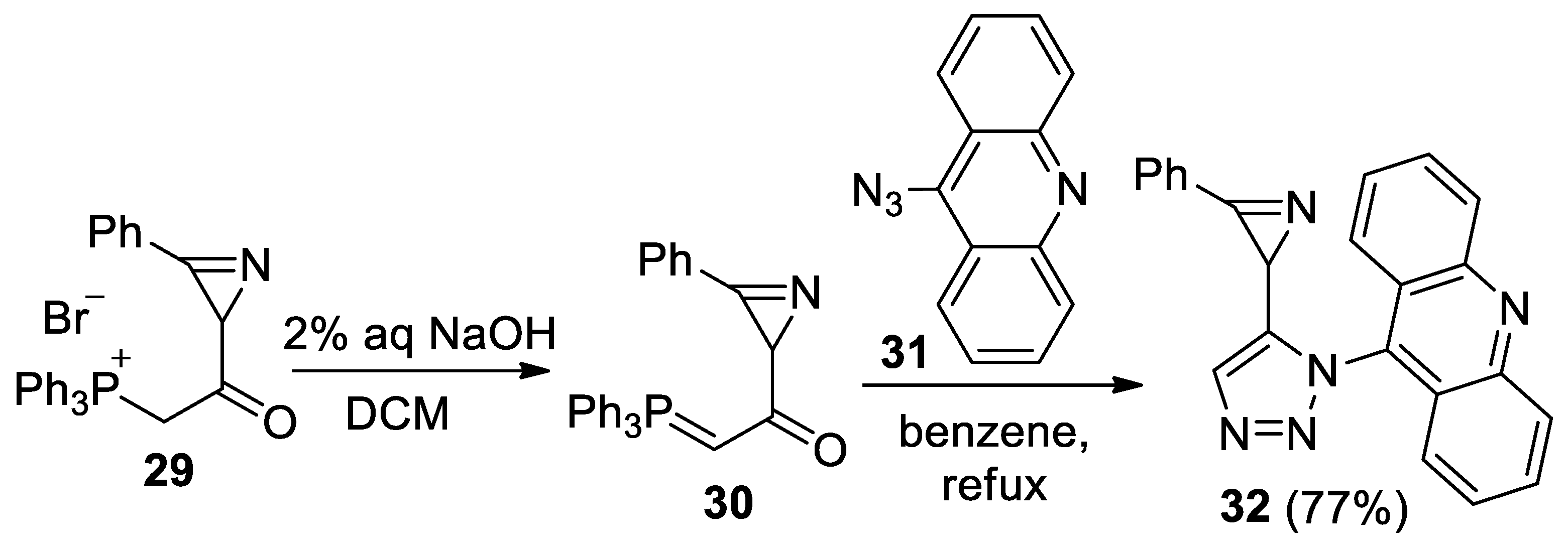
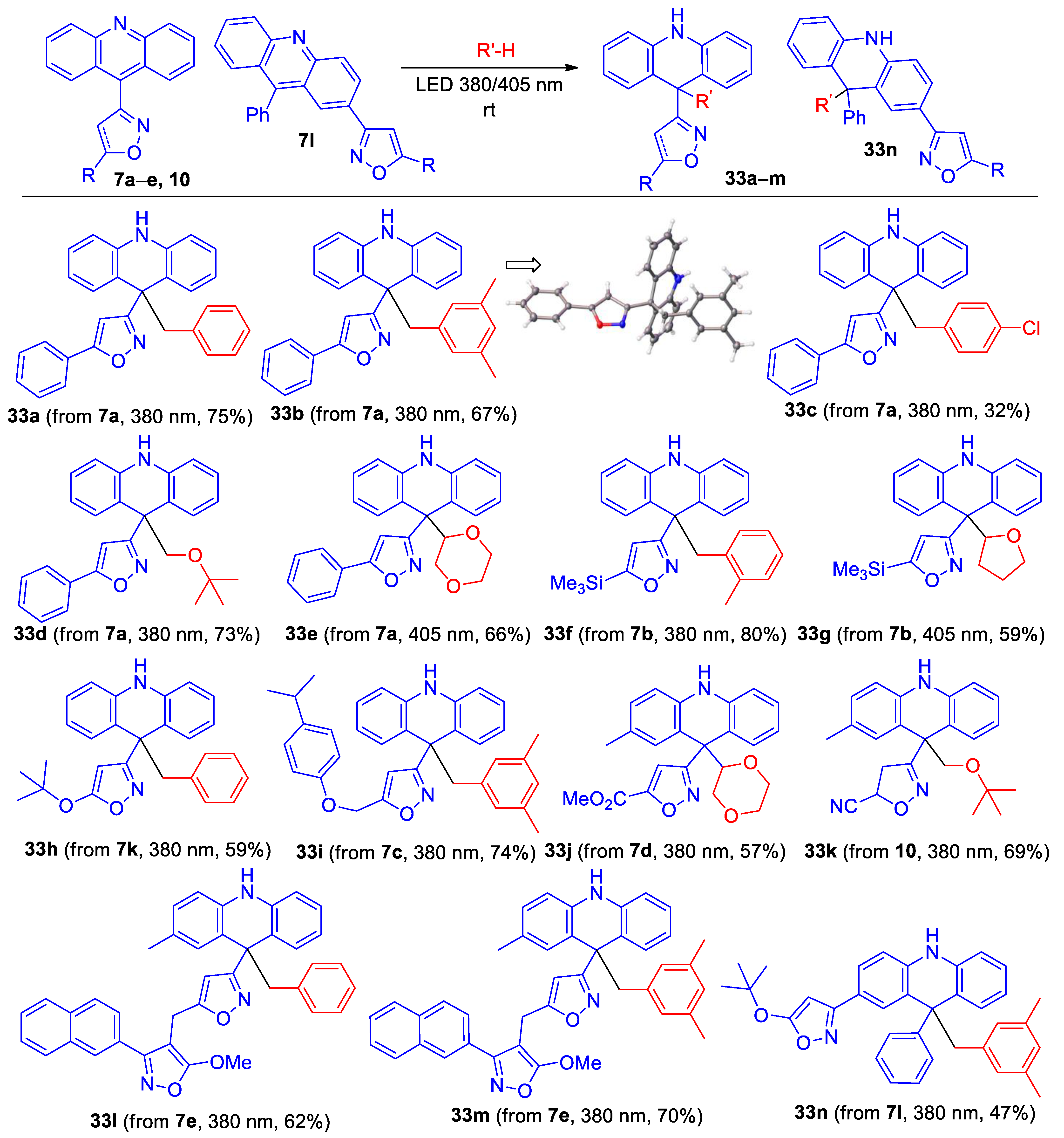
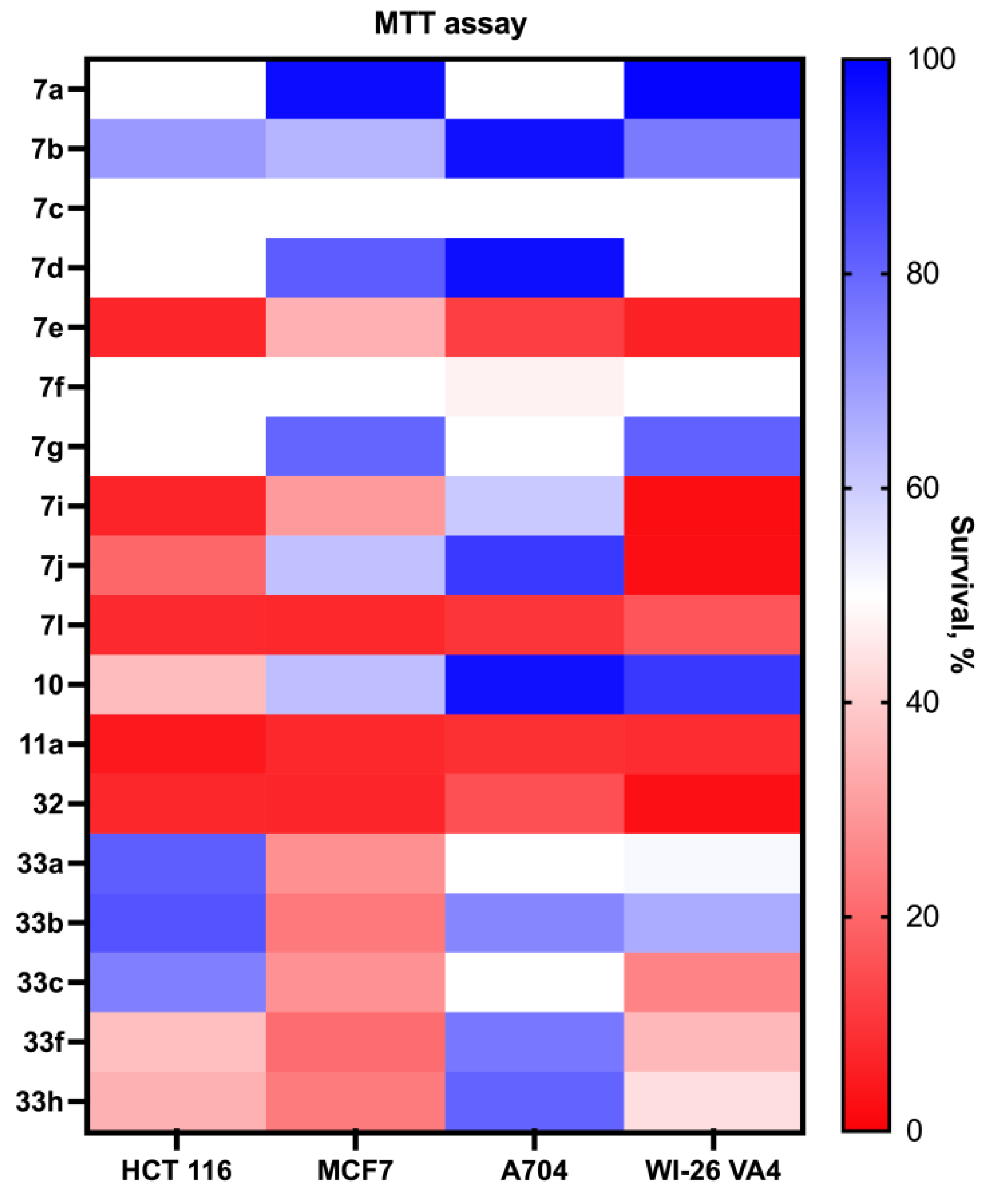
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).