
An Approach for Engineering Peptides for Competitive Inhibition of the SARS-COV-2 Spike Protein

 , ,
, ,  , , , , and
, , , , and
Abstract
1. Introduction
2. Results and Discussion
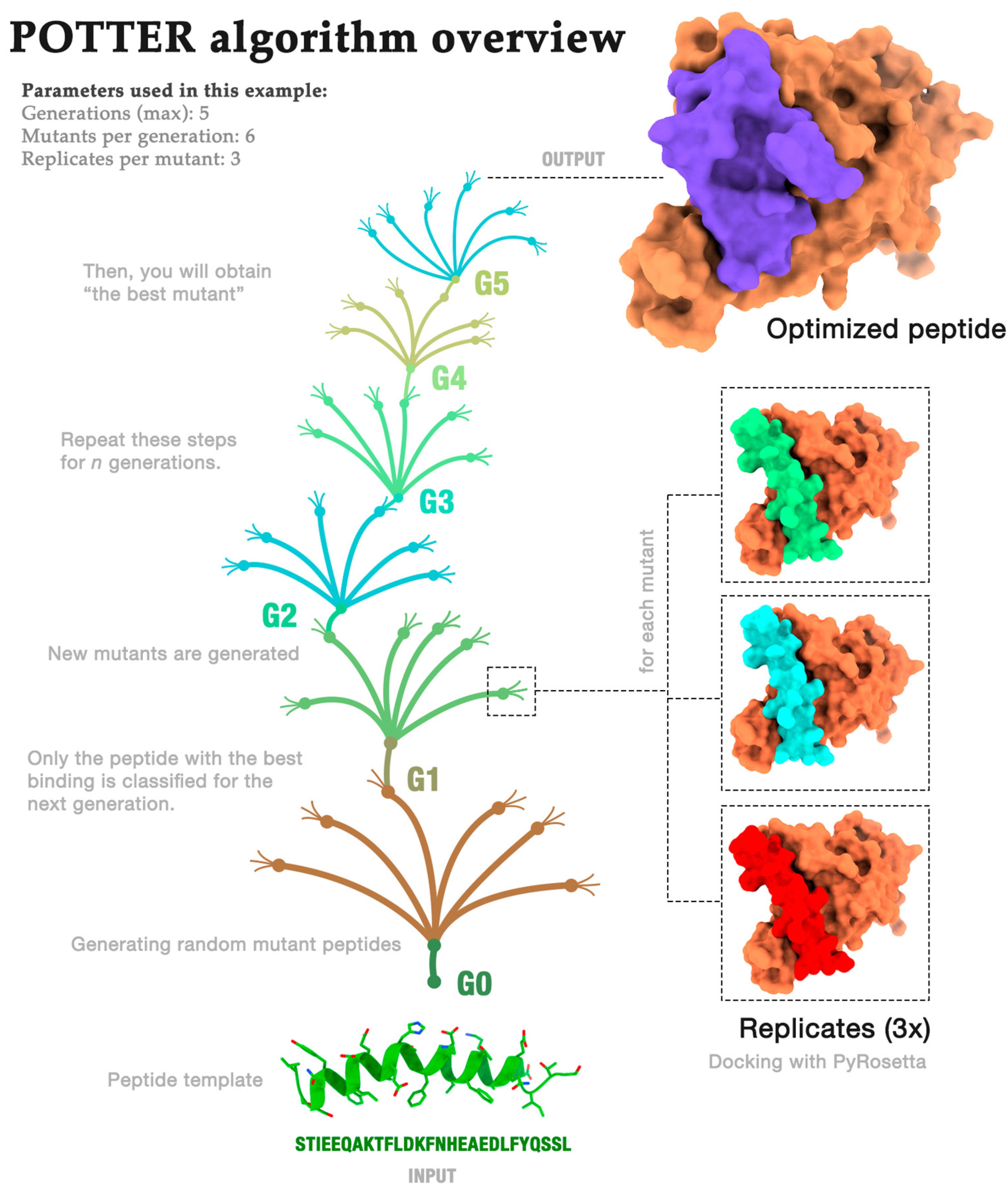
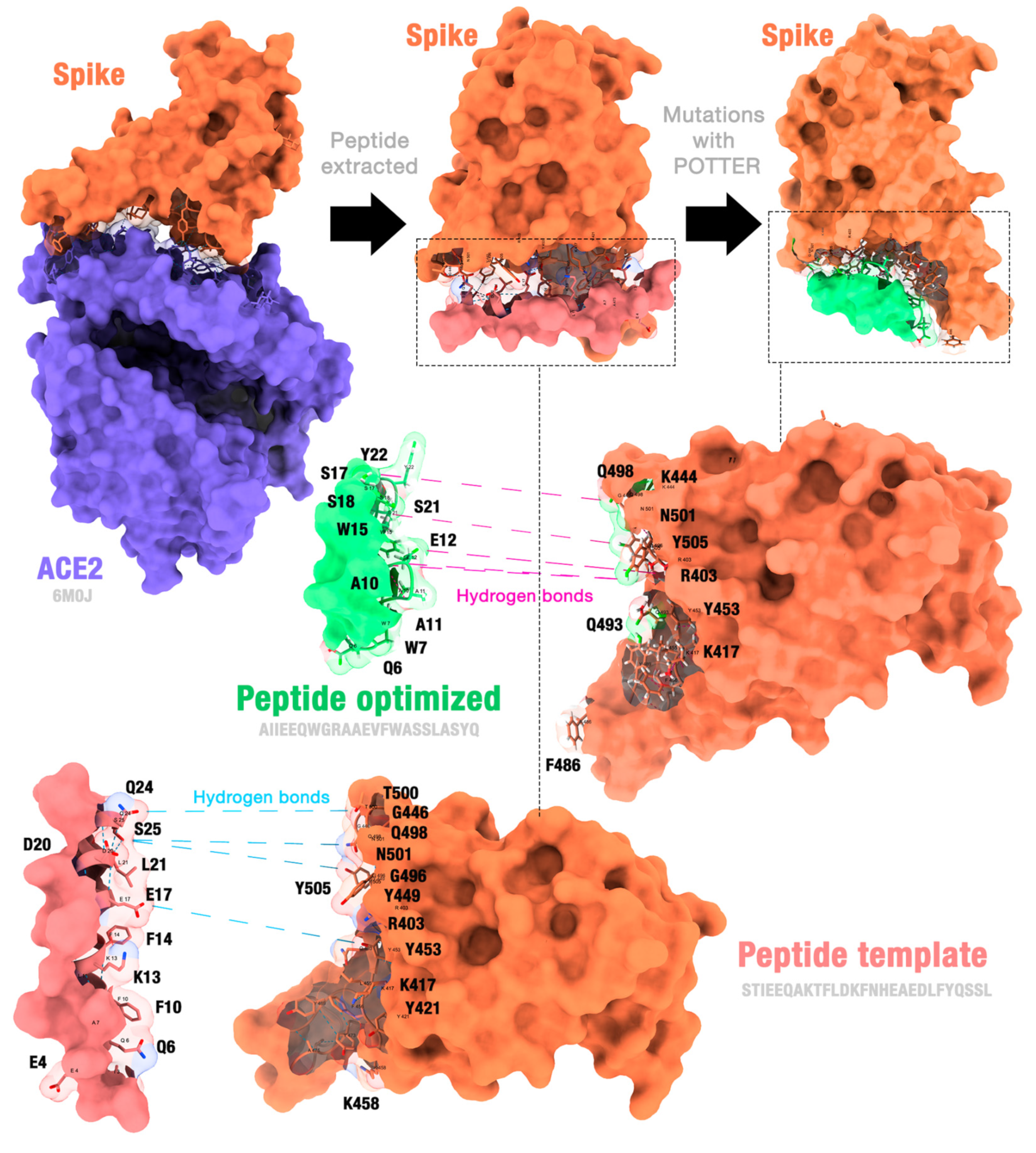
2.1. Algorithm Overview
- Generations (G): how often the process is repeated;
- Mutants (M): the number of mutants randomly generated in each generation;
- Repeats (R): the number of repeated dockings for each mutant.
Algorithm Demonstration
2.2. Case Studies
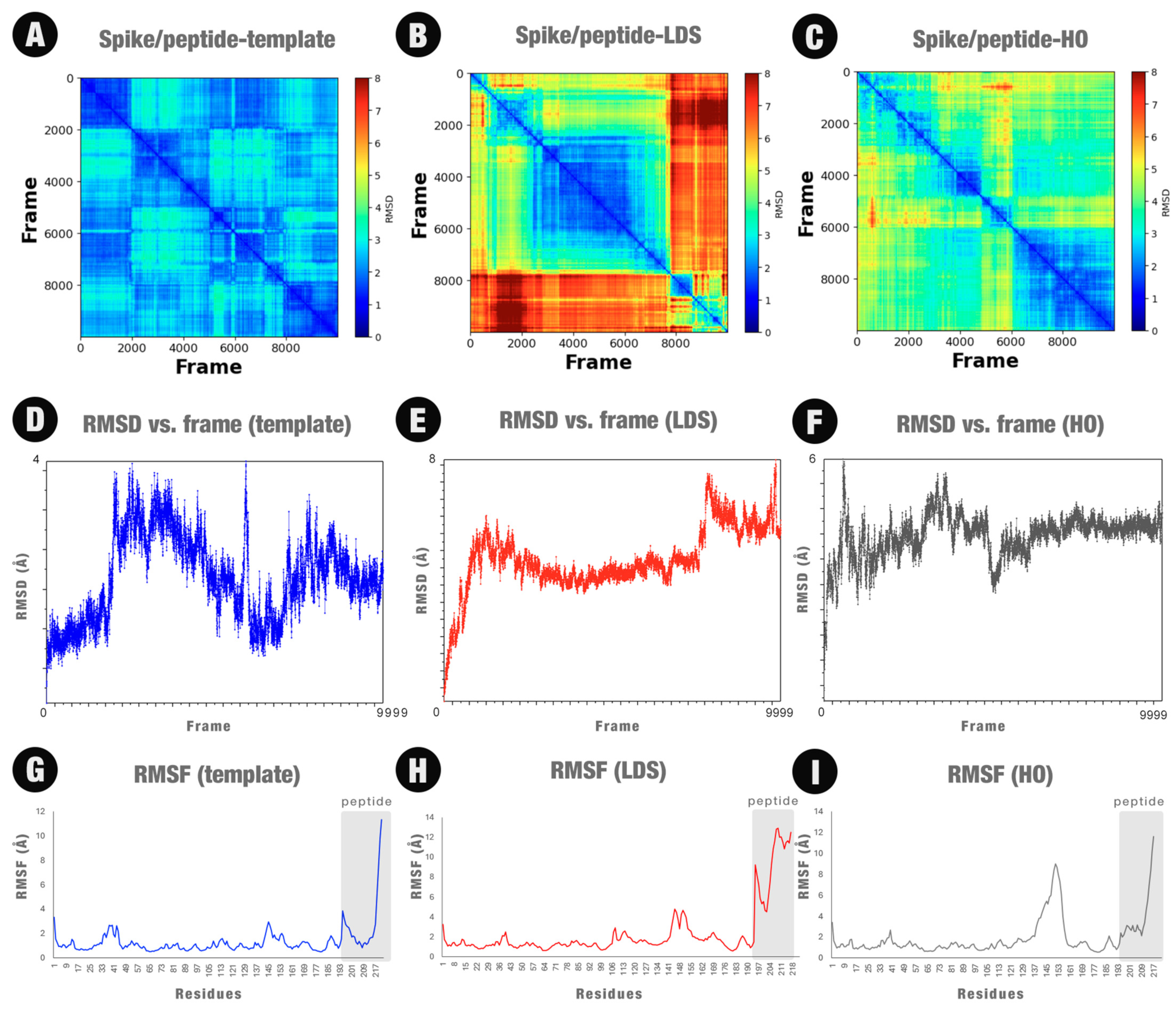
2.3. Molecular Dynamics Simulations
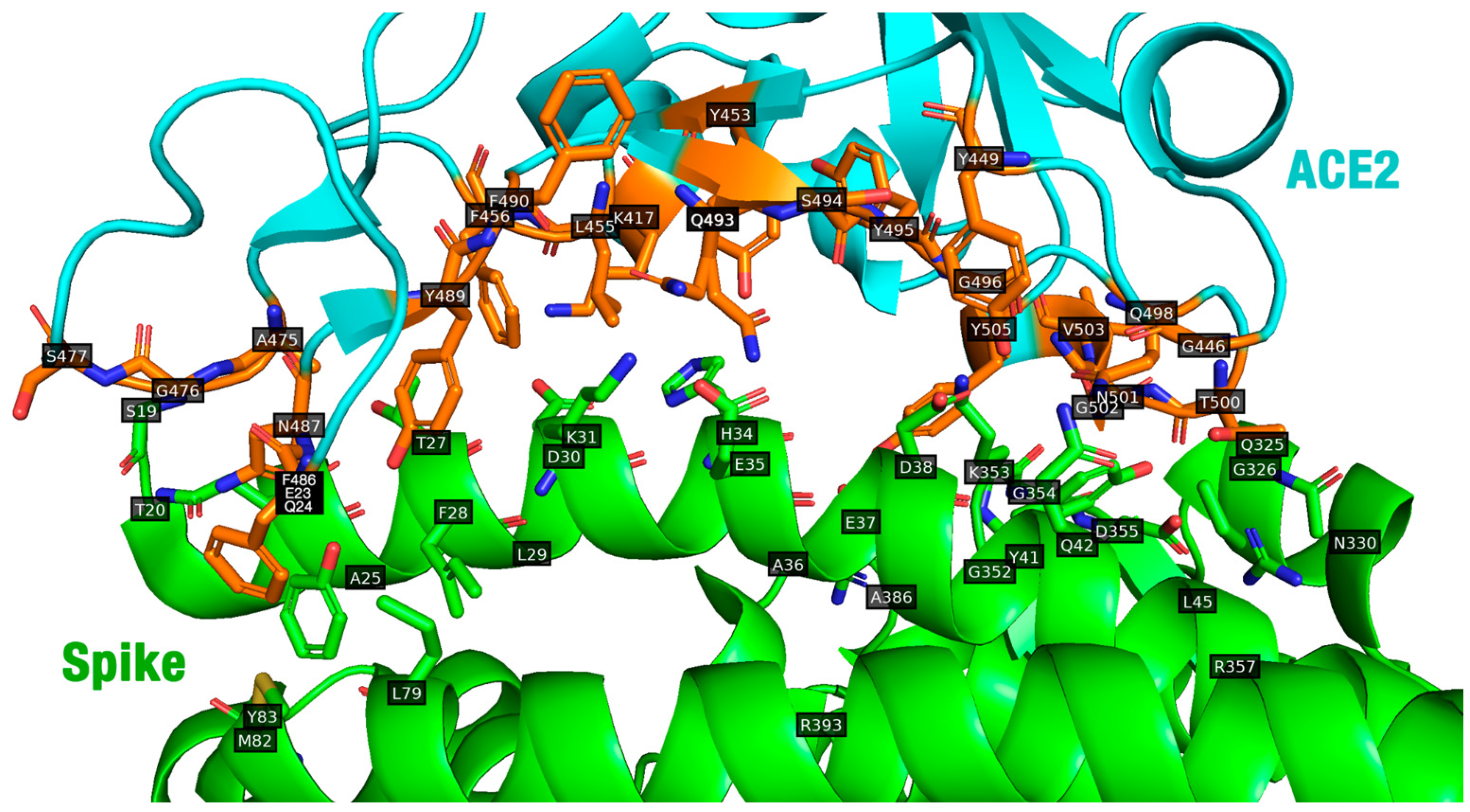
2.4. The Importance of Protein–Peptide Complexes
2.5. Comparison with Other Peptide Design Proposals Described in the Literature
2.6. Importance of POTTER Parameters
2.7. Insights Obtained by the MD Simulations
3. Materials and Methods
3.1. Data Collection
3.2. Algorithm Implementation
3.3. Molecular Modeling and Docking
3.4. Contacts
3.5. Case Studies
3.6. Molecular Dynamics
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- World Health Organization. Advice for the Public on COVID-19. Available online: https://www.who.int/emergencies/diseases/novel-coronavirus-2019/advice-for-public (accessed on 21 November 2023).
- Lan, J.; Ge, J.; Yu, J.; Shan, S.; Zhou, H.; Fan, S.; Zhang, Q.; Shi, X.; Wang, Q.; Zhang, L.; et al. Structure of the SARS-CoV-2 Spike Receptor-Binding Domain Bound to the ACE2 Receptor. Nature 2020, 581, 215–220. [Google Scholar] [CrossRef] [PubMed]
- Amoretti, M.; Amsler, C.; Bonomi, G.; Bouchta, A.; Bowe, P.; Carraro, C.; Cesar, C.L.; Charlton, M.; Collier, M.J.T.; Doser, M.; et al. Production and Detection of Cold Antihydrogen Atoms. Nature 2002, 419, 456–459. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, P.P.D.; Alves, N.A. Featuring ACE2 Binding SARS-CoV and SARS-CoV-2 through a Conserved Evolutionary Pattern of Amino Acid Residues. J. Biomol. Struct. Dyn. 2022, 40, 11719–11728. [Google Scholar] [CrossRef] [PubMed]
- Jackson, C.B.; Farzan, M.; Chen, B.; Choe, H. Mechanisms of SARS-CoV-2 Entry into Cells. Nat. Rev. Mol. Cell Biol. 2022, 23, 3–20. [Google Scholar] [CrossRef]
- Troyano-Hernáez, P.; Reinosa, R.; Holguín, Á. Evolution of SARS-CoV-2 Envelope, Membrane, Nucleocapsid, and Spike Structural Proteins from the Beginning of the Pandemic to September 2020: A Global and Regional Approach by Epidemiological Week. Viruses 2021, 13, 243. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.; Al Khodor, S. Pathophysiology and Treatment Strategies for COVID-19. J. Transl. Med. 2020, 18, 353. [Google Scholar] [CrossRef] [PubMed]
- Xia, S.; Zhu, Y.; Liu, M.; Lan, Q.; Xu, W.; Wu, Y.; Ying, T.; Liu, S.; Shi, Z.; Jiang, S.; et al. Fusion Mechanism of 2019-nCoV and Fusion Inhibitors Targeting HR1 Domain in Spike Protein. Cell. Mol. Immunol. 2020, 17, 765–767. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Yu, D.; Yan, H.; Chong, H.; He, Y. Design of Potent Membrane Fusion Inhibitors against SARS-CoV-2, an Emerging Coronavirus with High Fusogenic Activity. J. Virol. 2020, 94, e00635-20. [Google Scholar] [CrossRef] [PubMed]
- Ling, R.; Dai, Y.; Huang, B.; Huang, W.; Yu, J.; Lu, X.; Jiang, Y. In Silico Design of Antiviral Peptides Targeting the Spike Protein of SARS-CoV-2. Peptides 2020, 130, 170328. [Google Scholar] [CrossRef] [PubMed]
- Yan, R.; Zhang, Y.; Li, Y.; Xia, L.; Guo, Y.; Zhou, Q. Structural Basis for the Recognition of SARS-CoV-2 by Full-Length Human ACE2. Science 2020, 367, 1444–1448. [Google Scholar] [CrossRef] [PubMed]
- Tai, W.; He, L.; Zhang, X.; Pu, J.; Voronin, D.; Jiang, S.; Zhou, Y.; Du, L. Characterization of the Receptor-Binding Domain (RBD) of 2019 Novel Coronavirus: Implication for Development of RBD Protein as a Viral Attachment Inhibitor and Vaccine. Cell. Mol. Immunol. 2020, 17, 613–620. [Google Scholar] [CrossRef] [PubMed]
- Ho, C.; Lee, P.-C. COVID-19 Treatment—Current Status, Advances, and Gap. Pathogens 2022, 11, 1201. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Liu, Q.; Guo, D. Emerging Coronaviruses: Genome Structure, Replication, and Pathogenesis. J. Med. Virol. 2020, 92, 418–423. [Google Scholar] [CrossRef] [PubMed]
- Martins, P.M.; Santos, L.H.; Mariano, D.; Queiroz, F.C.; Bastos, L.L.; Gomes, I.d.S.; Fischer, P.H.C.; Rocha, R.E.O.; Silveira, S.A.; de Lima, L.H.F.; et al. Propedia: A Database for Protein–Peptide Identification Based on a Hybrid Clustering Algorithm. BMC Bioinform. 2021, 22, 1. [Google Scholar] [CrossRef] [PubMed]
- Martins, P.; Mariano, D.; Carvalho, F.C.; Bastos, L.L.; Moraes, L.; Paixão, V.; Cardoso de Melo-Minardi, R. Propedia v2.3: A Novel Representation Approach for the Peptide-Protein Interaction Database Using Graph-Based Structural Signatures. Front. Bioinform. 2023, 3, 1103103. [Google Scholar] [CrossRef] [PubMed]
- Lau, J.L.; Dunn, M.K. Therapeutic Peptides: Historical Perspectives, Current Development Trends, and Future Directions. Bioorg. Med. Chem. 2018, 26, 2700–2707. [Google Scholar] [CrossRef] [PubMed]
- Hamley, I.W. Peptides for Vaccine Development. ACS Appl. Bio Mater. 2022, 5, 905–944. [Google Scholar] [CrossRef] [PubMed]
- Wolfe, M.; Webb, S.; Chushak, Y.; Krabacher, R.; Liu, Y.; Swami, N.; Harbaugh, S.; Chávez, J. A high-throughput pipeline for design and selection of peptides targeting the SARS-Cov-2 Spike protein. Sci. Rep. 2021, 11, 21768. [Google Scholar] [CrossRef] [PubMed]
- Chaudhury, S.; Lyskov, S.; Gray, J.J. PyRosetta: A Script-Based Interface for Implementing Molecular Modeling Algorithms Using Rosetta. Bioinforma. Oxf. Engl. 2010, 26, 689–691. [Google Scholar] [CrossRef] [PubMed]
- French, S.; Robson, B. What Is a Conservative Substitution? J. Mol. Evol. 1983, 19, 171–175. [Google Scholar] [CrossRef]
- Robson, B. Computers and Viral Diseases. Preliminary Bioinformatics Studies on the Design of a Synthetic Vaccine and a Preventative Peptidomimetic Antagonist against the SARS-CoV-2 (2019-nCoV, COVID-19) Coronavirus. Comput. Biol. Med. 2020, 119, 103670. [Google Scholar] [CrossRef] [PubMed]
- Weng, G.; Gao, J.; Wang, Z.; Wang, E.; Hu, X.; Yao, X.; Cao, D.; Hou, T. Comprehensive Evaluation of Fourteen Docking Programs on Protein–Peptide Complexes. J. Chem. Theory Comput. 2020, 16, 3959–3969. [Google Scholar] [CrossRef]
- Neduva, V.; Linding, R.; Su-Angrand, I.; Stark, A.; Masi, F.d.; Gibson, T.J.; Lewis, J.; Serrano, L.; Russell, R.B. Systematic Discovery of New Recognition Peptides Mediating Protein Interaction Networks. PLoS Biol. 2005, 3, e405. [Google Scholar] [CrossRef] [PubMed]
- Glaser, F.; Steinberg, D.M.; Vakser, I.A.; Ben-Tal, N. Residue Frequencies and Pairing Preferences at Protein–Protein Interfaces. Proteins Struct. Funct. Bioinforma. 2001, 43, 89–102. [Google Scholar] [CrossRef]
- Valiente, P.A.; Wen, H.; Nim, S.; Lee, J.A.; Kim, H.J.; Kim, J.; Perez-Riba, A.; Paudel, Y.P.; Hwang, I.; Kim, K.-D.; et al. Computational Design of Potent D-Peptide Inhibitors of SARS-CoV-2. J. Med. Chem. 2021, 64, 14955–14967. [Google Scholar] [CrossRef] [PubMed]
- Sitthiyotha, T.; Chunsrivirot, S. Computational design of SARS-CoV-2 peptide binders with better predicted binding affinities than the human ACE2 receptor. Sci. Rep. 2021, 11, 15650. [Google Scholar] [CrossRef]
- Sun, J.; Liu, X.; Zhang, S.; Li, M.; Zhang, Q.; Chen, J. Molecular Insights and Optimization Strategies for the Competitive Binding of Engineered ACE2 Proteins: A Multiple Replica Molecular Dynamics Study. Phys. Chem. Chem. Phys. 2023, 41, 28479–28496. [Google Scholar] [CrossRef] [PubMed]
- Liang, S.; Liu, X.; Zhang, S.; Li, M.; Zhang, Q.; Chen, J. Binding mechanism of inhibitors to SARS-CoV-2 main protease deciphered by multiple replica molecular dynamics simulations. Phys. Chem. Chem. Phys. 2022, 24, 1743–1759. [Google Scholar] [CrossRef]
- Robson, B. Preliminary Bioinformatics Studies on the Design of Synthetic Vaccines and Preventative Peptidomimetic Antagonists against the Wuhan Seafood Market Coronavirus. Possible Importance of the KRSFIEDLLFNKV Motif. 2020; preprint. [Google Scholar] [CrossRef]
- Robson, B. COVID-19 Coronavirus Spike Protein Analysis for Synthetic Vaccines, a Peptidomimetic Antagonist, and Therapeutic Drugs, and Analysis of a Proposed Achilles’ Heel Conserved Region to Minimize Probability of Escape Mutations and Drug Resistance. Comput. Biol. Med. 2020, 121, 103749. [Google Scholar] [CrossRef] [PubMed]
- Li, F. Structure, Function, and Evolution of Coronavirus Spike Proteins. Annu. Rev. Virol. 2016, 3, 237–261. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Delano, W.L. The PyMOL Molecular Graphics System. 2002. Available online: http://www.pymol.org/ (accessed on 21 November 2023).
- Meng, E.C.; Goddard, T.D.; Pettersen, E.F.; Couch, G.S.; Pearson, Z.J.; Morris, J.H.; Ferrin, T.E. UCSF ChimeraX: Tools for structure building and analysis. Protein Sci. 2023, 32, e4792. [Google Scholar] [CrossRef]
- Goddard, T.D.; Huang, C.C.; Meng, E.C.; Pettersen, E.F.; Couch, G.S.; Morris, J.H.; Ferrin, T.E. UCSF ChimeraX: Meeting Modern Challenges in Visualization and Analysis. Protein Sci. 2018, 27, 14–25. [Google Scholar] [CrossRef]
- Bisong, E. Google Colaboratory. In Building Machine Learning and Deep Learning Models on Google Cloud Platform; Apress: Berkeley, CA, USA, 2019; pp. 59–64. ISBN 978-1-4842-4470-8. [Google Scholar]
- Raveh, B.; London, N.; Zimmerman, L.; Schueler-Furman, O. Rosetta FlexPepDock Ab-Initio: Simultaneous Folding, Docking and Refinement of Peptides onto Their Receptors. PLoS ONE 2011, 6, e18934. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://repositorio.ufmg.br/handle/1843/BUBD-AB8FHM (accessed on 21 November 2023).
- Fassio, A.; Santos, L.; Silveira, S.; Ferreira, R.; Melo-Minardi, R. nAPOLI: A Graph-Based Strategy to Detect and Visualize Conserved Protein-Ligand Interactions in Large-Scale. IEEE/ACM Trans. Comput. Biol. Bioinform. 2019, 17, 1317–1328. [Google Scholar] [CrossRef]
- Pimentel, V.; Mariano, D.; Cantão, L.X.S.; Bastos, L.L.; Fischer, P.; de Lima, L.H.F.; Fassio, A.V.; Melo-Minardi, R.C.d. VTR: A Web Tool for Identifying Analogous Contacts on Protein Structures and Their Complexes. Front. Bioinform. 2021, 1, 730350. [Google Scholar] [CrossRef] [PubMed]
- Pimentel, V.; Mariano, D.; Cantão, L.X.S.; Bastos, L.L.; Fischer, P.; de Lima, L.H.F.; Fassio, A.V.; de Melo-Minardi, R.C. VTR: An Algorithm for Identifying Analogous Contacts on Protein Structures and Their Complexes. 2020; preprint. [Google Scholar] [CrossRef]
- 43. Phillips, J.C.; Hardy, D.J.; Maia, J.D.; Stone, J.E.; Ribeiro, J.V.; Bernardi, R.C.; Buch, R.; Fiorin, G.; Hénin, J.; Jiang, W.; et al. Scalable molecular dynamics on CPU and GPU architectures with NAMD. J. Chem. Phys. 2020, 153, 044130. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Jurrus, E.; Engel, D.; Star, K.; Monson, K.; Brandi, J.; Felberg, L.E.; Brookes, D.H.; Wilson, L.; Chen, J.; Liles, K.; et al. Improvements to the APBS Biomolecular Solvation Software Suite. Protein Sci. 2018, 27, 112–128. [Google Scholar] [CrossRef] [PubMed]
- Nelson, M.T.; Humphrey, W.; Gursoy, A.; Dalke, A.; Kalé, L.V.; Skeel, R.D.; Schulten, K. NAMD: A Parallel, Object-Oriented Molecular Dynamics Program. Int. J. Supercomput. Appl. High Perform. Comput. 1996, 10, 251–268. [Google Scholar] [CrossRef]
- Gowers, R.J.; Linke, M.; Barnoud, J.; Reddy, T.J.E.; Melo, M.N.; Seyler, S.L.; Domanski, J.; Dotson, D.L.; Buchoux, S.; Kenney, I.M.; et al. MDAnalysis: A Python Package for the Rapid Analysis of Molecular Dynamics Simulations; Los Alamos National Lab. (LANL): Los Alamos, NM, USA, 2019. [Google Scholar]
- Michaud-Agrawal, N.; Denning, E.J.; Woolf, T.B.; Beckstein, O. MDAnalysis: A Toolkit for the Analysis of Molecular Dynamics Simulations. J. Comput. Chem. 2011, 32, 2319–2327. [Google Scholar] [CrossRef] [PubMed]
- Harris, C.R.; Millman, K.J.; van der Walt, S.J.; Gommers, R.; Virtanen, P.; Cournapeau, D.; Wieser, E.; Taylor, J.; Berg, S.; Smith, N.J.; et al. Array Programming with NumPy. Nature 2020, 585, 357–362. [Google Scholar] [CrossRef] [PubMed]
- NASA/ADS. Matplotlib: A 2D Graphics Environment. Available online: https://ui.adsabs.harvard.edu/abs/2007CSE.9.90H/abstract (accessed on 11 December 2023).




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Amino Acid | Substitution Allowed | Amino Acid | Substitution Allowed |
|---|---|---|---|
| A | E, S, T | M | F, W, Y |
| C | S, T, V | N | G, D, Q |
| D | E | P | G |
| E | A, D | Q | N, E |
| F | M, W, Y | R | H, K |
| G | N, P | S | A, T |
| H | K, R | T | A, I, S |
| I | L, V | V | A, I, L |
| K | H, R | W | F, M, Y |
| L | I, V | Y | F, M, W |
| Case Study | CS1 | CS2 | CS3 |
|---|---|---|---|
| PDB | 6M0J | 6M0J | 6M0J |
| Receptor | Spike (6M0J:E) | Spike (6M0J:E) | Spike (6M0J:E) |
| Initial peptide sequence | STIEEQAKTFLDKFNHEAEDLFYQSSL | STIEEQAKTFLDKFNHEAEDLFYQSSL | STIEEQAKTFLDKFNHEAEDLFYQSSL |
| Sequence length | 27 | 27 | 27 |
| Occupancy | 55% | 47% | 42% |
| Docking score | 54 | 170 | 78 |
| Parameters | |||
| Replicates (per docking) | 50 | 3 | 3 |
| Mutants (per generation) | 50 | 100 | 1000 |
| Generations (max) | 17 | 100 | 14 |
| Manual curation (MC) | |||
| Best peptide (MC) | AIIEEQWGRAAEVFWASSLASYQ | ATVEENSRTYIDHFNRATDDYWATVETFD | TEEQAKTFLDFDLFWQSSLN |
| Generation (MC) | 6 | 14 | 2 |
| Docking score (MC) | −194.71 | −177.7 | −224.52 |
| Occupancy (MC) | 41% | 37% | 33% |
| Identity | 29% | 29% | 57% |
| Higher occupancy (HO) | |||
| Best peptide (HO) | ESIDAGFPRAAELFYESALSSMN | TTIEAQAHSMIERWPRDTAEWWTAIATMD | TEENAKSFVDFDLFYQATLQ |
| Generation (HO) | 16 | 87 | 10 |
| Docking score (HO) | 1432 | 302 | 123 |
| Occupancy (HO) | 71% | 68% | 67% |
| Identity | 23% | 17% | 43% |
| # | PDB | Initial Peptide Sequence | Replicates | Mutants | Generations |
|---|---|---|---|---|---|
| 1 | 6M0J | STIEEQAKTFLDKFNHEAEDLFYQSSL | 50 | 50 | 17 |
| 2 | 6M0J | STIEEQAKTFLDKFNHEAEDLFYQSSL | 3 | 100 | 100 |
| 3 | 6M0J | STIEEQAKTFLDKFNHEAEDLFYQSSL | 3 | 1000 | 14 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
de Abreu, A.P.; Carvalho, F.C.; Mariano, D.; Bastos, L.L.; Silva, J.R.P.; de Oliveira, L.M.; de Melo-Minardi, R.C.; Sabino, A.d.P. An Approach for Engineering Peptides for Competitive Inhibition of the SARS-COV-2 Spike Protein. Molecules 2024, 29, 1577. https://doi.org/10.3390/molecules29071577
de Abreu AP, Carvalho FC, Mariano D, Bastos LL, Silva JRP, de Oliveira LM, de Melo-Minardi RC, Sabino AdP. An Approach for Engineering Peptides for Competitive Inhibition of the SARS-COV-2 Spike Protein. Molecules. 2024; 29(7):1577. https://doi.org/10.3390/molecules29071577
Chicago/Turabian Stylede Abreu, Ana Paula, Frederico Chaves Carvalho, Diego Mariano, Luana Luiza Bastos, Juliana Rodrigues Pereira Silva, Leandro Morais de Oliveira, Raquel C. de Melo-Minardi, and Adriano de Paula Sabino. 2024. "An Approach for Engineering Peptides for Competitive Inhibition of the SARS-COV-2 Spike Protein" Molecules 29, no. 7: 1577. https://doi.org/10.3390/molecules29071577
APA Stylede Abreu, A. P., Carvalho, F. C., Mariano, D., Bastos, L. L., Silva, J. R. P., de Oliveira, L. M., de Melo-Minardi, R. C., & Sabino, A. d. P. (2024). An Approach for Engineering Peptides for Competitive Inhibition of the SARS-COV-2 Spike Protein. Molecules, 29(7), 1577. https://doi.org/10.3390/molecules29071577