Abstract
The fascaplysin and homofascaplysin class of marine natural products has a characteristic 12H-pyrido[1,2-a:3,4-b′]diindole pentacyclic structure. Fascaplysin was isolated in 1988 from the marine sponge Fascaplysinopsis bergquist sp. The analogs of fascaplysin, such as homofascaplysins A, B, and C, were discovered late in the Fijian sponge F. reticulate, and also have potent antimicrobial activity and strong cytotoxicity against L-1210 mouse leukemia. In this review, the total synthesis of fascaplysin and its analogs, such as homofascaplysins A, B, and C, will be reviewed, which will offer useful information for medicinal chemistry researchers who are interested in the exploration of marine alkaloids.
1. Introduction
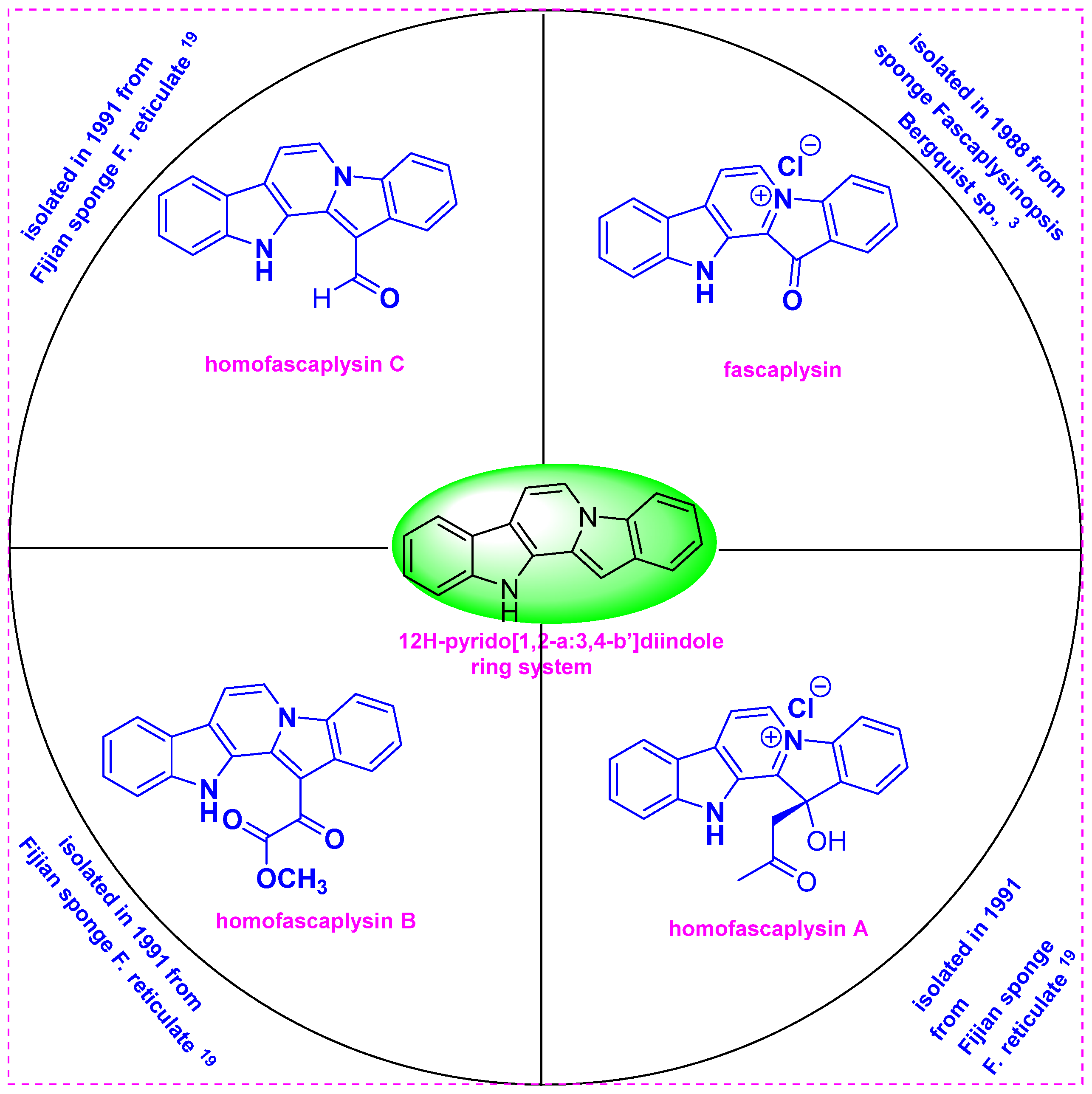
The β-carboline group is a valuable structural motif found in many plants, marine organisms, insects, and mammals, and compounds in this family show a broad range of biological activities [1,2,3,4]. In particular, the 12H-pyrido[1,2-a:3,4-b′]diindole ring system is an important component of various marine alkaloids, such as fascaplysin and homofascaplysins A–C (Figure 1) [5]. Among compounds derived from the sponge Fascaplysinopsis bergquist sp., fascaplysin, which was first isolated in 1988, is the most studied [6]. This natural product was later isolated by several other researchers from Hyrtios erecta [7] as well as from Thorectandra sp. tunicate [8], Didemnum sp. [9]., and sponge Fascaplysinopsis [10].
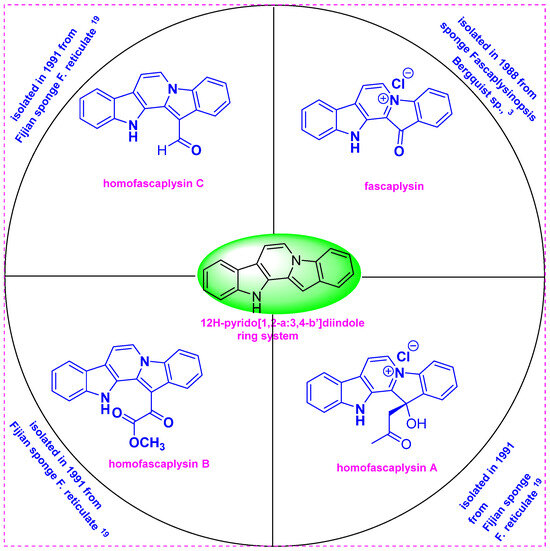
Figure 1.
Structures of fascaplysin and homofascaplysins A–C.
In 1988, Ireland et al. first isolated and determined the structure of the red pigment fascaplysin, a pentacyclic quaternary salt from the Fijian sponge Fascaplysinopsis bergquist sp., which was collected near Dravuni Island, Fiji [6]. The lyophilized sponge was extracted with methanol, and then fractionation was performed using the Kupchan partitioning method. The above methanol/water fraction was washed with CCl4 and then extracted with chloroform. This CHCl3 partition exhibited substantial cytotoxicity and high antimicrobial activity. The chloroform fraction was chromatographed over Sephadex LH-20 using methanol, and subsequent purification by crystallization from methanol yielded fascaplysin. The structure of fascaplysin (13-oxo-12,13-dihydropyrido[1,2-a:3,4-b′] diindol-5-ium chloride) was determined using 1H and 13C NMR spectroscopy, X-ray analysis, and El and FAB mass spectrometry (Figure 1).
In 1990, Crews et al. isolated the bioactive constituents from F. reticulata collected from the Benga Lagoon, Fiji. F. reticulata was found to contain new 12H-pyrido[1,2-a:3,4-b′]diindole alkaloids such as homofascaplysins A–C as well as the known natural products fascaplysin and (+)-octopamine [11]. Four separate specimens of F. reticulata sponge were collected by SCUBA at 10–20 m between 1985 and 1989 from the Benga Lagoon, Fiji. The specimens were preserved for long periods before extraction. The viscous crude oil obtained by methanol extraction of the sponge specimens contained three alkaloids as major constituents, as determined by 1H and 13C NMR spectroscopy. The crude oil was then successively partitioned using equal volumes of aqueous methanol and a series of solvents including hexanes, CCl4, and dichloromethane. The CCl4 fraction was chromatographed over Sephadex LH20 using methanol and then purified by repeated reversed-phase HPLC. This fraction afforded a mixture of homofascaplysins A–C.
Fascaplysin inhibits the growth of several microbes [12] and has been widely studied for the selective inhibition of CDK4, a cyclin-dependent kinase (CDK), as reported by Sony et al. [13]. The inhibition of CDKs by small molecules is an area of recent interest in anticancer research [14]. Fascaplysin is the first biologically active planar, toxic anticancer agent. Unfortunately, fascaplysin cannot be used as an anticancer drug because it is extremely toxic due to its planar structure that intercalates with DNA [15]. As a key element in the cell division cycle, CDKs are essential for cell proliferation and healthy cell growth. Consequently, various nonplanar analogs of fascaplysin have been synthesized and studied. Amongst these compounds, the CDK4 inhibitors BPT and CA224 inhibit tubulin polymerization in vitro and exhibit antitumor activity in the colon cancer tumor model HCT-116 in vivo [16].
Chen et al. revealed that fascaplysin inhibits angiogenesis by suppressing the vascular endothelial growth factor (VEGF) and inducing apoptosis in human umbilical vein endothelial cells (HUVECs) [17]. In addition, Oh et al. demonstrated that fascaplysin inhibits VEGFR2, TRKA, HIF-1α, and survivin, thereby preventing cancer cell growth [18]. Hamilton et al. examined the cytotoxic effects of fascaplysin against non-small cell and small cell lung cancer. Fascaplysin was found to induce tumor cell apoptosis through several mechanisms, including G1/0 cell cycle arrest [19]. Furthermore, fascaplysin significantly upregulates the expression of PD-L1 in lung cancer cells, thus improving the sensitivity of anti-PD-1 immunotherapy in vivo.
The enzyme acetylcholinesterase (AChE) is responsible for the death of neurons in Alzheimer’s disease. Fascaplysin inhibits AChE noncompetitively, with IC50 and Ki values of 1.49 and 2.28 µM, respectively [20]. At 1 mM, fascaplysin displays promising P-gp activation and AChE inhibition, which together with good medication safety demonstrates the potential of this compound as an anti-Alzheimer agent [21]. Furthermore, fascaplysin shows potential antiplasmodial activity, inhibiting Plasmodium falciparum strains NF54 and K1 with IC50 values of 34 and 50 ng/mL, respectively [22]. Thus, fascaplysin is a potent in vitro inhibitor of chloroquine-resistant P. falciparum and chloroquine-susceptible (NF54) strains. Due to the potent antiplasmodial activity, fascaplysin demonstrates the potential to be a leading structure. In addition, the fascaplysin analogs homofascaplysins A–C, which were isolated from the Fijian sponge Fascaplysinopsis reticulate in 1991, have strong cytotoxicity against L-1210 mouse leukemia cells and potent antimicrobial activity [11].
Although fascaplysin has a wide-ranging bioactive spectrum, its advance study has been disturbed by the limited number of compounds isolated from marine microorganism sources. In the future, modifying the planar structure of fascaplysin to reduce its toxicity could play a huge role in chemistry discoveries. These encouraging biological activities linked with fascaplysin have already led to the discovery of a few synthetic targets, and there is much unexplored medicinal chemistry space that may further lead to the discovery of novel biological active lead compounds. Investigations on nonplanar fascaplysin derivatives can form the starting point for elaboration of novel lead compounds, and therefore require finding effective procedures for their syntheses. As a result, many research groups have enthusiastically put their effort into total synthesis. This review may help to find the suitable total synthesis method for a nontoxic, nonplanar structure of fascaplysin derivatives in future synthesis.
Many methods have been established for the synthesis of fascaplysin, but there are limited reports on the synthesis of homofascaplysins B and C. Moreover, to the best of our knowledge, the synthetic methods for obtaining fascaplysin and related analogs, such as homofascaplysins A–C, have not yet been comprehensively reviewed. Herein, we systematically review important advances in synthetic strategies for obtaining fascaplysin and its metabolites homofascaplysins A–C, focusing on a selection of representative examples.
2. Discussion
2.1. Total and Formal Synthesis of Fascaplysin
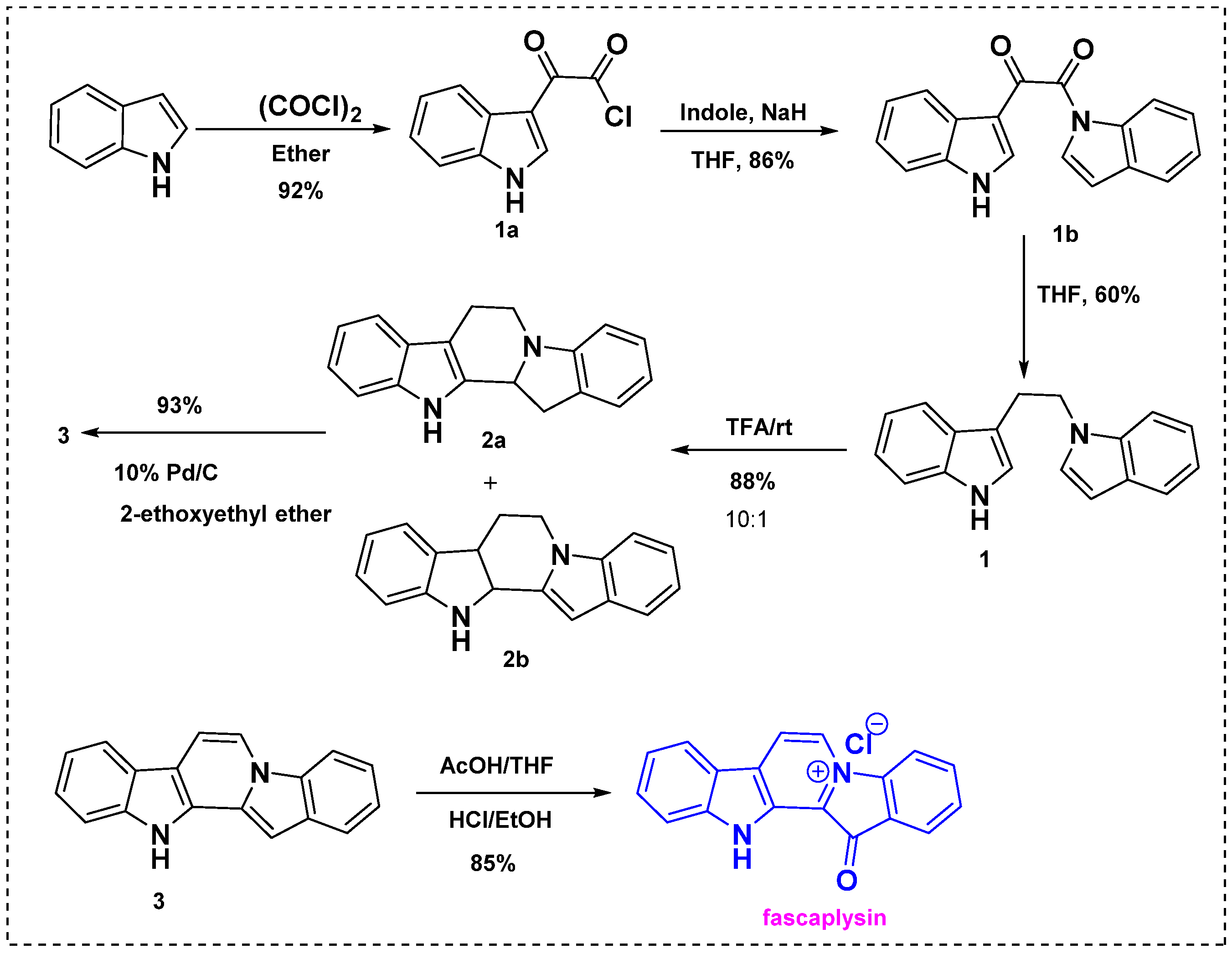
In 1990, Gribble et al. described the first total synthesis of fascaplysin from indole (Scheme 1) [23]. This approach involved peracid oxidation followed by reaction with oxalyl chloride/methanol or Vilsmeier formylation of the key diindole intermediate. Initially, the indole was treated with oxalyl chloride in ether and gave 3-indolyl glyoxylyl chloride 1a at 92%, as shown in Scheme 1. Then, 3-indolylglyoxylyl chloride 1a was permitted to react with I-indolylsodium to give keto amide 1b in an 86% yield. The next step was reduction of 1b with sodium (mono)trifluoroacetoxyborohydride, which afforded diindole 1 in a 60% yield and a 47% overall yield from the starting indole. Further, diindole 1 was then treated with trifluoro acetic acid to obtain a mixture of cyclized products 2a and 2b in a 10:1 ratio. Direct oxidation of this mixture with 10% Pd/C in refluxing 2-ethoxyethyl ether gave fully aromatic pentacycle 3 as a pale green solid. The reaction of 3 with peracetic acid in cold THF followed by treatment with con. HCl/EtOH afforded fascaplysin in an 85% yield. Thus, fascaplysin was efficiently synthesized from indole in seven steps with an overall yield of 65%. The main advantage of this method is that none of the steps, except the final one, involve chromatography, and the overall yield was very high.
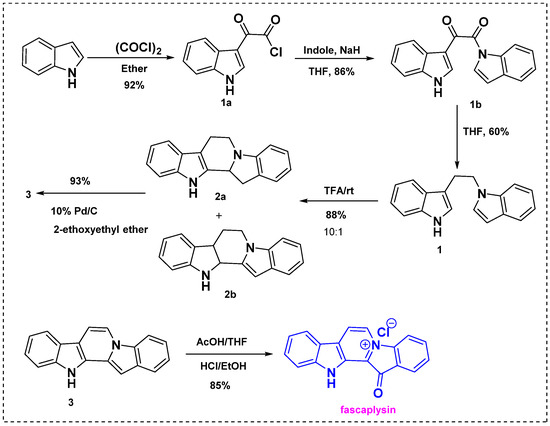
Scheme 1.
First total synthesis of fascaplysin by Gribble et al. [23].
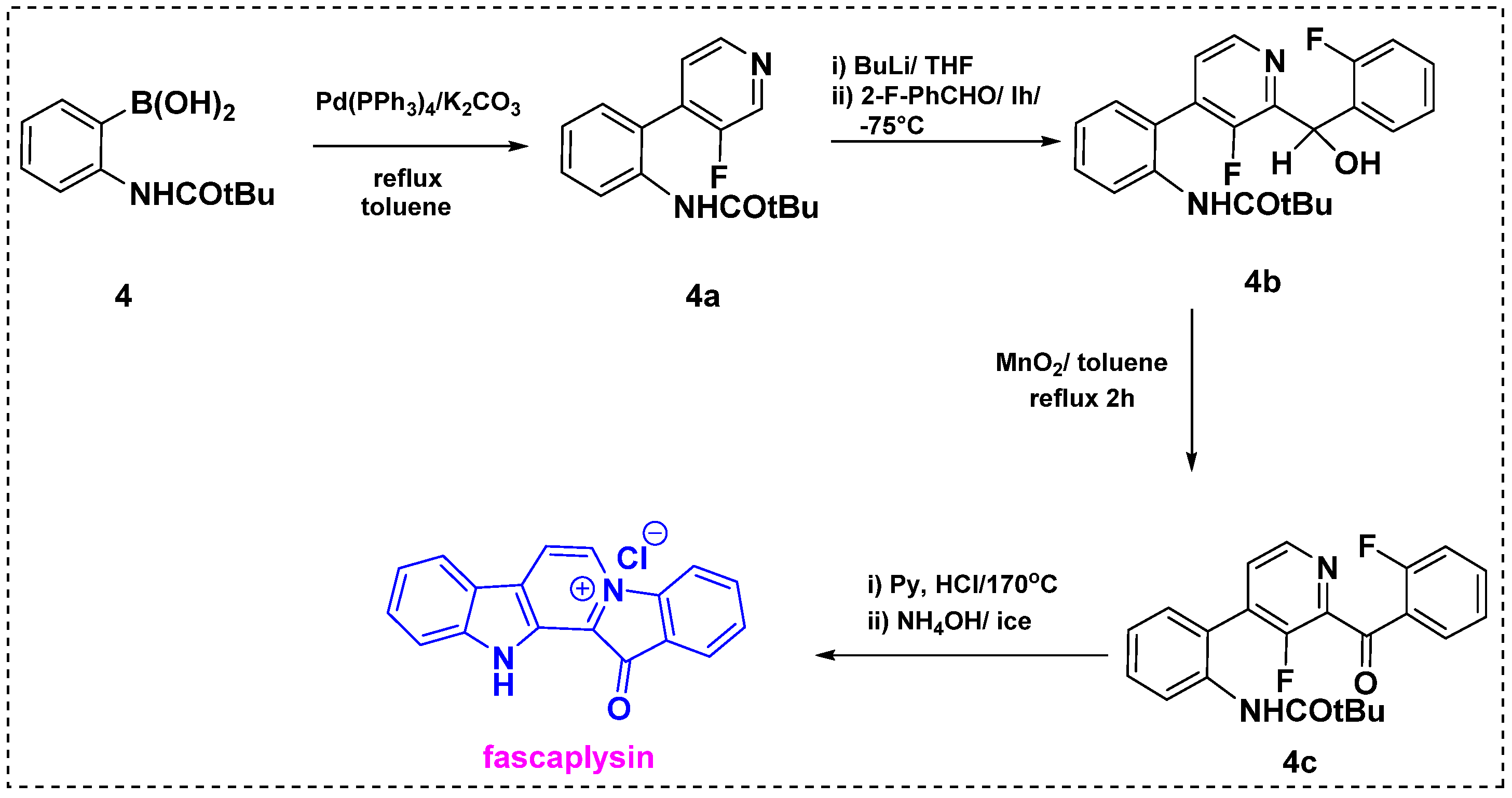
In 1993, Qukguiner et al. developed a strategy for the total synthesis of fascaplysin from simple benzene and pyridine derivatives (Scheme 2) [24]. Palladium-catalyzed Suzuki cross-coupling between boronic acid 4 and iodopyridine gave bis aryl product 4a. Regioselective coupling of 4a with 2-fluorobenzaldehyde in the presence of n-BuLi afforded corresponding pyridine 4b in a 95% yield. Further, oxidation of 4b with MnO2 in refluxing toluene provided carbonyl derivative 4c in a 99% yield. The one-pot double cyclization of 4c by treatment with pyridinium chloride at 170 °C followed by a basic workup gave fascaplysin in an 82% yield. The reported synthesis of fascaplysin relies on key steps such as cross-coupling, metalation, and cyclization. This method is fully regioselective and provides a 76% overall yield from starting material 4.
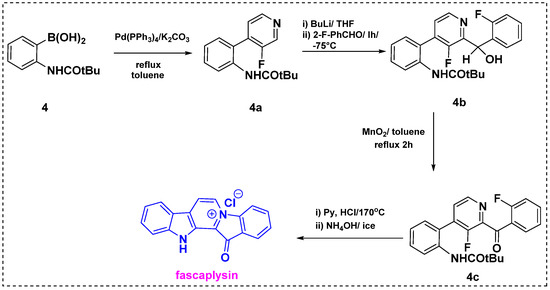
Scheme 2.
Total synthesis of fascaplysin by Qukguiner et al. [24].
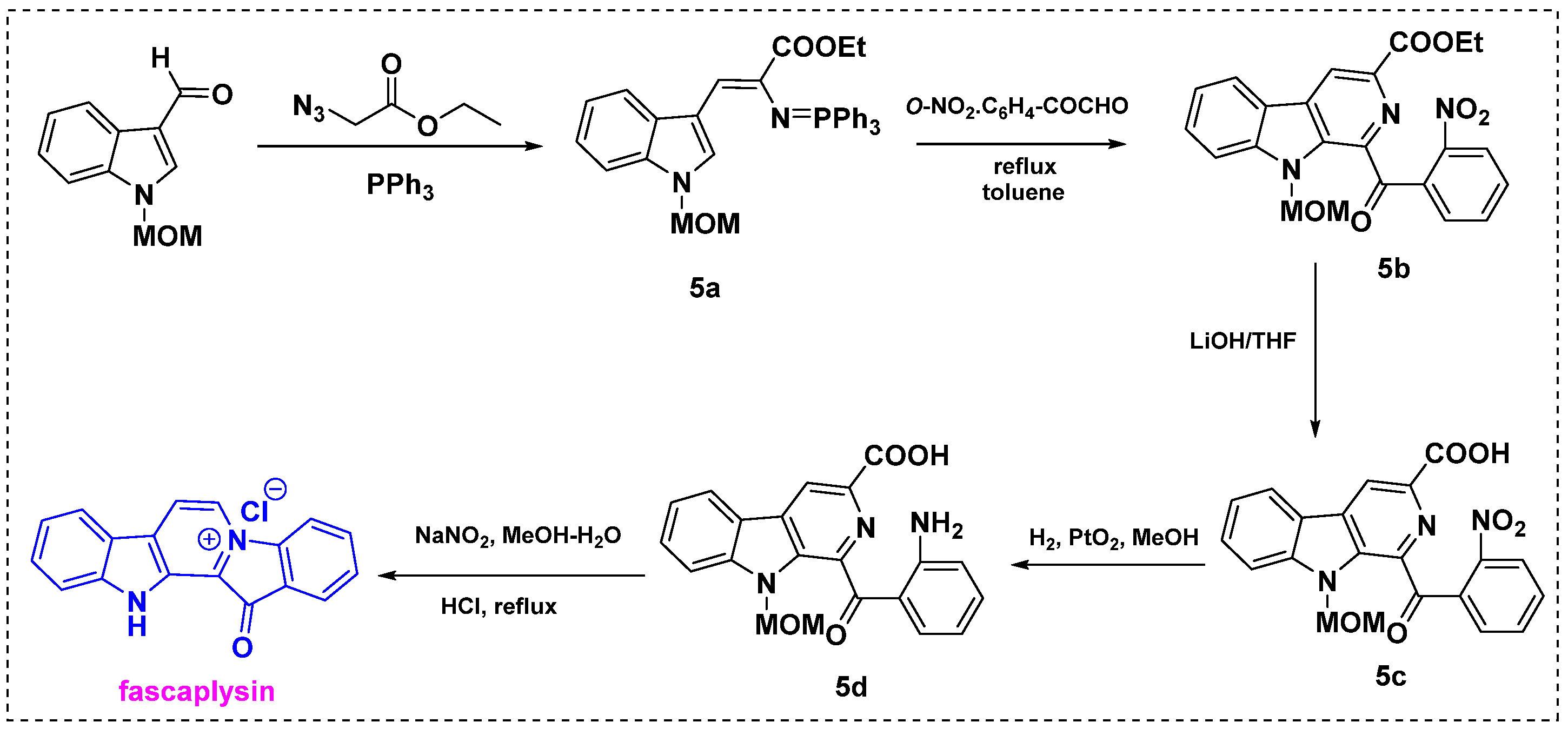
In 1994, Molina et al. described the synthesis of fascaplysin from N-methoxymethyl-3-formylindole in four steps (Scheme 3) [25]. N-Methoxymethyl-3-formylindole was converted into intermediate iminophosphorane 5a by treatment with ethyl azidoacetate followed by triphenylphosphine. Iminophosphorane 5a was treated with a nitro-substituted aryl glyoxal in toluene at 160 °C in a sealed tube to give 1-aroyl-β-carboline derivative 5b in a 60–65% yield. Subsequent hydrolysis of 5b with LiOH/THF at room temperature provided 5c in a 90% yield. The nitro group was then selectively reduced by PtO2/catalytic hydrogenation to give 5d in an 80% yield. Finally, deprotection and decarboxylation by diazotization and further heating afforded fascaplysin in a 60% yield. This method provides a 72% overall yield from the starting material.
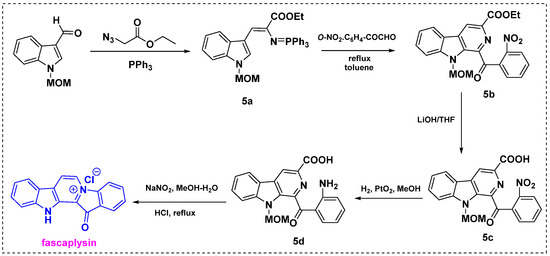
Scheme 3.
Total synthesis of fascaplysin by Molina et al. [25].
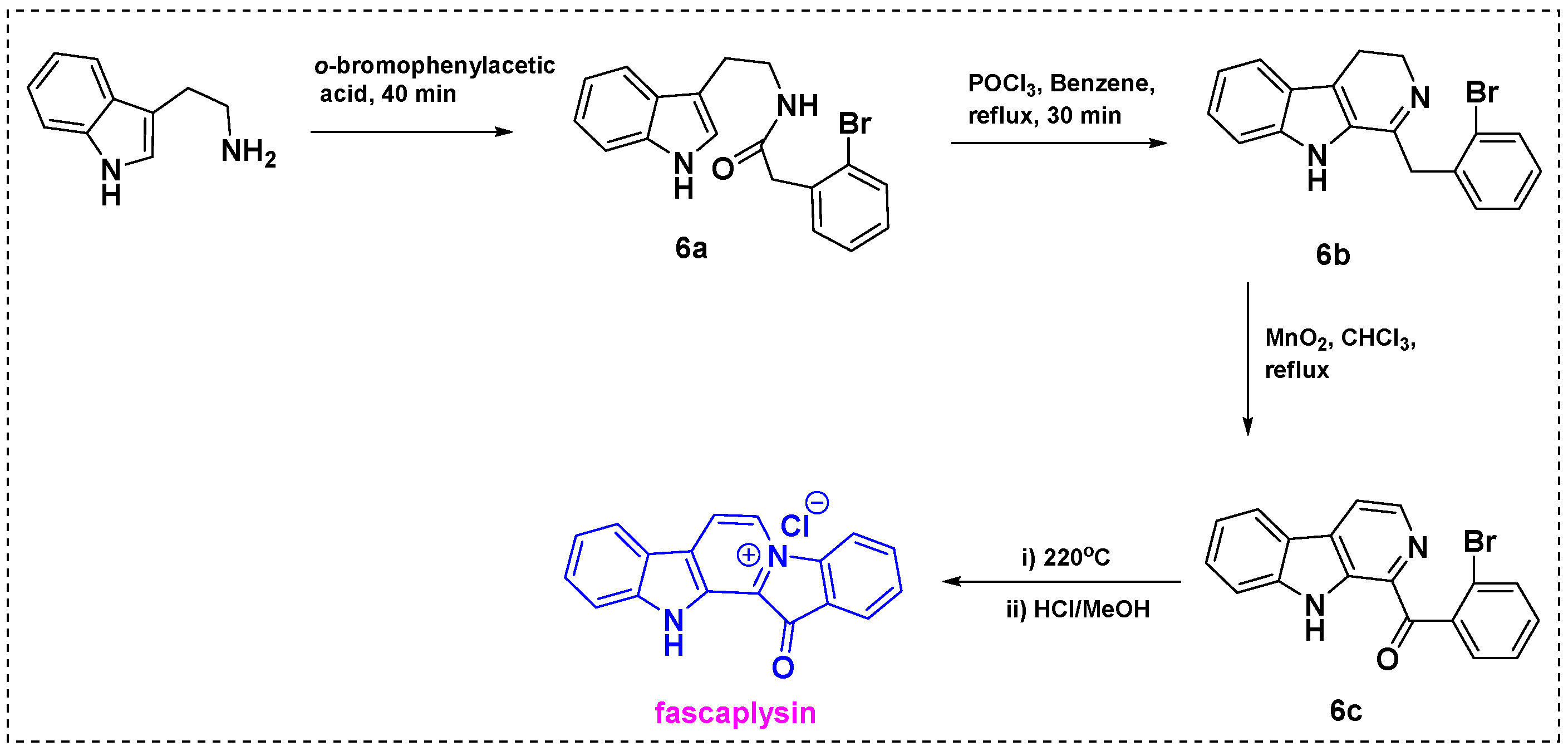
In 1997, Novikov et al. obtained a 44% overall yield for the total synthesis of fascaplysin from tryptamine in five steps (Scheme 4) [26]. Acylation of tryptamine with o-bromophenylacetic acid gave corresponding amide 6a, which was converted into dihydro-13-carboline 6b using POCl3. Subsequent reaction of 6b with MnO2 in CHCl3 under reflux conditions gave α-acyl-substituted β-carboline 6c. Heating of 6c yielded pyridodiindole quaternary salt 6d, which was treated with dry HCl in methanol to give fascaplysin in an overall yield of 44%.
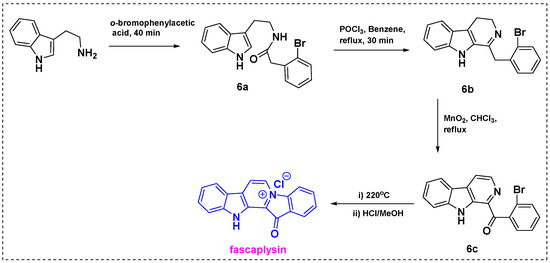
Scheme 4.
Total synthesis of fascaplysin from tryptamine.
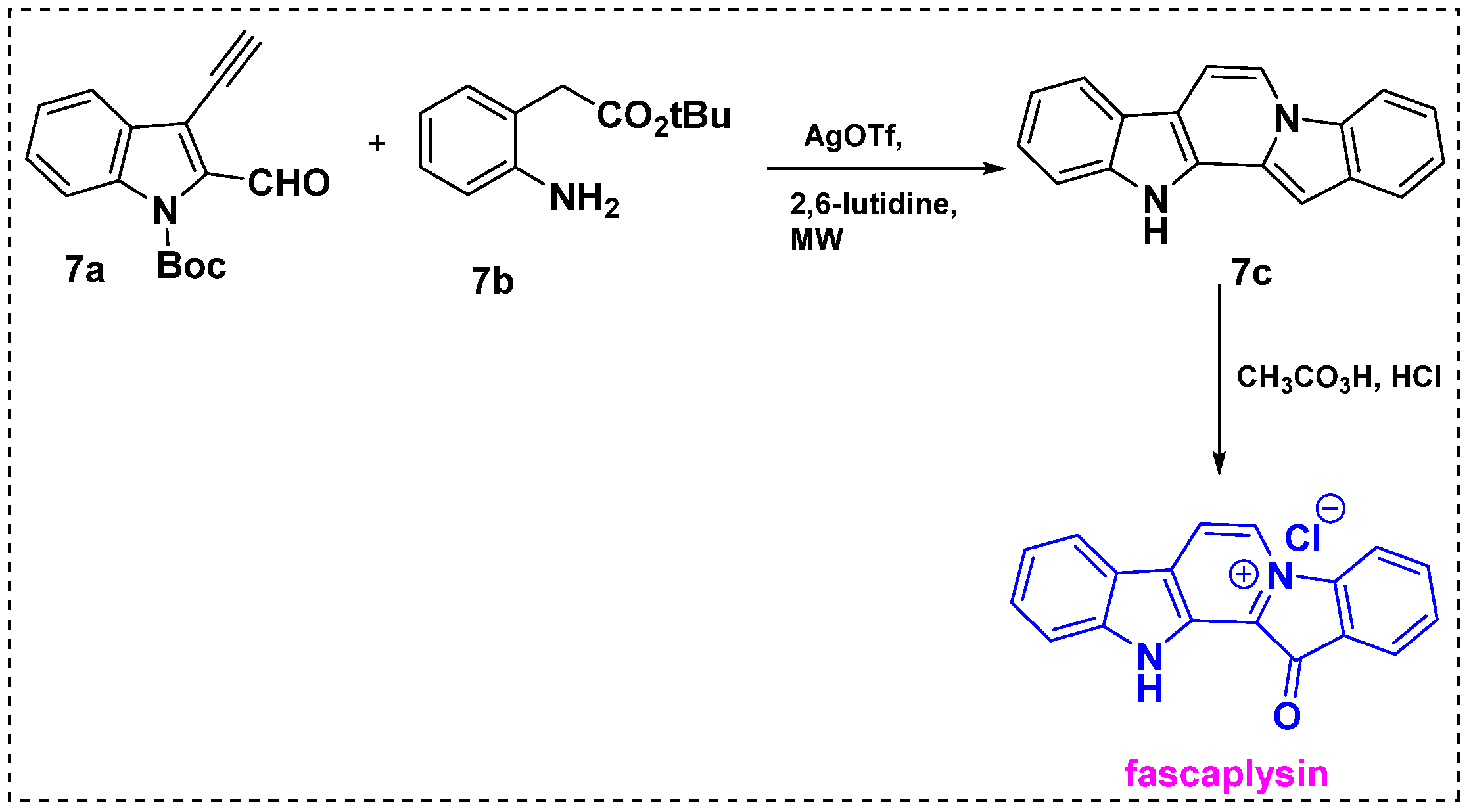
In 2010, Waldmann et al. developed a silver-catalyzed cascade reaction, which was then applied in the total synthesis of fascaplysin (Scheme 5) [27]. In this method, commercially available Boc-protected 3-ethynyl-indole-2-carbaldehyde 7a was used as a precursor. The microwave-assisted silver-catalyzed cascade cyclization of 7a with aniline 7b yielded pentacyclic core 7c in a high yield after acidic workup. Peracetic acid oxidation of diindole 7c followed by salt formation with con. HCl provided a 52% overall yield.
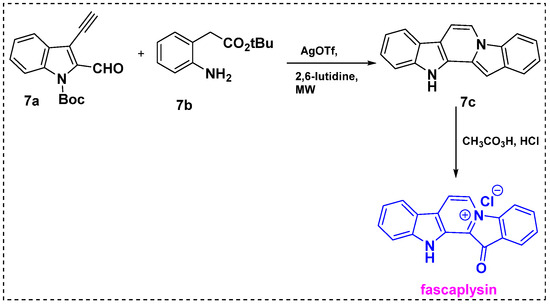
Scheme 5.
Total synthesis of fascaplysin via a silver-catalyzed cascade reaction.
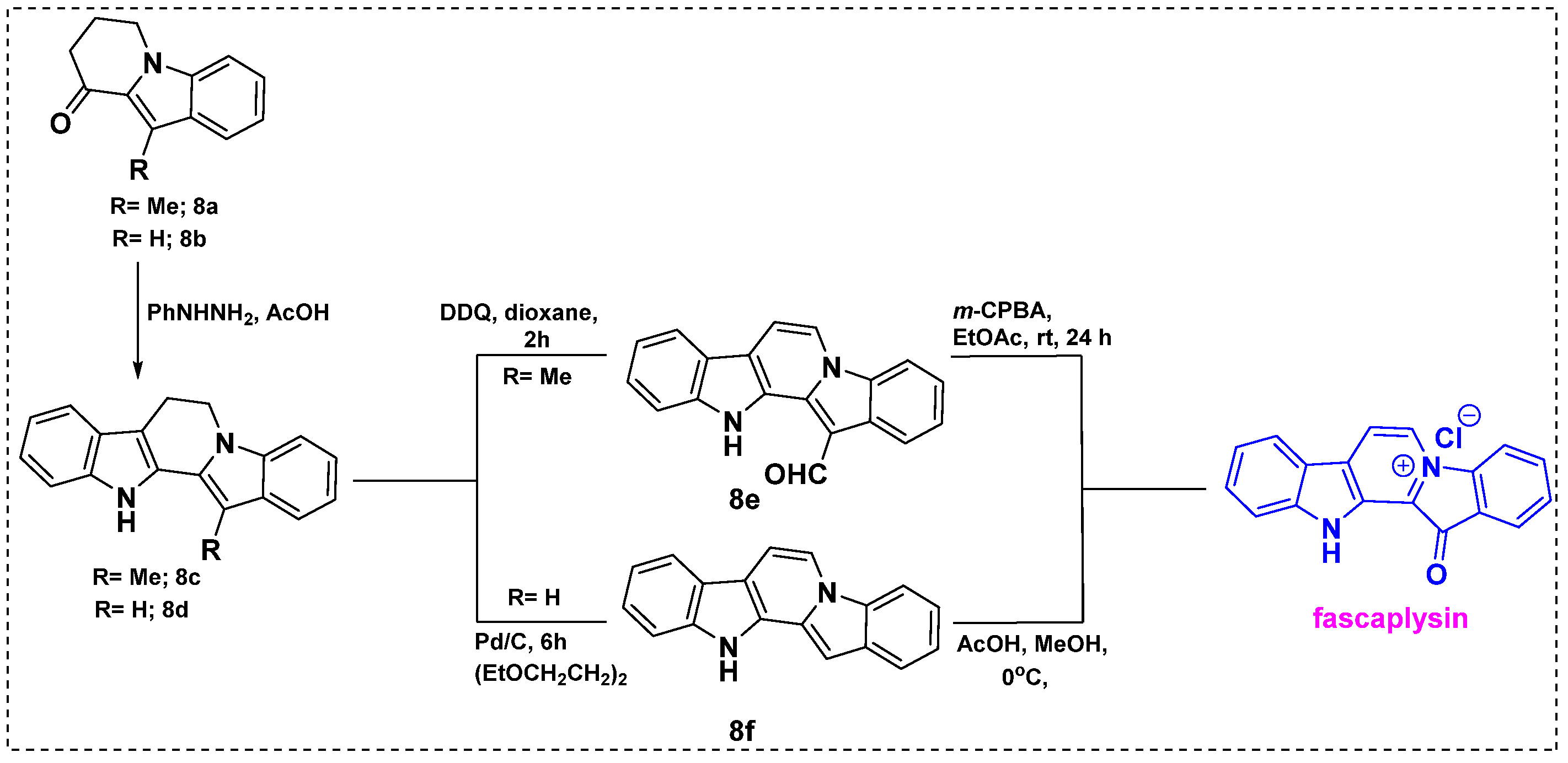
Also in 2010, Zhidkov et al. developed a short synthetic route to fascaplysin from 3-methyl indole/indole ketone 8a/8b using the popular Fischer indole synthesis reaction as a key step (Scheme 6) [28]. Fischer cyclization of 8a or 8b gave corresponding bisindole 8c or 8d in a 90–91% yield, which were further converted into 8e by 2,3-dichloro-5,6-dicyanobenzoquinone (DDQ) oxidation or 8f by dehydrogenation over Pd/C. The yield was 50% for 8e and 75% for 8f. Finally, the reaction of 8e with mCPBA provided fascaplysin in a 67% yield and 8f with acetic acid in methanol gave fascaplysin in 85% yields. Therefore, fascaplysin can be synthesized from commercially available ethyl indole-2-carboxylate in seven steps with overall yields of 69% through 8e and 82% through intermediate 8f.
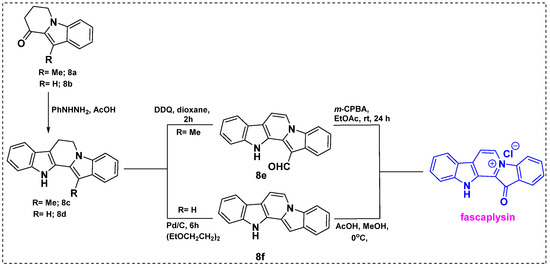
Scheme 6.
Total synthesis of fascaplysin using Fischer cyclization as a key step.
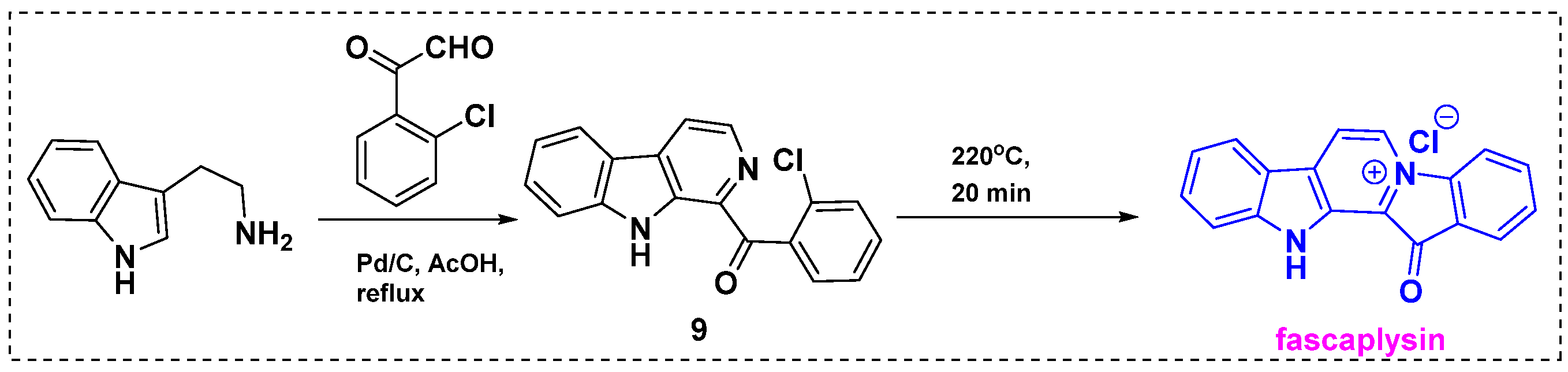
In 2012, Bharate et al. described a two-step total synthesis of fascaplysin from commercially available tryptamine in a 68% overall yield (Scheme 7) [29]. As well as being shorter, less expensive, and more efficient than previously reported synthetic methods, this approach has potential for the large-scale synthesis of fascaplysin. A key step in this strategy is tandem dehydrative condensation between ortho-chloro-substituted glyoxal and tryptamine followed by dehydrogenation. Initially, tryptamine was reacted with glyoxal in acetic acid in the presence of Pd/C to give cyclized product 9 in an 85% yield. Next, β-carboline 9 was heated at 220 °C for 20 min to induce ring closure and produce fascaplysin in an 80% yield.
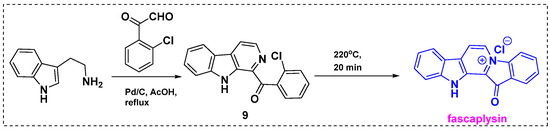
Scheme 7.
Total synthesis of fascaplysin from tryptamine.
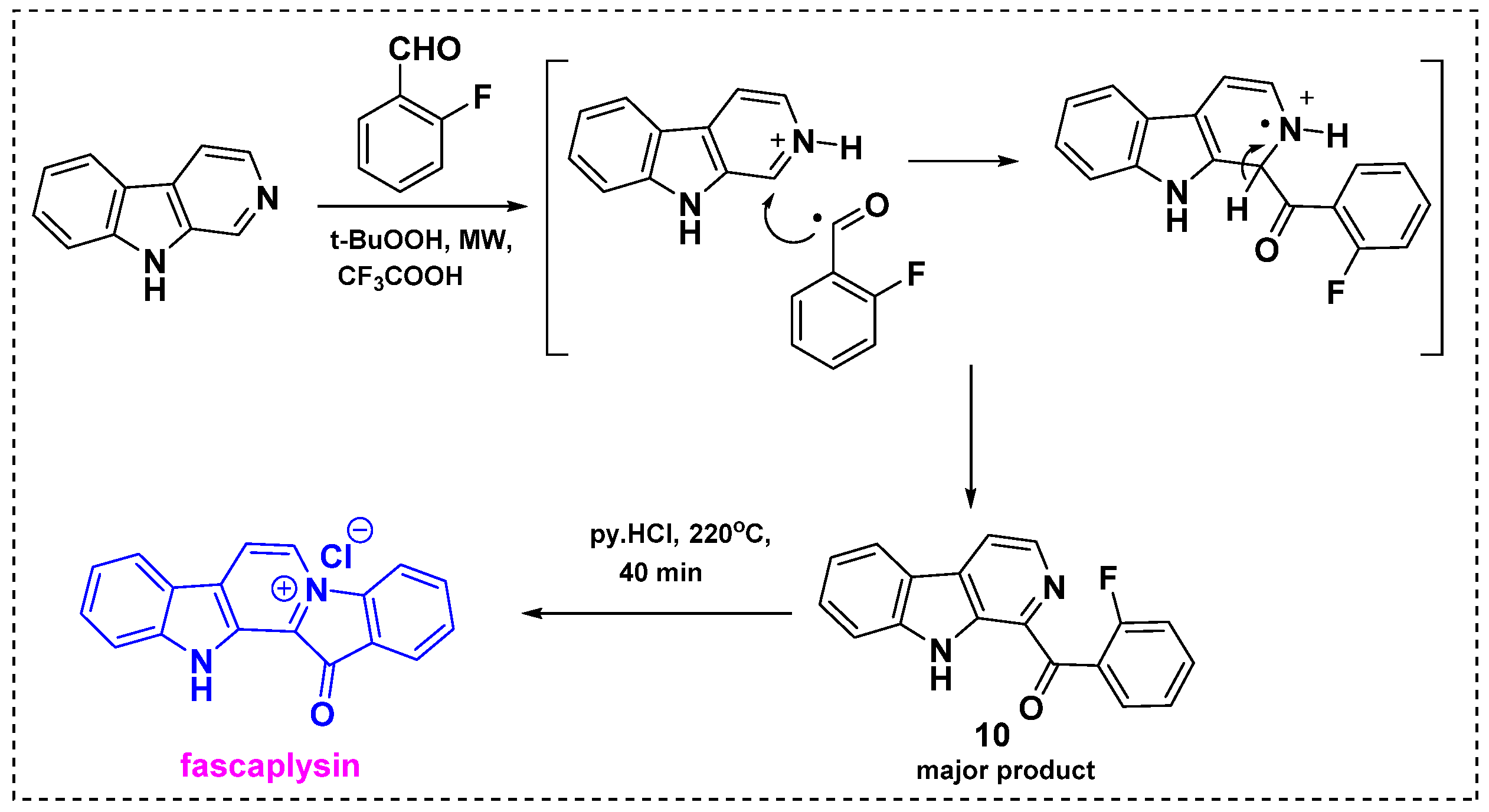
In 2013, Zhidkov et al. developed a two-step method for the synthesis of fascaplysin via a microwave-assisted Minisci reaction of β-carboline followed by intramolecular quaternization (Scheme 8) [30]. Initially, the reaction of β-carboline with o-fluorobenzaldehyde and t-BuOOH in TFA under microwave irradiation gave β-carboline 10 in a 65% yield as well as trace amounts of byproducts. This report presents the first microwave-assisted Minisci reaction. In this reaction, TFA can act as an acid catalyst and t-BuOOH is involved in the radical generation in the aldehyde moiety. Therefore, TFA was catalyzed to initiate nucleophilic radical substitution to an electron-deficient aromatic compound of β-carboline, resulting in the introduction of an alkyl group of the aldehyde moiety to a nitrogen containing β-carboline, as shown in Scheme 8. Heating of 10 with pyridinium chloride at 220 °C and a subsequent workup gave fascaplysin in an 80% yield. This method provides fascaplysin in a 72% overall yield from commercially available β-carboline.
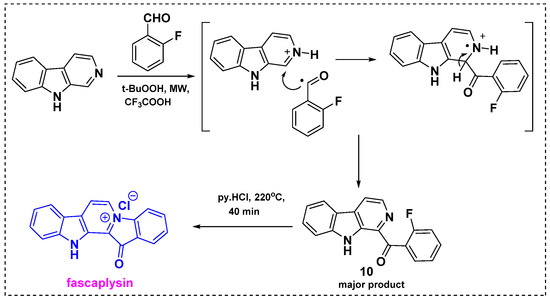
Scheme 8.
Two-step synthesis of fascaplysin under microwave conditions.
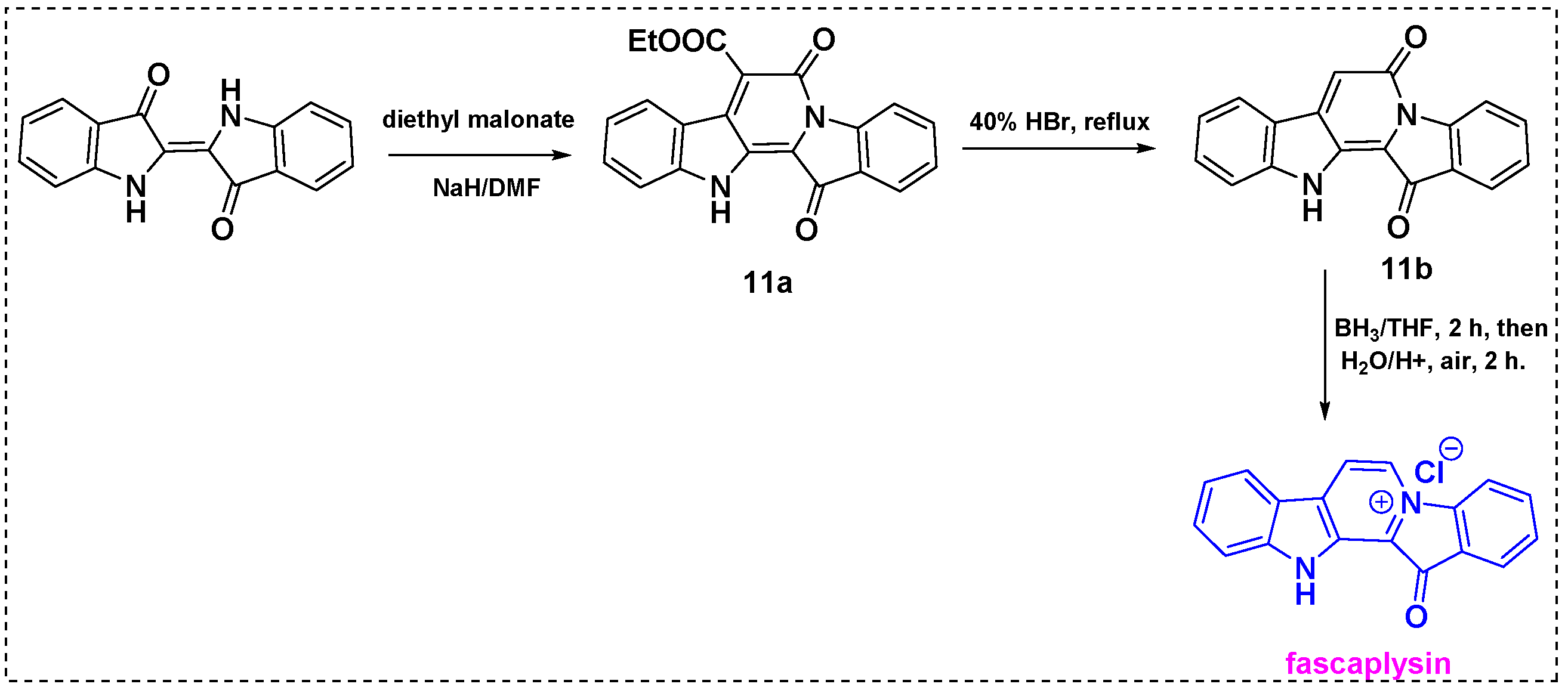
The same research group reported a novel three-step method involving indigo and methylene active compounds for the total synthesis of fascaplysin and its derivatives in 2018 (Scheme 9) [31]. Indigo was reacted with diethyl malonate in DMF/NaH to produce 11a in a 75% yield. Subsequent hydrolysis and decarboxylation of 11a via refluxing in 40% hydrobromic acid for 2 h gave 11b in a 95% yield. Finally, 11b was refluxed with a BH3/THF complex for 2 h and then oxidized in air to afford fascaplysin in a 43% yield and, overall, a 71% yield from indigo.
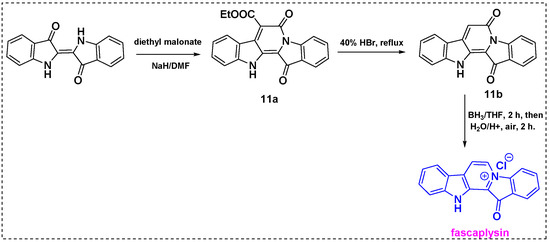
Scheme 9.
Total synthesis of fascaplysin from indigo.
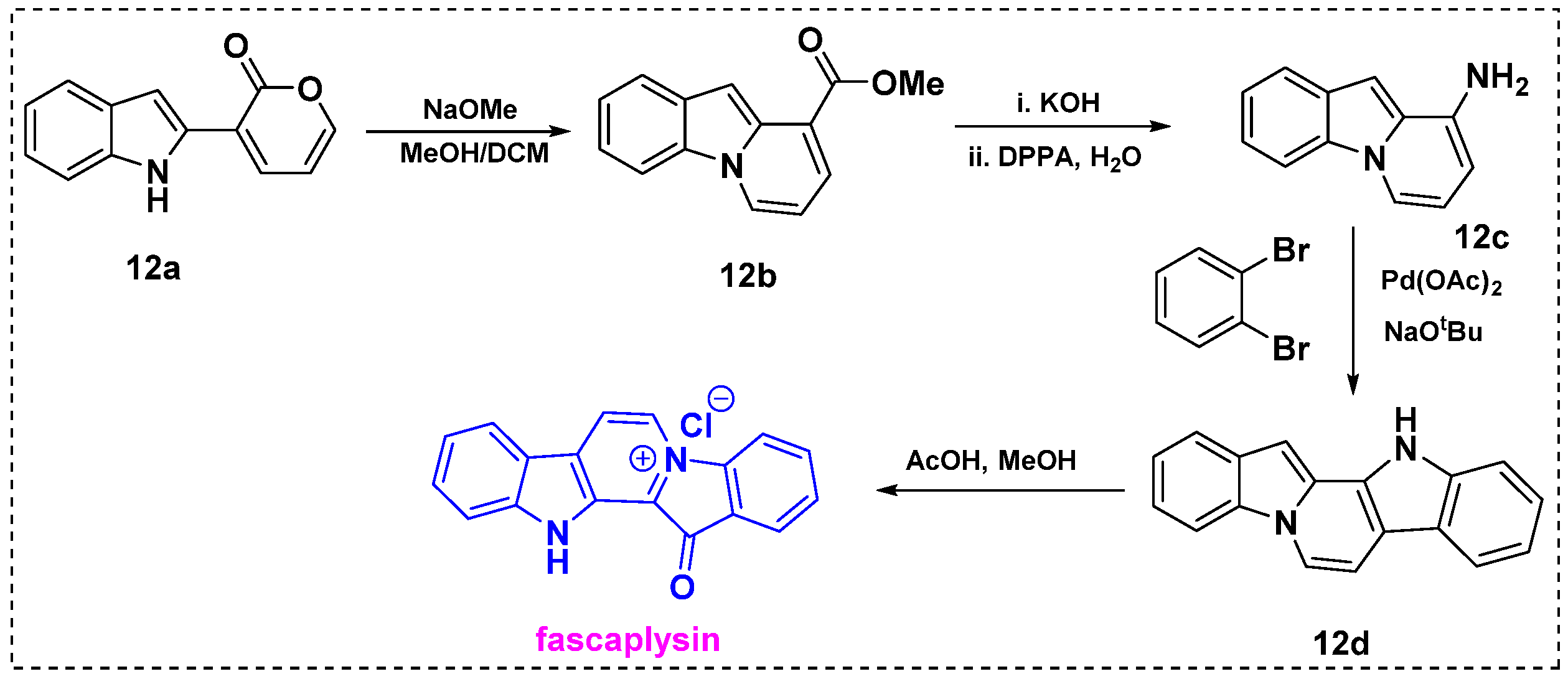
In 2020, Sarpong et al. described the synthesis of diverse N-fused heterocycles, including the pyrido[1,2-a] indole scaffold, using an effective pyrone remodeling strategy [32]. To demonstrate its utility, this methodology was applied in the synthesis of fascaplysin (Scheme 10). Initially, indole–pyrone 12a was reacted with sodium methoxide as a nucleophile in dichloromethane/methanol to furnish 12b in a 61% yield. Then, hydrolysis of ester 12a with KOH provided an intermediate carboxylic acid in a 99% yield, which efficiently underwent Curtius rearrangement in the presence of DPPA/H2O to generate amine 12c in a 94% yield. Next, the palladium-catalyzed amination/C–H arylation domino coupling reaction of 12c and 1,2-dibromobenzene in the presence of dppf ligand gave the product 12d at 55%, which was further converted to fascaplysin via a previously reported procedure using acetic acid/methanol.
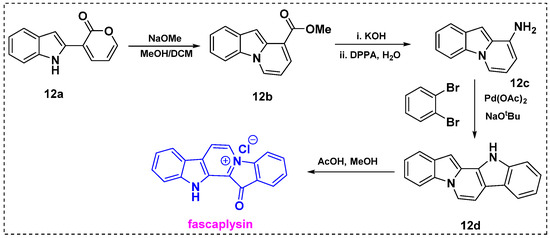
Scheme 10.
Formal synthesis of fascaplysin via pyrone remodeling.
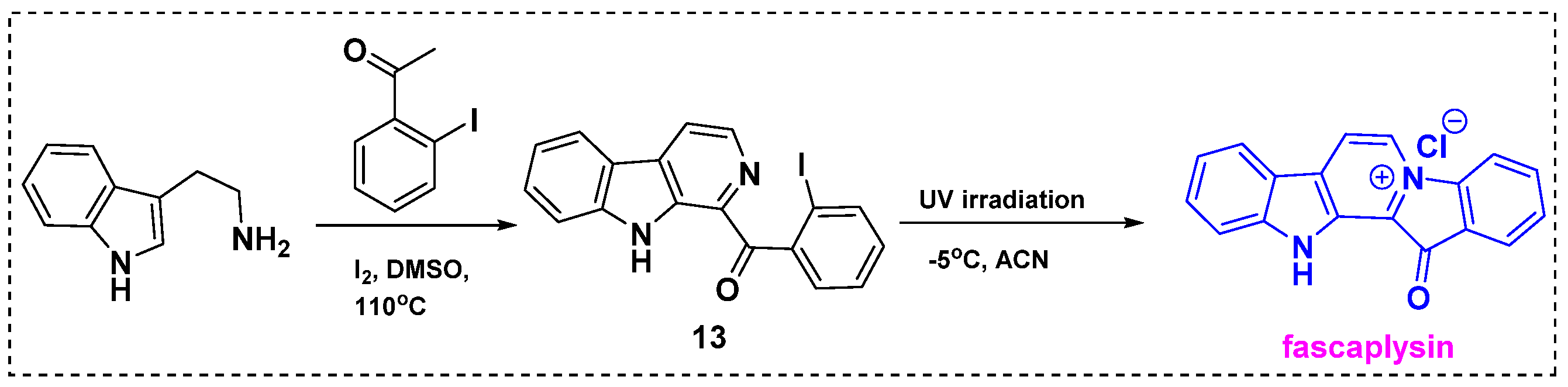
In 2023, Tryapkin et al. described a novel two-step synthesis of fascaplysin derivatives based on low-temperature UV quaternization (Scheme 11) [33]. The first step of this method was a modification of the synthetic routes from tryptamine and acetophenones previously reported by Zhu et al. and Battini et al. The reaction of tryptamine with 2-iodoacetophenone in DMSO/I2 at 110 °C gave isoquinoline 13 in a 40% yield. Next, UV quaternization was performed in acetonitrile at −5 °C to prevent side product formation, and fascaplysin was obtained in a 50% yield. The usage of acetonitrile with the temperature decreasing to −50 °C hinders the side reactions to allow obtaining fascaplysin in a good yield. Notably, only the starting material and quaternization product (fascaplysin) were observed in the reaction mixture. After fascaplysin was isolated, three-times-repeated irradiation of the starting material remaining in the reaction mixture increased the product yield to 91%.
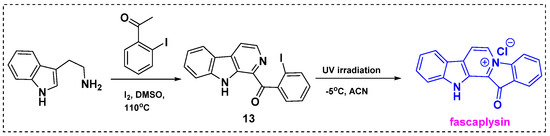
Scheme 11.
Total synthesis of fascaplysin from tryptamine by UV irradiation.
2.2. Total and Formal Synthesis of Homofascaplysins B–C
The therapeutic potential of homofascaplysins A–C is based on a broad range of bioactivities, including anticancer, antibacterial, antifungal, antiviral, and antimalarial properties. Homofascaplysins A–C have effective cytotoxicity against L-1210 mouse leukemia cells and potent antimicrobial activity [7,34]. Although fascaplysin has been synthesized by several groups, as detailed in Section 1, only a few syntheses of homofascaplysins B and C have been reported. A total of five synthetic routes have been reported for total synthesis of fascaplysin C and three synthetic routes for homfascaplysin B since 1992. Here, we are going to briefly discuss each and every total synthesis of homofascaplysins A–C reported in the literature up to 2023.
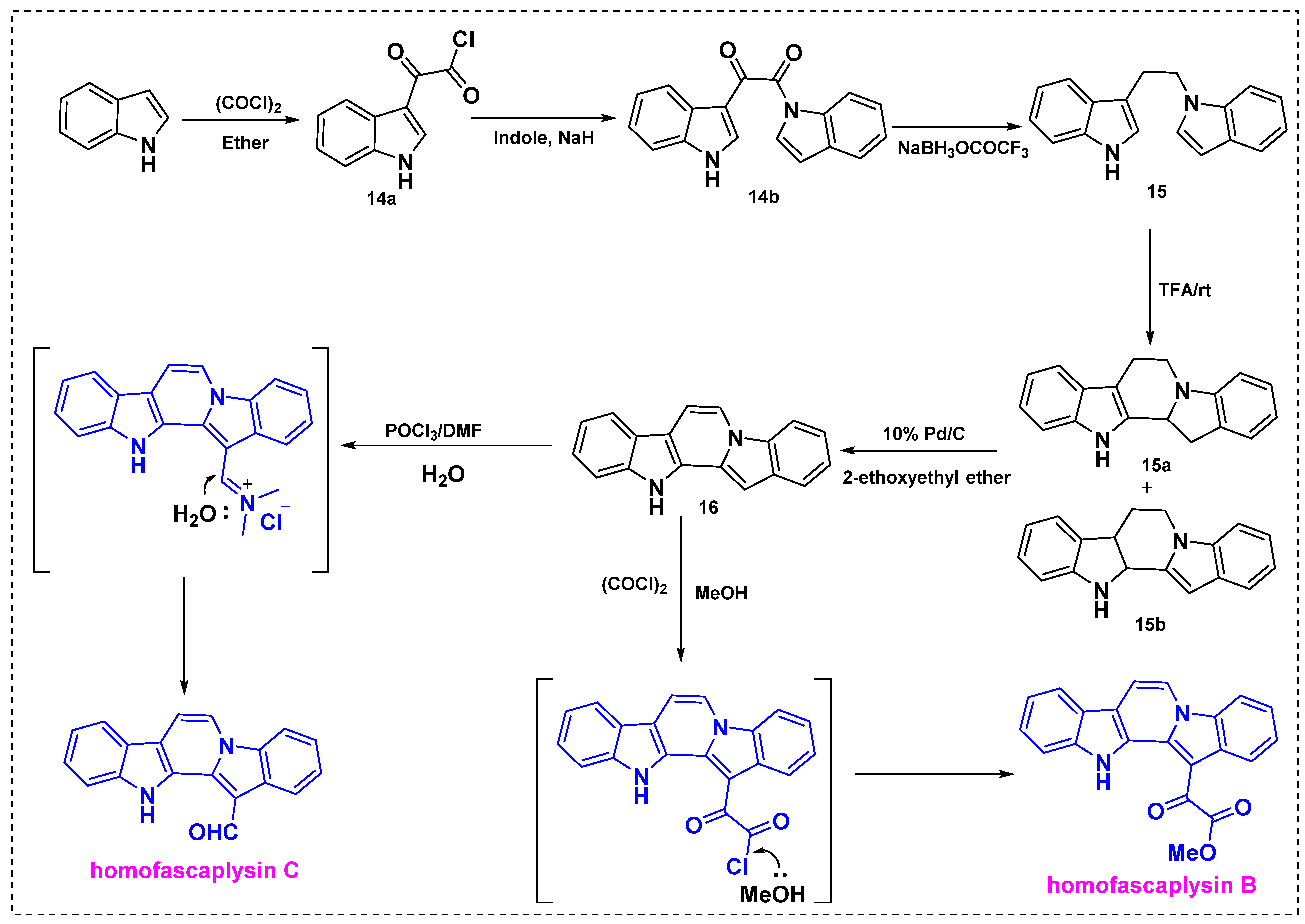
The first total synthesis was described in 1992 by Gribble et al., who obtained homofascaplysins B and C from indole in seven steps with overall yields of 76% and 67%, respectively (Scheme 12) [35]. These compounds were synthesized by peracid oxidation followed by reaction of the key diindole intermediate with oxalyl chloride/methanol or Vilsmeier formylation. In the first step, indole was reacted with oxalyl chloride under mild reaction conditions to give 3-indolyl glyoxylyl chloride 14a, which was further treated with the sodium salt of indole to afford keto amide 14b in an 86% yield. Treatment of keto amide 14b with sodium trifluoroacetoxyborohydride provided diindole 15 in a 47% overall yield. Next, the neat reaction of diindole 15 in CF3CO2H at room temperature provided a 10:1 mixture of 15a and 15b in a good yield. The crude mixture of 15a and 15b was dehydrogenated with 10% Pd/C in EtOAc under reflux to give diindole 16 in a 98% yield. Finally, 16 was treated with the Vilsmeier reagent POCl3/DMF to obtain homofascaplysin C in an 88% yield or with oxalyl chloride followed by methanol to obtain homofascaplysin B in a 99% yield. In both reactions, intermediate 16 provided the regioselective products homofascaplysins B and C.
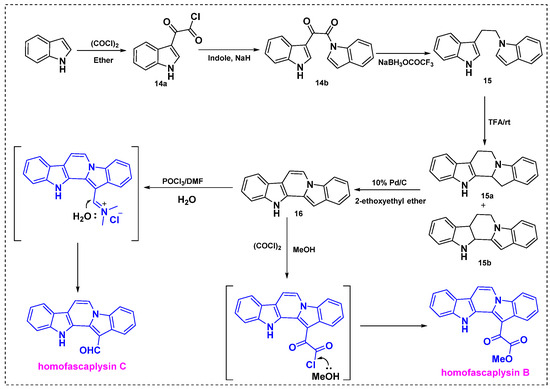
Scheme 12.
First total synthesis of homofascaplysins B and C from indole.
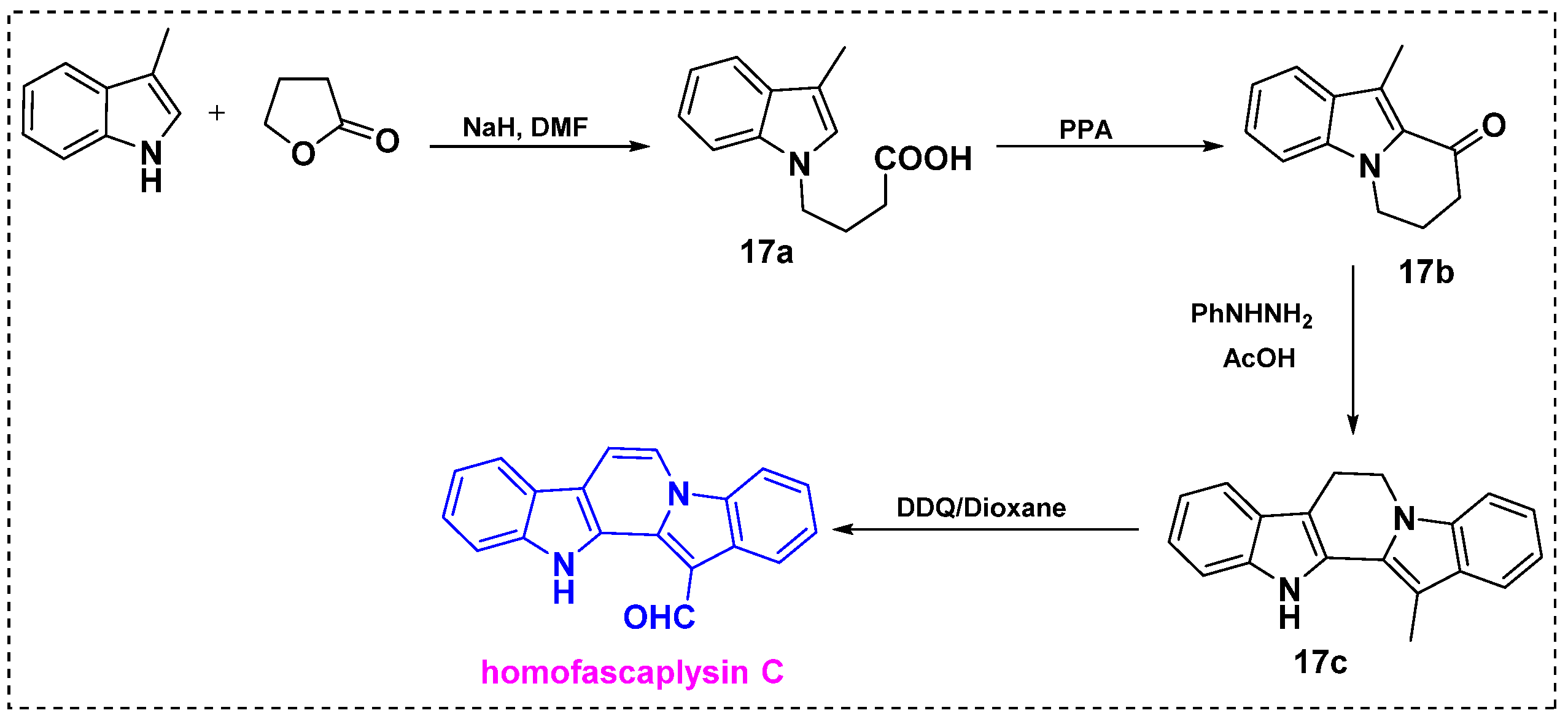
In 1996, Dubovitskii et al. developed an efficient protocol for the total synthesis of homofascaplysin C from commercially available 3-methylindole in only four steps (Scheme 13) [36]. In the first step, the reaction of 3-methylindole with γ-butyrolactone in the presence of NaH/DMF provided acid 17a in a 95% yield. Next, cyclization of acid 17a in PPA at 100 °C gave pyrido[1,2-a] indole 17b in a 90% yield. Using the Fischer indole synthesis method, 17b was treated with phenylhydrazine hydrochloride in refluxing acetic acid to obtain 6,7-dihydro-13-methyl-12H-pyrido[1,2-a:3,4-b′]diindole 17c in a 91% yield. Finally, oxidative dehydrogenation and oxidation of the methyl group to a formyl group by DDQ in dioxane under reflux for 2 h gave homofascaplysin C in a 50% yield and an 81% overall yield from the 3-methyl indole in four steps. This route is highly favorable for scaling up reactions because of the easy availability of the starting materials, like 3-methyl-indole.
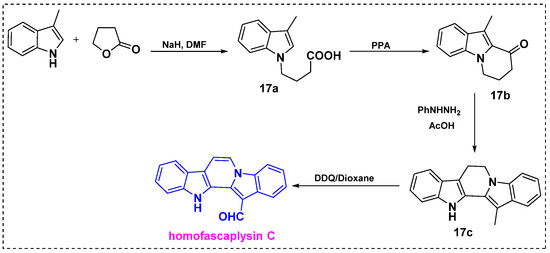
Scheme 13.
Total synthesis of homofascaplysin C from 3-methylindole.
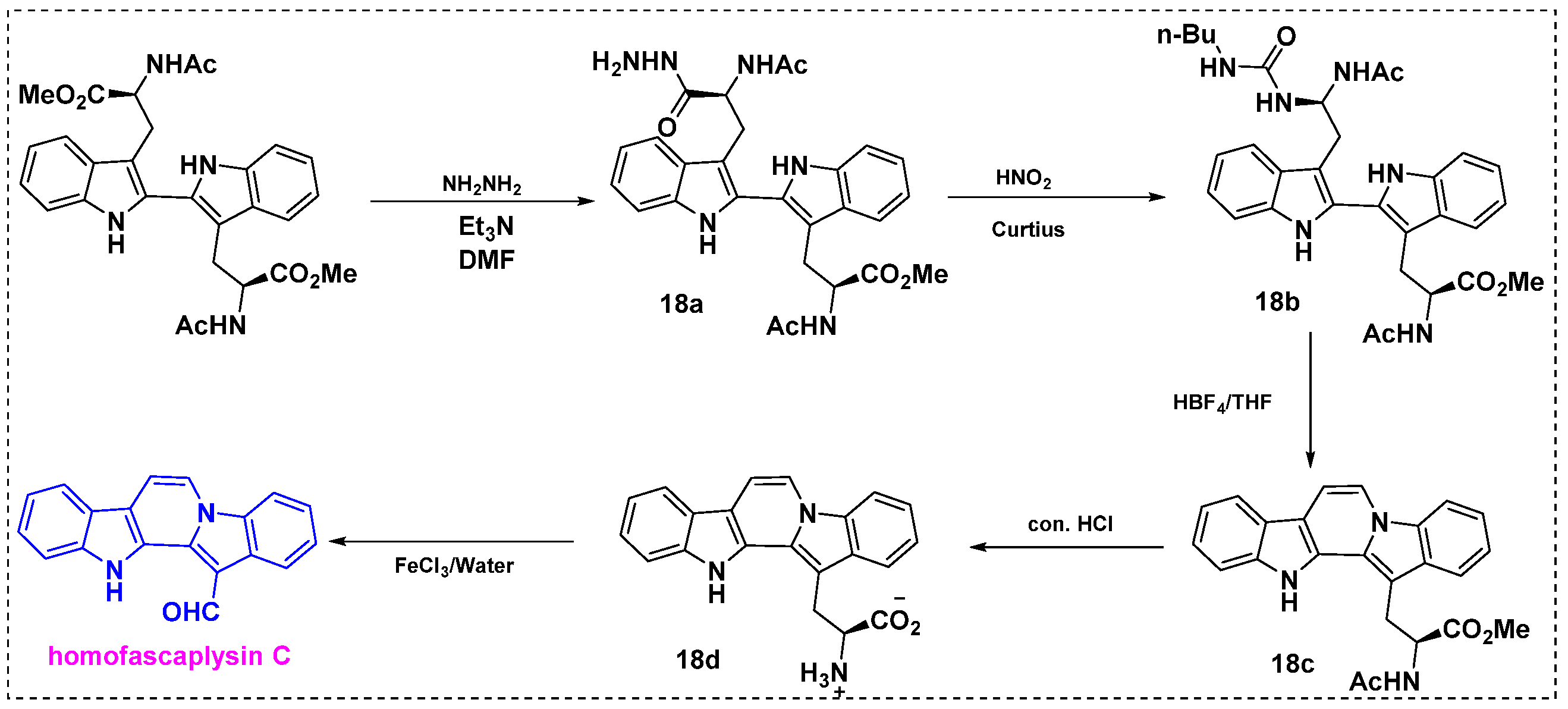
In 1999, Van Vranken et al. described the total synthesis of homofascaplysin C from peptides via the Mannich dimerization of tryptophan side chains (Scheme 14) [37]. This novel and efficient method relies on peptide chemistry. Initially, ditryptophan was converted into monohydrazide 18a using 1.5 equiv of hydrazine in DMF, and the product yield was 30%. Triethylamine was added to the reaction mixture to facilitate proton-transfer steps. Subsequently, acyl hydrazide 18a was converted into an acyl azide using a mixture of NaNO2, 1 N HCl, and glacial acetic acid in chloroform. After extraction and drying, the acyl azide was trapped by n-butylamine to give urea 18b in a 48% yield. Unexpectedly, ionization of 18b in THF with anhydrous HBF4 gave the cyclized product 18c in an 83% yield instead of the expected product of uncyclized aldehyde. The structure of 18c was interesting because it contained the indolo[1,2-a] carbazole ring system found in fascaplysins. Finally, compound 18c was hydrolyzed to amino acid 18d. The reaction was first performed under basic conditions, giving 18d only in a 25% yield, but it was later found that the corresponding carboxamide hydrolyses under acidic conditions could give 18d in a 43% yield, and then oxidative cleavage of the amino acid moiety with aqueous ferric chloride provided homofascaplysin C in a 23% yield, and the overall yield was 45% from tryptophan.
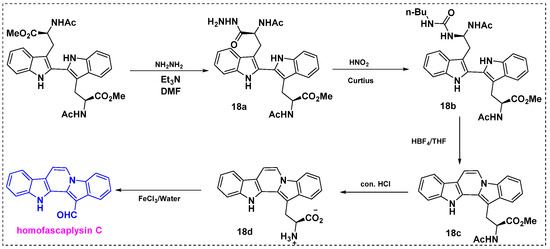
Scheme 14.
Total synthesis of homofascaplysin C from ditryptophan esters.
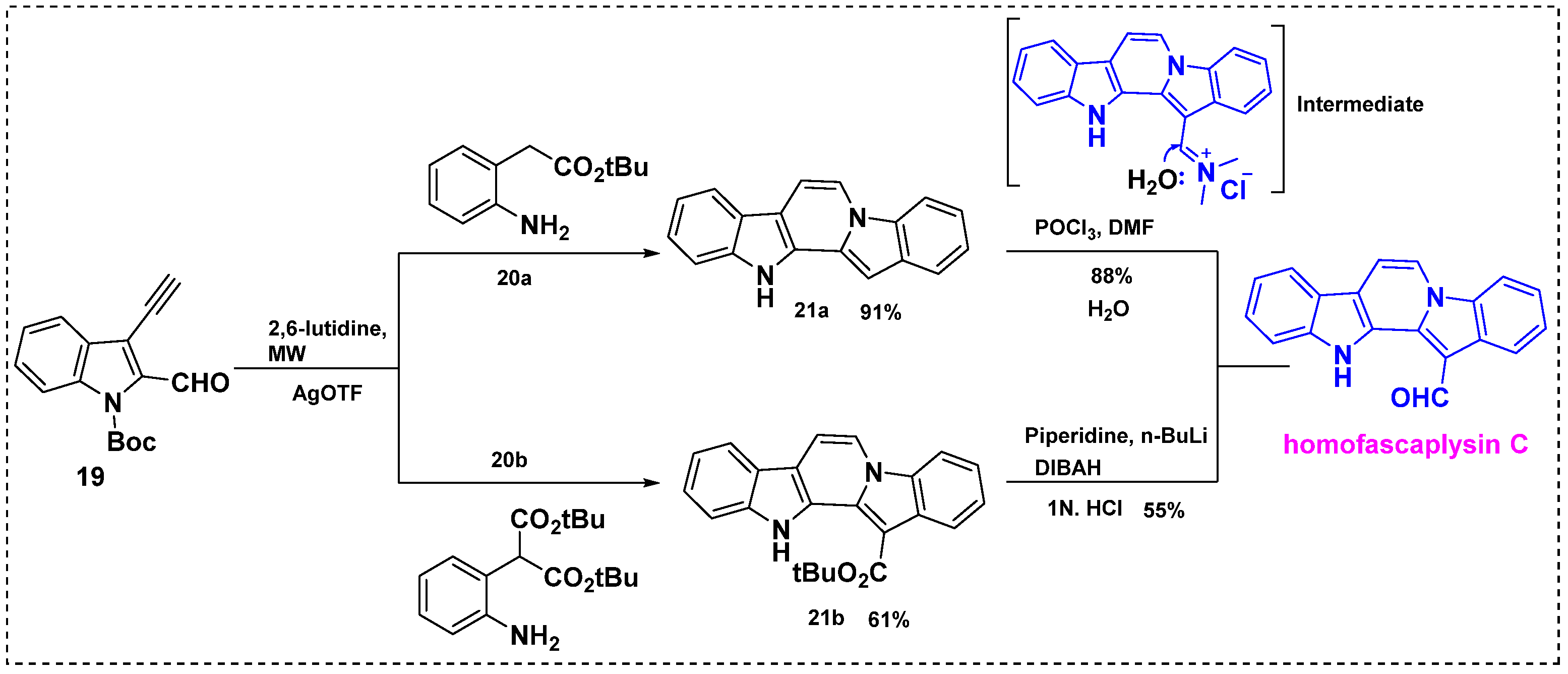
In 2010, Waldmann et al. described a silver-catalyzed cascade reaction and its application in the total synthesis of homofascaplysin C from Boc-protected 3-ethynyl-indole-2-carbaldehyde 19 (Scheme 15) [27]. The microwave-assisted silver-catalyzed cascade cyclization of 19 with aniline 20a or 20b yielded pentacyclic core 21a or 21b in a 91% yield or a 61% yield, respectively. Partial reduction of the tert-butyl ester of 21b with in situ generated lithium diisobutyl piperidino hydroaluminate provided homofascaplysin C in a 55% yield and a 58% overall yield from 19 (two steps). In contrast, diindole 21a was effectively converted into homofascaplysin C in an 88% yield by formylation with POCl3 in DMF, and the overall yield was 89% from 19 (two steps). This reaction proceeded regioselectively to produce homofascaplysin C, and no other isomers were observed, as shown in Scheme 15.
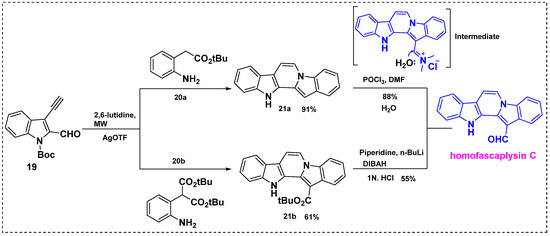
Scheme 15.
Total synthesis of homofascaplysin C via a silver-catalyzed cascade reaction.
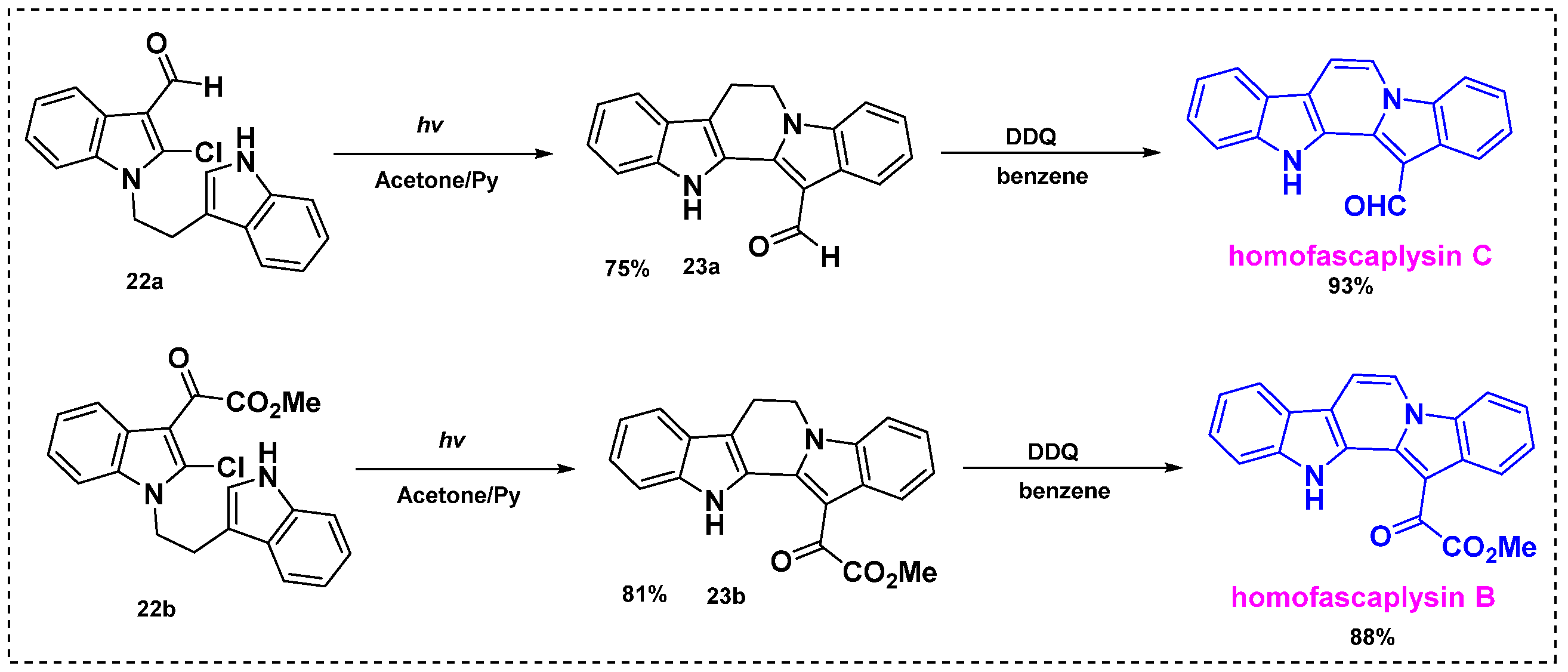
In 2012, Zhang et al. developed a two-step photocyclization/dehydrogenation reaction for the synthesis of homofascaplysins B and C from indole derivatives (Scheme 16) [38]. Irradiation of 2-chloro-1-[2-(indol-3-yl)-ethyl] indole-3-carbaldehyde 22a or 22b in deaerated anhydrous acetone containing pyridine at an ambient temperature for 12–13 h provided photocyclized products 23a in a 75% yield or 23b in an 81% yield. Subsequent dehydrogenation of 23a or 23b by DDQ in benzene for 2–3 h at room temperature gave homofascaplysins C and B in 93% and 88% yields, respectively. The overall yield was 84% for homofascaplysin C from 22a and 84% for homofascaplysin C from 22b. This group also developed a one-step sequential photocyclization and photochemical dehydrogenation reaction in Cu(OAc)2- and air-saturated acetone to synthesize homofascaplysins C and B directly from 22a or 22b, but the yields were very low. Therefore, the two-step photocyclization/dehydrogenation route is more efficient for the synthesis of homofascaplysins B and C and other analogs.
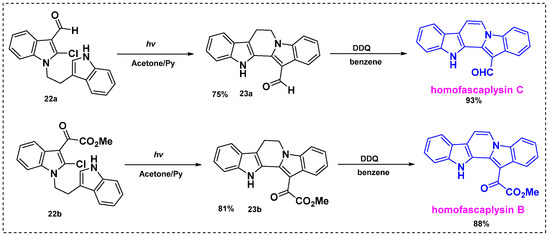
Scheme 16.
Total synthesis of homofascaplysins B and C through photocyclization/dehydrogenation reactions.
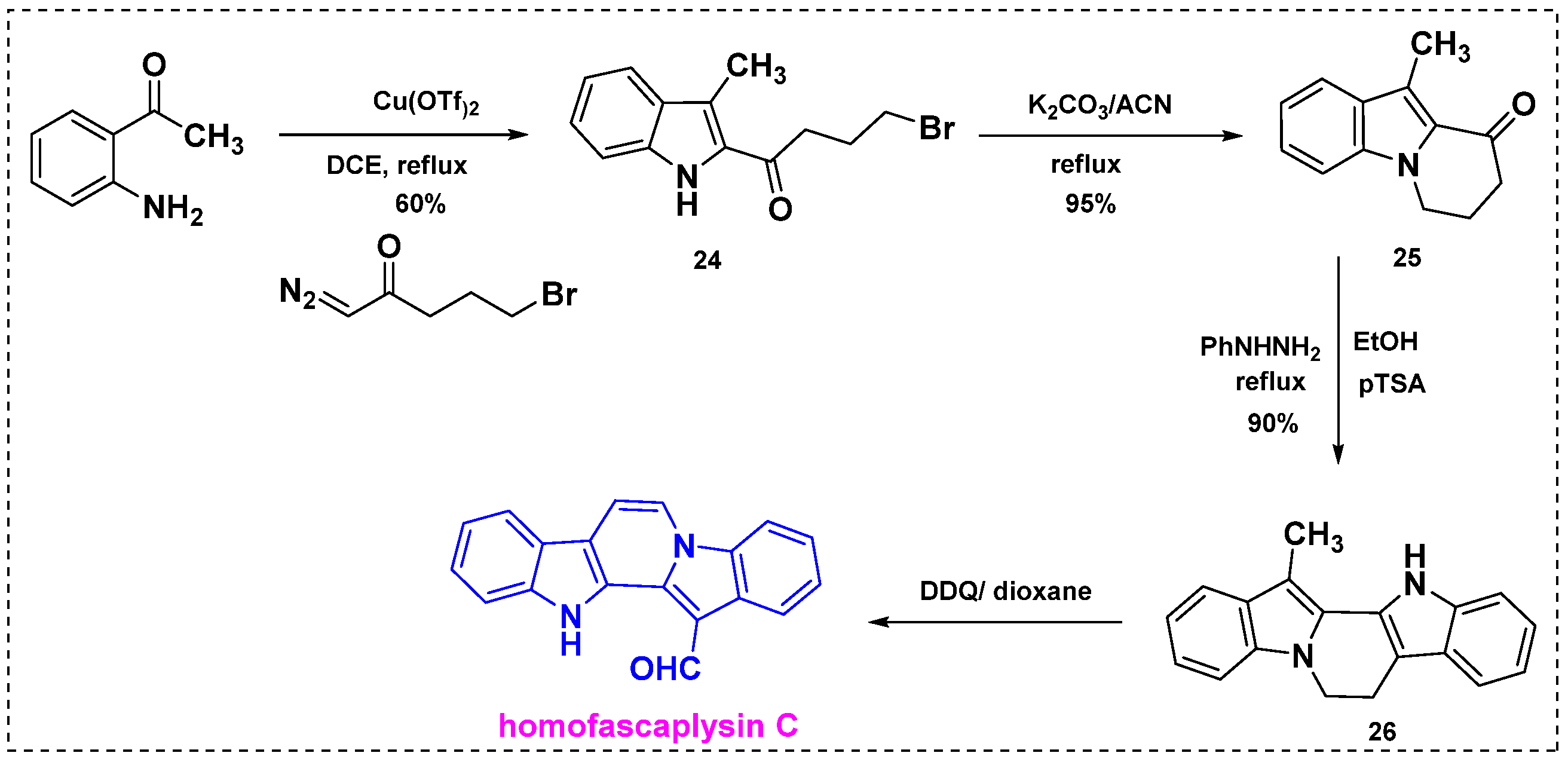
In 2013, Reddy et al. applied an α-diazoketone-based methodology to the formal synthesis of homofascaplysin C [39]. Initially, 2,4,5-trisubstituted pyrrole derivatives were synthesized by coupling α-diazoketones with β-enaminoketones and esters using 10 mol% Cu(OTf)2. In addition, a wide range of 2,3-disubstituted indole derivatives were prepared from α-diazoketones and 2-aminoaryl or alkyl ketones. Finally, homofascaplysin C was synthesized by coupling an alkyl diazoketone with 2-aminoacetophenone (Scheme 17). Specifically, the coupling of 2-aminoacetophenone with bromo butyric diazoketone in the presence of 10% Cu(OTf)2 afforded 2,3-disubstituted indole 24 in a 60% yield. Subsequent intramolecular cyclization of 24 in K2CO3/acetonitrile under reflux afforded compound 25 in a 95% yield. Further treatment of 25 with phenylhydrazine in the presence of pTSA in ethanol at 80 °C gave Fischer indole product 26 regioselectively in a 90% yield. The oxidation of 26 under the conventional DDQ/dioxane conditions reported in the literature provided homofascaplysin C in a 50% yield and a 74% overall yield from 2-amino acetophenone in four steps.
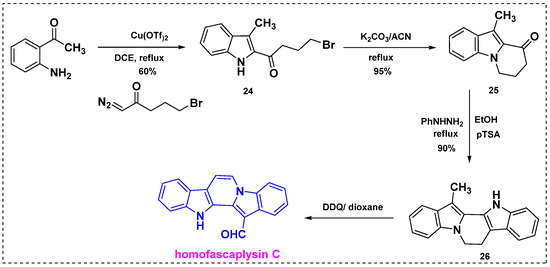
Scheme 17.
Total synthesis of homofascaplysin C from α-diazoketones.
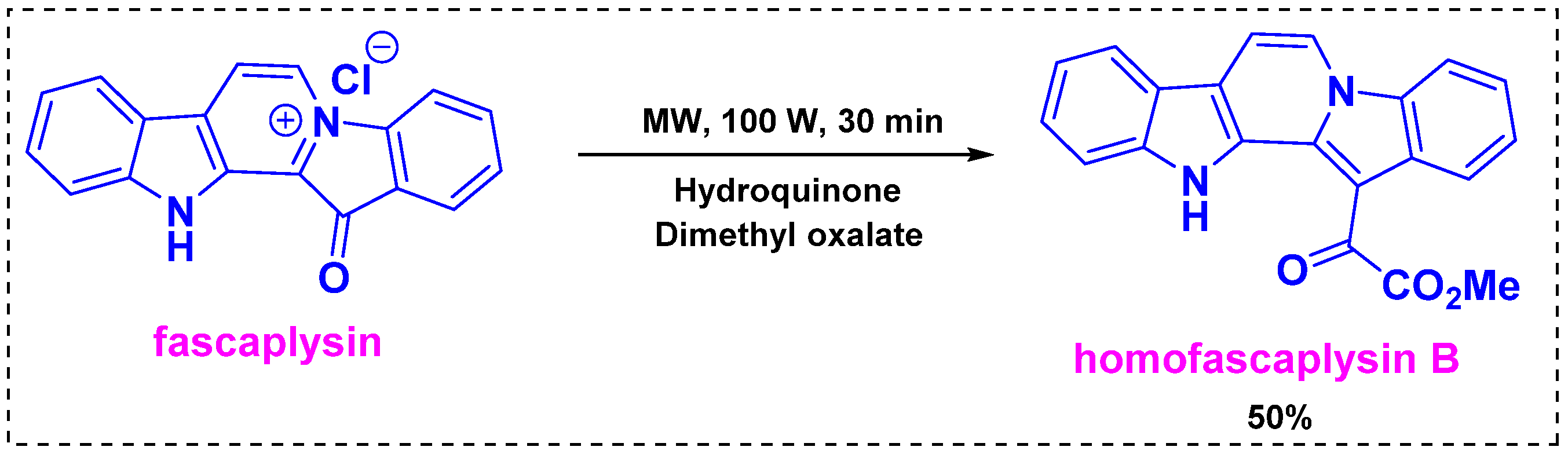
In 2018, Zhidkov et al. developed a one-step transformation of the marine alkaloid fascaplysin into homofascaplysin B using traditional heating or microwave conditions [40]. In the heating method, the reaction of fascaplysin with excess dimethyl oxalate and Na2S2O3 (10 equiv) in an autoclave at 200 °C for 30 min produced homofascaplysin B in a 30% yield. The presence of Na2S2O3 (10 equiv) increased the yield from 24% to 30%. To further improve the yield, microwave irradiation was employed. Under irradiation at 100 W, the reaction of fascaplysin with dimethyl oxalate was complete after 30 min and provided a similar yield of homofascaplysin B. Subsequently, fascaplysin was reacted with excess dimethyl oxalate under microwave irradiation at 100 W for 30 min in the presence of dihydroquinone as a reducing agent, and these optimal conditions provided homofascaplysin B in a 50% yield (Scheme 18). The mechanism of this reaction was not investigated properly in the manuscript, but the reaction proceeded regioselectively.
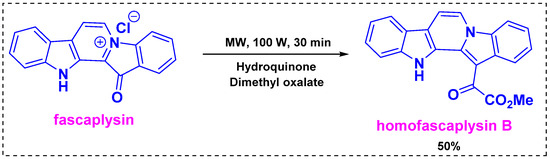
Scheme 18.
One-step transformation of the marine alkaloid fascaplysin into homofascaplysin B.
3. Conclusions
Owing to its anticancer and antimicrobial properties, fascaplysin and its derivatives have potential for therapeutic applications. Moreover, fascaplysin exhibits potent antiplasmodial, antioxidant, and anti-inflammatory activities, highlighting new paths for the development of biomedical applications and paving the way for developing novel fascaplysin analogs. All the above discussed methods except Bharate et al.’s method have some drawbacks like harsh reaction conditions, difficulty scaling up, and limited fascaplysin analog synthesis. The total synthesis developed by Bharate et al. has significant scope for scaling up to the kilogram scale and synthesis of various fascaplysin derivatives using commercially available starting materials.
However, owing to its unusual planar structure, fascaplysin can intercalate in DNA, which results in extreme toxicity and may affect drug product formation. To reduce these toxic effects, the development of related targeted drugs via the structural modification of fascaplysin is essential. Surprisingly, no synthetic methods have yet been reported for homofascaplysin A. In our opinion, development of a new synthetic route to total synthesis of homofascaplysin A and its derivatives is worth further study in terms of its DNA intercalation and toxic effects. This review, which summarizes the advances in the total synthesis of fascaplysin and its metabolites over the past 30 years, will aid in promoting the development of efficient, scalable, and ecofriendly methods for generating these compounds as well as new derivatives.
Author Contributions
This manuscript was written and edited by all authors. All authors have read and agreed to the published version of the manuscript.
Funding
University of Cincinnati, James L. Winkle College of Pharmacy.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
References are given to support the data presented in this study.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Rosillo, M.; Gomez, A.G.; Dominguez, G.; Pérez-Castells, J. Chemistry of Biologically Active β-Carbolines. Targets Heterocycl. Syst. 2008, 12, 212–257. [Google Scholar]
- Lounasmaa, M.; Hanhinen, P.; Westersund, M. The Sarpagine Group of Indole Alkaloids. Alkaloids Chem. Biol. 1999, 52, 103–195. [Google Scholar]
- Hu, J.F.; Haman, R.T.; Hill, R.; Kelly, M. The manzamine alkaloids. Alkaloids Chem. Biol. 2003, 60, 207. [Google Scholar]
- Hesse, M. Alkaloids: Nature’s Curse or Blessing; Wiley–VCH: Weinheim, Germany, 2002. [Google Scholar]
- Bharate, S.B.; Manda, S.; Mupparapu, N.; Battini, N.; Vishwakarma, R.A. Chemistry and Biology of Fascaplysin, a Potent Marine-Derived CDK-4 Inhibitor. MiniRev. Med. Chem. 2012, 12, 650–664. [Google Scholar] [CrossRef] [PubMed]
- Roll, D.M.; Ireland, C.M.; Lu, H.S.M.; Clardy, J. Fascaplysin, an unusual antimicrobial pigment from the marine sponge Fascaplysinopsis sp. J. Org. Chem. 1988, 53, 3276–3278. [Google Scholar] [CrossRef]
- Kirsch, G.; Kong, G.M.; Wright, A.D.; Kaminsky, R. A new bioactive sesterterpene and antiplasmodial alkaloids from the marine sponge hyrtios cf. erecta. J. Nat. Prod. 2000, 63, 825–829. [Google Scholar] [CrossRef] [PubMed]
- Charan, R.D.; McKee, T.C.; Gustafson, K.R.; Pannell, L.K.; Boyd, M.R. Thorectandramine, a novel β-carboline alkaloid from the marine sponge Thorectandra sp. Tetrahedron Lett. 2002, 43, 5201–5204. [Google Scholar] [CrossRef]
- Segraves, N.L.; Robinson, S.J.; Garcia, D.; Said, S.A.; Fu, X.; Schmitz, F.J.; Pietraszkiewicz, H.; Valeriote, F.A.; Crews, P.J. Comparison of fascaplysin and related alkaloids: A study of structures, cytotoxicities, and sources. Nat. Prod. 2004, 67, 783–792. [Google Scholar] [CrossRef]
- Jimenez, C.; Crews, P. Novel marine sponge derived amino acids 13. Additional psammaplin derivatives from Psammaplysilla purpurea. Tetrahedron 1991, 47, 2097–2102. [Google Scholar] [CrossRef]
- Jiménez, C.; Quiñoá, E.; Adamczeski, M.; Hunter, L.M.; Crews, P. Novel sponge-derived amino acids. 12. Tryptophan-derived pigments and accompanying sesterterpenes from Fascaplysinopsis reticulata. J. Org. Chem. 1991, 56, 3403. [Google Scholar] [CrossRef]
- Soni, R.; Muller, L.; Furet, P.; Schoepfer, J.; Stephan, C.; Mecker, S.Z.; Fretz, H.; Chaudhuri, B. Inhibition of cyclin-dependent kinase 4 (Cdk4) by fascaplysin, a marine natural product. Biochem. Biophys. Res. Commun. 2000, 275, 877–884. [Google Scholar] [CrossRef] [PubMed]
- Soni, R.; O’Reilly, T.; Furet, P.; Muller, L.; Stephan, C.; Zumstein-Mecker, S.; Fretz, H.; Fabbro, D.; Chaudhuri, B. Selective In Vivo and In Vitro Effects of a Small Molecule Inhibitor of Cyclin-Dependent Kinase 4. J. Natl. Cancer Inst. 2001, 93, 436–446. [Google Scholar] [CrossRef]
- Huwe, A.; Mazitschek, R.; Giannis, A. Small Molecules as Inhibitors of Cyclin-Dependent Kinases. Angew. Chem. Int. Ed. 2003, 42, 2122–2138. [Google Scholar] [CrossRef]
- Hormann, A.; Chaudhuri, B.; Fretz, H. DNA binding properties of the marine sponge pigment fascaplysin. Bioorg. Med. Chem. 2001, 9, 917–921. [Google Scholar] [CrossRef] [PubMed]
- Alberts, B.; Johnson, A.; Lewis, J.; Raff, M.; Roberts, K.; Walter, P. Molecular Biology of the Cell, 4th ed.; Garland Science: New York, NY, USA, 2002; Chapter 17. [Google Scholar]
- Wang, F.; Chen, H.; Yan, X.; Zheng, Y. Fascaplysin Sensitizes Cells to TRAIL-Induced Apoptosis through Upregulating DR5 Expression. Chin. J. Oceanol. Limnol. 2013, 31, 560–569. [Google Scholar] [CrossRef]
- Oh, T.I.; Lee, Y.M.; Nam, T.J.; Ko, Y.S.; Mah, S.; Kim, J.; Kim, Y.; Reddy, R.H.; Kim, Y.J.; Hong, S.; et al. Fascaplysin Exerts Anti-Cancer Effects through the Downregulation of Survivin and HIF-1α and Inhibition of VEGFR2 and TRKA. Int. J. Mol. Sci. 2017, 29, 2074. [Google Scholar] [CrossRef]
- Hamilton, G. Cytotoxic Effects of Fascaplysin against Small Cell Lung Cancer Cell Lines. Mar. Drugs. 2014, 12, 1377–1389. [Google Scholar] [CrossRef]
- Manda, S.; Sharma, S.; Wani, A.; Joshi, P.; Kumar, V.; Guru, S.K.; Bharate, S.S.; Bhushan, S.; Vishwakarma, R.A.; Kumar, A. Discovery of a Marine-Derived Bis-Indole Alkaloid Fascaplysin, as a New Class of Potent P-Glycoprotein Inducer and Establishment of Its Structure–Activity Relationship. Eur. J. Med. Chem. 2016, 107, 1–11. [Google Scholar] [CrossRef]
- Wang, C.; Wang, S.; Li, H.; Hou, Y.; Cao, H.; Hua, H.H.; Li, D. Marine-Derived Lead Fascaplysin: Pharmacological Activity, Total Synthesis, and Structural Modification. Mar. Drugs. 2023, 21, 226. [Google Scholar] [CrossRef]
- Gul, W.; Hamann, M. Indole alkaloid marine natural products: An established source of cancer drug leads with considerable promise for the control of parasitic, neurological and other diseases. Life Sci. 2005, 78, 442–453. [Google Scholar] [CrossRef]
- Pelcman, B.; Gribble, G.W. Total synthesis of the marine sponge pigment fascaplysin. Tetrahedron Lett. 1990, 31, 2381. [Google Scholar] [CrossRef]
- Rocca, P.; Marsais, F.; Godard, A.; Qukguiner, G. A short synthesis of the antimicrobial marine sponge pigment fascaplysin. Tetrahedron Lett. 1993, 34, 7917–7918. [Google Scholar] [CrossRef]
- Molina, P.; Fresneda, P.M.; Zafra, S.G.; Almendros, P. Iminophosphorane-mediated syntheses of the fascaplysin alkaloid of marine origin and nitramarine. Tetrahedron Lett. 1994, 35, 8851–8854. [Google Scholar] [CrossRef]
- Radchenko, O.S.L.; Novikov, V.L.; Elyakov, G.B. A simple and practical approach to the synthesis of the marine sponge pigment fascaplysin and related compounds. Tetrahedron Lett. 1997, 38, 5339. [Google Scholar] [CrossRef]
- Waldmann, H.; Eberhardt, L.; Wittsteinab, K.; Kumar, K. Silver catalyzed cascade synthesis of alkaloid ring systems: Concise total synthesis of fascaplysin, homofascaplysin C and analogues. Chem. Commun. 2010, 46, 4622–4624. [Google Scholar] [CrossRef]
- Zhidkov, M.E.; Baranova, O.V.; Kravchenko, N.S.; Dubovitskii, S.V. A new method for the synthesis of the marine alkaloid fascaplysin. Tetrahedron Lett. 2010, 51, 6498–6499. [Google Scholar] [CrossRef]
- Bharate, S.B.; Manda, S.; Joshi, P.; Singh, B.; Vishwakarma, R. Total synthesis and anti-cholinesterase activity of marine-derived bis-indole alkaloid fascaplysin. Med. Chem. Commun. 2012, 3, 1098–1103. [Google Scholar] [CrossRef]
- Zhidkov, M.E.; Kaminskii, V.A. A new method for the synthesis of the marine alkaloid fascaplysin based on the microwave-assisted Minisci reaction. Tetrahedron Lett. 2013, 54, 3530–3532. [Google Scholar] [CrossRef]
- Zhidkov, M.E.; Kantemirov, A.V.; Koisevnikov, A.V.; Andin, A.N.; Kuzmich, A.S. Syntheses of the marine alkaloids 6-oxofascaplysin, fascaplysin and their derivatives. Tetrahedron Lett. 2018, 59, 708–711. [Google Scholar] [CrossRef]
- Palani, V.; Perea, M.A.; Gardner, K.E.; Sarpong, R. A pyrone remodeling strategy to access diverse heterocycles: Application to the synthesis of fascaplysin natural products. Chem. Sci. 2021, 12, 1528–1534. [Google Scholar] [CrossRef]
- Tryapkin, O.A.; Kantemirov, A.V.; Dyshlovoy, S.A.; Prassolov, V.S.; Spirin, P.V.; Amsberg, G.V.; Sidorova, M.A.; Zhidkov, M.E. A New Mild Method for Synthesis of Marine Alkaloid Fascaplysin and Its Therapeutically Promising Derivatives. Mar. Drugs 2023, 21, 424. [Google Scholar] [CrossRef]
- Popov, A.M.; Stonik, V.A. Physiological activity of fascaplysine, an unusual pigment from tropic sea sponges. Antibiot. Khimioter. 1991, 36, 12–14. [Google Scholar]
- Gribble, G.W.; Pelcman, B. Total syntheses of the marine sponge pigments fascaplysin and homofascaplysin B and C. J. Org. Chem. 1992, 57, 3636–3642. [Google Scholar] [CrossRef]
- Dubovitskii, S.V. Method for synthesis of 12H-pyrido[1,2-a:3,4-b′]diindoles. Total synthesis of homofascaplysin C. Tetrahedron Lett. 1996, 37, 5207–5208. [Google Scholar] [CrossRef]
- Carter, D.S.; Van Vranken, D.L. Synthesis of Homofascaplysin C and Indolo[2,3-a] carbazole from Ditryptophans. J. Org. Chem. 1999, 64, 8537–8545. [Google Scholar] [CrossRef]
- Dai, Y.; Zhang, W.; Wang, K.; Wang, W.; Zhang, W. Synthesis of homofascaplysin B, C, and analogues by the photocyclization of 3-acyl-2-chloro-1-[2-(indol-3-yl) ethyl] indoles. Tetrahedron 2013, 69, 1912–1918. [Google Scholar] [CrossRef]
- Reddy, B.V.S.; Reddy, M.R.; Rao, Y.G.; Yadav, J.S.; Sridhar, B. Cu(OTf)2-Catalyzed Synthesis of 2,3-Disubstituted Indoles and 2,4,5-Trisubstituted Pyrroles from α-Diazoketones. Org. Lett. 2013, 15, 464–467. [Google Scholar] [CrossRef]
- Zhidkov, M.E.; Sidorova, M.A.; Lyakhova, I.A. One-step transformation of the marine alkaloid fascaplysin into homofascaplysins B and B-1. The first syntheses of 3-bromohomofascaplysin B and 3–bromohomofascaplysin B-1. Tetrahedron Lett. 2018, 59, 1417–1420. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).