
Chemoenzymatic Synthesis of ABC-Type Enantiostructured Triacylglycerols by the Use of the p-Methoxybenzyl Protective Group

 , ,
, ,  and
and
Abstract
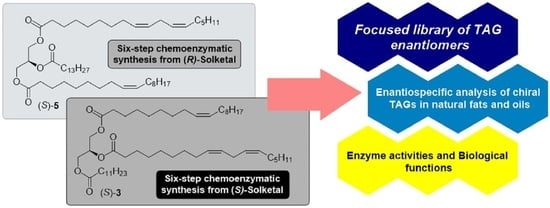
:
1. Introduction
2. Results and Discussion
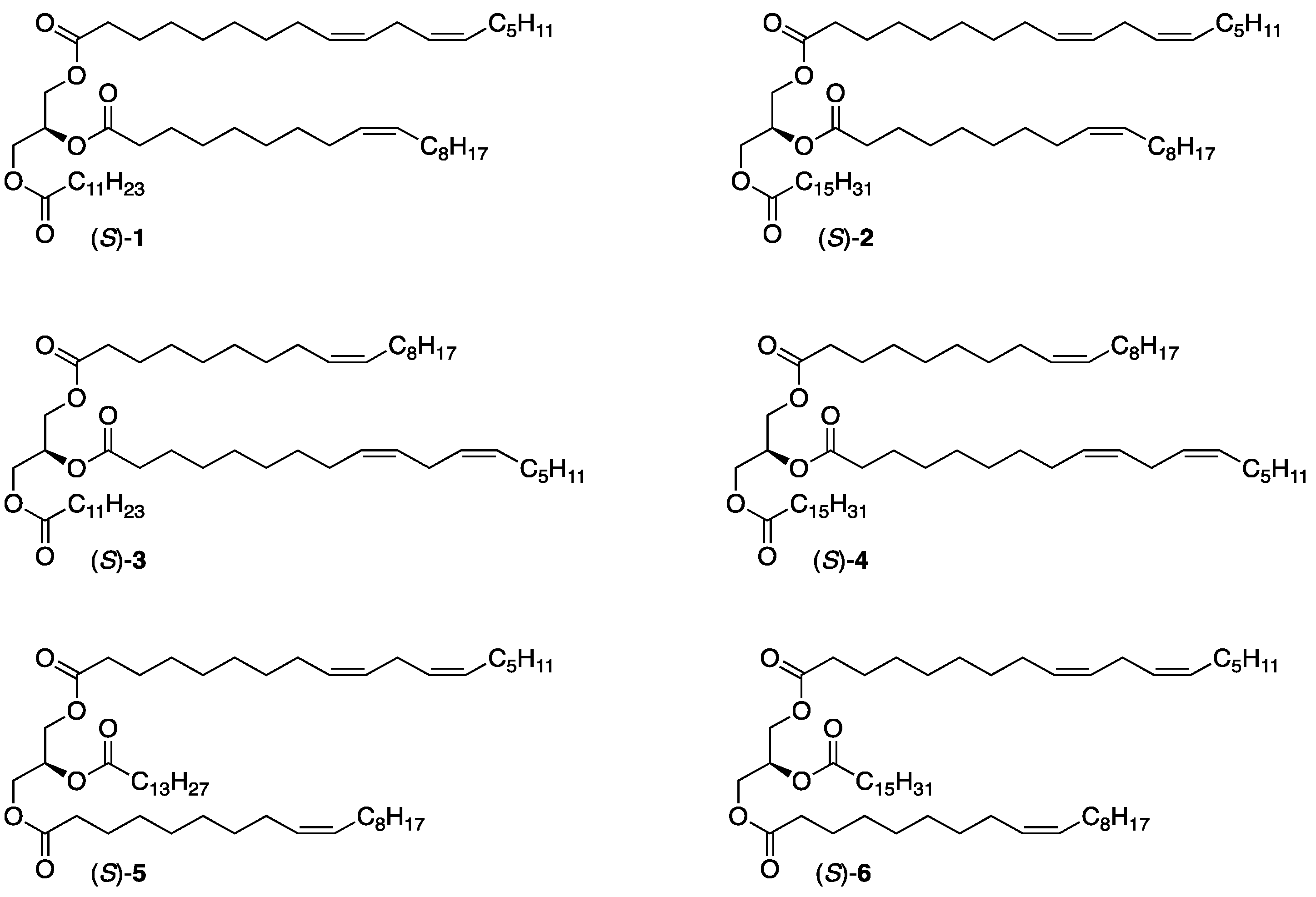
2.1. Chemoenzymatic Synthesis of the SUU’ Subclass Category TAGs, (S)-1 and 2
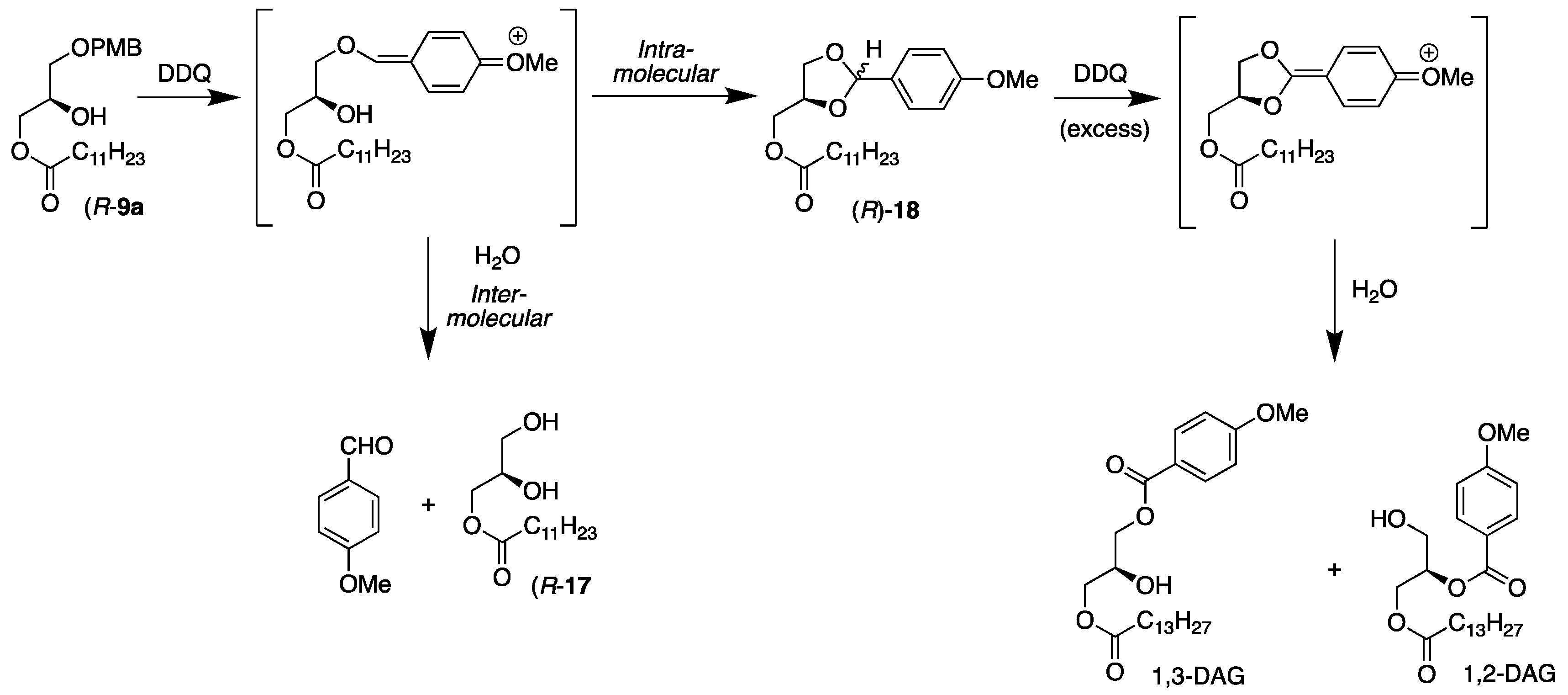
2.1.1. DDQ Deprotection with an Incorporated MUFA Present
2.1.2. DDQ Deprotection with an Incorporated PUFA Present
2.1.3. Chemical Coupling of the Final Fatty Acid
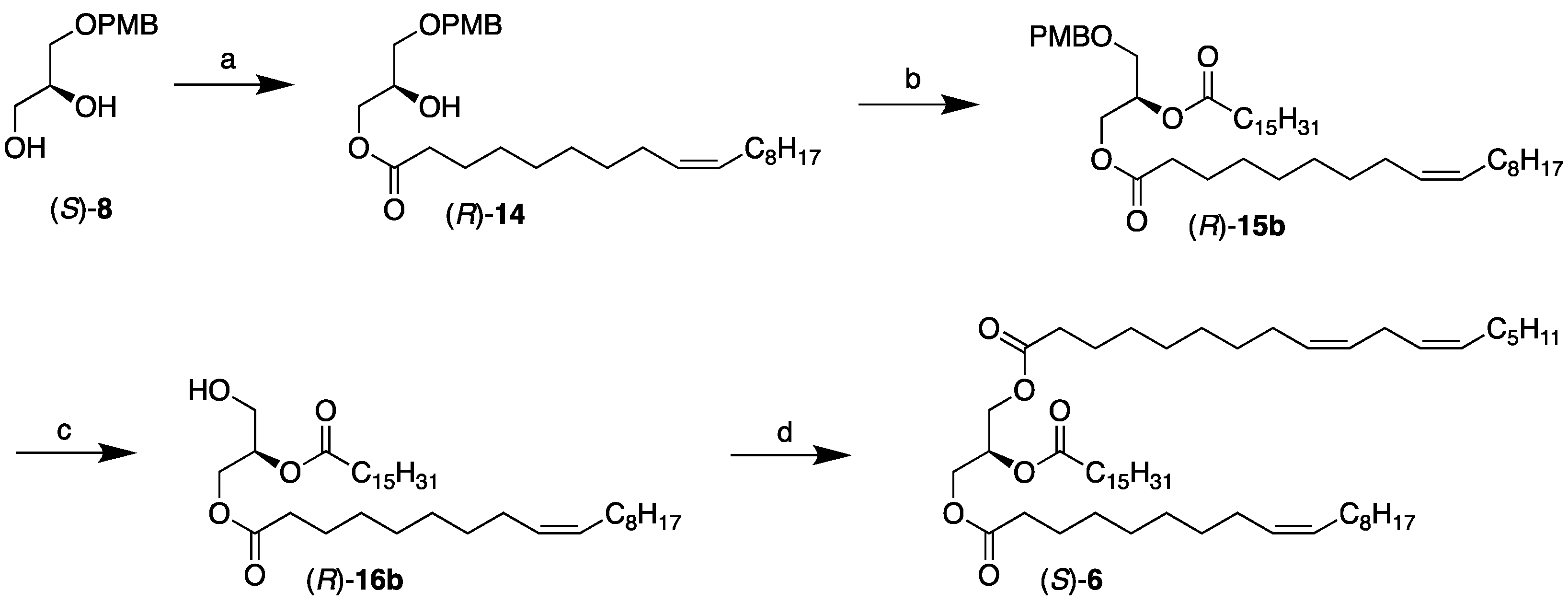
2.2. Chemoenzymatic Synthesis of the USU’ Subclass Category TAGs, (S)-5 and 6
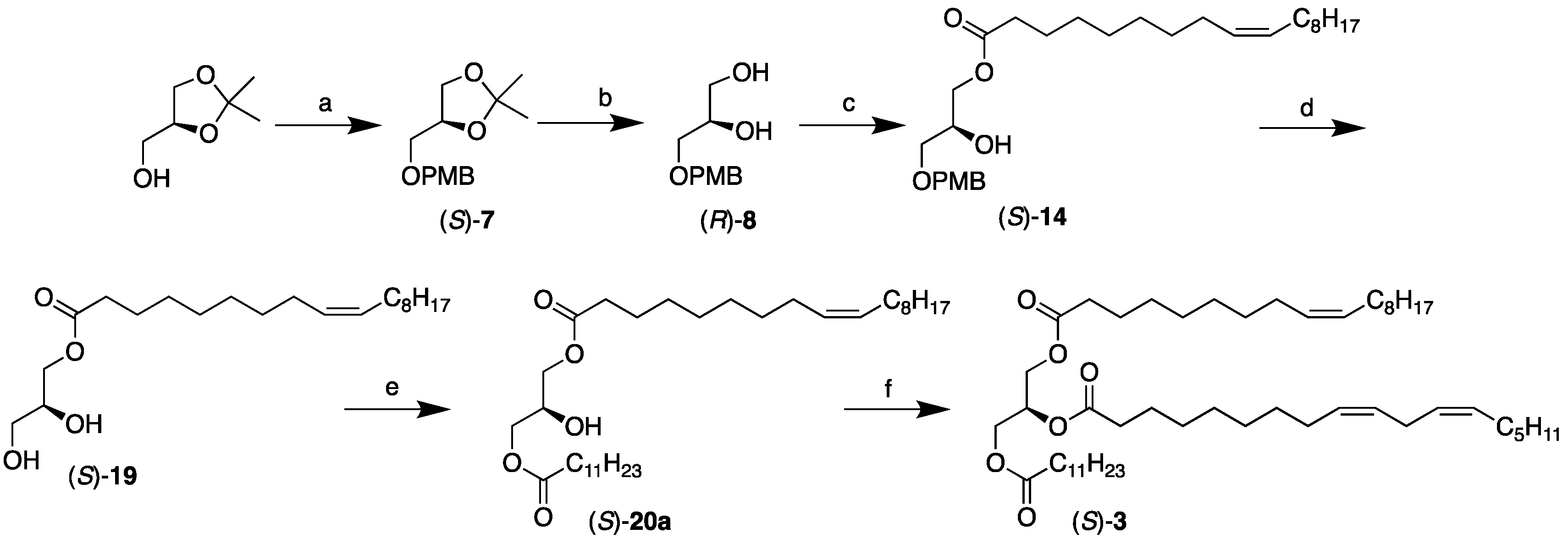
2.3. Chemoenzymatic Synthesis of the Remaining SUU’ Subclass Category TAGs, (S)-3 and 4
2.3.1. Deprotection with an Open sn-2 Position
2.3.2. TAG Synthesis via a Double-Lipase-Step Method
3. Materials and Methods
3.1. General Information
3.2. Synthesis of the SUU’ Subclass Category TAGs, (S)-1 and 2
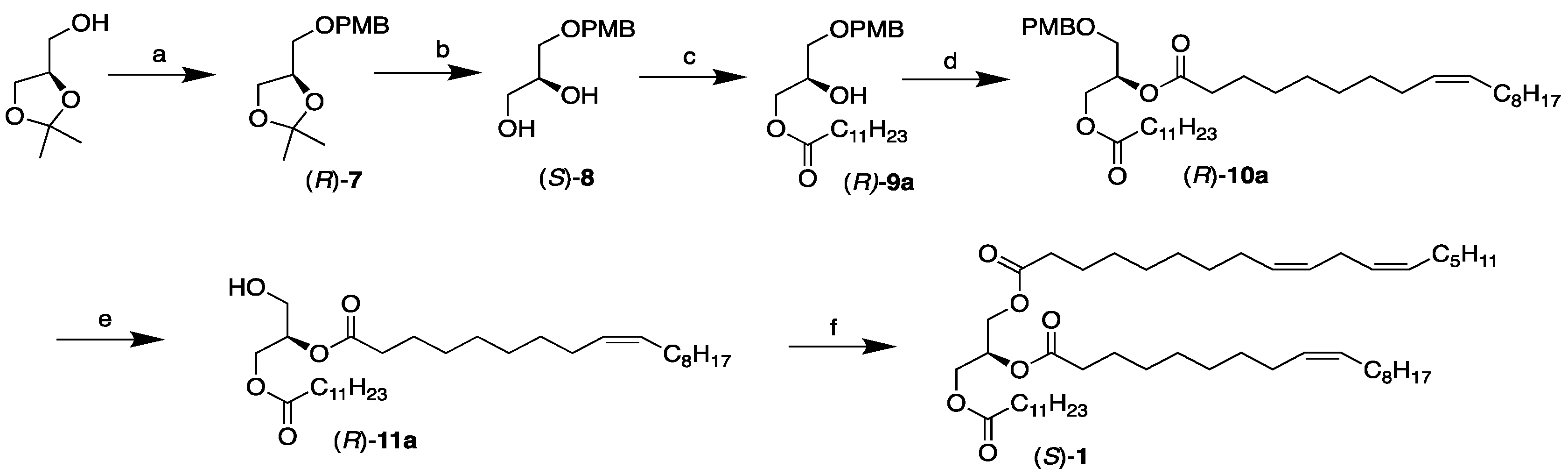
3.2.1. Synthesis of 2,3-O-Isopropylidene-1-O-(p-methoxybenzyl)-sn-glycerol, (R)-7
3.2.2. Synthesis of 1-O-(p-Methoxybenzyl)-sn-glycerol, (S)-8
3.2.3. Synthesis of 3-Dodecanoyl-1-O-(p-methoxybenzyl)-sn-glycerol, (R)-9a
3.2.4. Synthesis of (R)-9b and (R)-9c
3.2.5. Synthesis of 3-Dodecanoyl-1-O-(p-methoxybenzyl)-2-[(9Z)-octadec-9-enoyl)]-sn-glycerol, (R)-10a
3.2.6. Synthesis of (R)-10b, (R)-10c, (R)-10d, and (R)-10e
3.2.7. Synthesis of 3-Dodecanoyl-2-[(9Z)-octadec-9-enoyl)]-sn-glycerol, (R)-11a
3.2.8. Synthesis of 2-[(9Z)-Octadec-9-enoyl)]-3-tetradecanoyl-sn-glycerol, (R)-11b and (R)-11c
3.2.9. Synthesis of 3-Hexadecanoyl-2-[(9Z,12Z)-octadeca-9,12-dienoyl)]-sn-glycerol, (R)-11e
3.2.10. Synthesis of 3-Dodecanoyl-1-[(9Z,12Z)-octadeca-9,12-dienoyl)]-2-[(9Z)-octadec-9-enoyl)]-sn-glycerol, (S)-1
3.2.11. Synthesis of (S)-2, (R)-12a, (R)-12b, (R)-12c, (R)-12d, (R)-12e, and (R)-12f
3.3. Synthesis of the USU’ Subclass Category TAGs, (S)-5 and 6
3.3.1. Synthesis of Oleic Acid Acetoxime Ester, 13
3.3.2. Synthesis of 1-O-(p-Methoxybenzyl)-3-[(9Z)-octadec-9-enoyl)]-sn-glycerol, (R)-14
3.3.3. Synthesis of (R)-15a, (R)-15b, (R)-16a, (R)-16b, (S)-5, and (S)-6
3.4. Synthesis of the SUU’ Subclass Category TAGs, (S)-3 and 4
3.4.1. Synthesis of 3-Dodecanoyl-sn-glycerol, (R)-17
3.4.2. Synthesis of [2-(p-methoxyphenyl)-1,3-dioxolan-4-yl]methyl Dodecanoate, (R)-18
3.4.3. Conversion from Acetal, (R)-18, to (R)-17
3.4.4. Synthesis of (R)-8, (S)-14, (S)-19, (S)-20a, (S)-20b, (S)-3, and (S)-4.
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gunstone, F.D. The Chemistry of Oils and Fats. Sources, Composition, Properties and Uses; Blackwell Publishing: Oxford, UK, 2004; pp. 50–75. [Google Scholar]
- IUPAC-IUB Commission on Biochemical Nomenclature. The nomenclature of lipids. Eur. J. Biochem. 1977, 79, 11–21. [Google Scholar] [CrossRef]
- Lísa, M.; Holčapek, M. Characterization of triacylglycerol enantiomers using chiral HPLC/APCI-MS and synthesis of enantiomeric triacylglycerols. Anal. Chem. 2013, 85, 1852–1859. [Google Scholar] [CrossRef] [PubMed]
- Kalpio, M.; Nylund, M.; Linderborg, K.M.; Yang, B.; Kristinsson, B.; Haraldsson, G.G.; Kallio, H. Enantioselective chromatography in analysis of triacylglycerols common in edible fats and oils. Food Chem. 2015, 172, 718–724. [Google Scholar] [CrossRef] [PubMed]
- Řezanka, T.; Kolouchová, I.; Nedbalová, L.; Sigler, K. Enantiomeric separation of triacylglycerols containing very long chain fatty acids. J. Chromatogr. A 2018, 1557, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Nagai, T.; Kinoshita, T.; Kasamatsu, E.; Yoshinaga, K.; Mizobe, H.; Yoshida, A.; Itabashi, Y.; Gotoh, N. Simultaneous separation of triacylglycerol enantiomers and positional isomers by chiral high performance liquid chromatography coupled with mass spectrometry. J. Oleo Sci. 2019, 68, 1019–1026. [Google Scholar] [CrossRef] [PubMed]
- Kalpio, M.; Magnússon, J.D.; Gudmundsson, H.G.; Linderborg, K.M.; Kallio, H.; Haraldsson, G.G.; Yang, B. Synthesis and enantiospecific analysis of enantiostructured triacylglycerols containing n-3 polyunsaturated fatty acids. Chem. Phys. Lipids 2020, 231, 104937. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.J.; Zhou, X.H.; Han, B.; Yu, Z.; Yi, H.X.; Jiang, S.L.; Li, Y.Y.; Pan, J.C.; Zhang, L.W. Regioisomeric and enantiomeric analysis of primary triglycerides in human milk by silver ion and chiral HPLC atmospheric pressure chemical ionization-MS. J. Dairy Sci. 2020, 103, 7761–7774. [Google Scholar] [CrossRef]
- Kallio, H.; Yli-Jokipii, K.; Kurvinen, J.P.; Sjövall, O.; Tahvonen, R. Regioisomerism of Triacylglycerols in Lard, Tallow, Yolk, Chicken Skin, Palm Oil, Palm Olein, Palm Stearin, and a Transesterified Blend of Palm Stearin and Coconut Oil Analyzed by Tandem Mass Spectrometry. J. Agric. Food Chem. 2001, 49, 3363–3369. [Google Scholar] [CrossRef]
- Sun, C.; Wei, W.; Zou, X.; Huang, J.; Jin, Q.; Wang, X. Evaluation of Triacylglycerol Composition in Commercial Infant Formulas on the Chinese Market: A Comparative Study Based on Fat Source and Stage. Food Chem. 2018, 252, 154–162. [Google Scholar] [CrossRef]
- Zhang, X.; Qi, C.; Zhang, Y.; Wei, W.; Jin, Q.; Xu, Z.; Tao, G.; Wang, X. Identification and Quantification of Triacylglycerols in Human Milk Fat Using Ultra-Performance Convergence Chromatography and Quadrupole Time-of-Flight Mass Spectrometery with Supercritical Carbon Dioxide as a Mobile Phase. Food Chem. 2019, 275, 712–720. [Google Scholar] [CrossRef]
- Kristinsson, B.; Linderborg, K.M.; Kallio, H.; Haraldsson, G.G. Synthesis of enantiopure structured triacylglycerols. Tetrahedron Asymmetry 2014, 25, 125–132. [Google Scholar] [CrossRef]
- Gudmundsson, H.G.; Linderborg, K.M.; Kallio, H.; Yang, B.; Haraldsson, G.G. Synthesis of Enantiopure ABC-Type Triacylglycerols. Tetrahedron 2020, 76, 130813. [Google Scholar] [CrossRef]
- Pariyani, R.; Zhang, Y.; Haraldsson, G.G.; Chen, K.; Linderborg, K.M.; Yang, B. Metabolomic investigation of brain and liver in rats fed docosahexaenoic acid in regio- and enantiopure triacylglycerols. Mol. Nutr. Food Res. 2024, 68, 2300341. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, A.; Linderborg, K.M.; Zhao, A.; Kallio, H.; Haraldsson, G.G.; Zhang, Y.; Yang, B. Influence of dietary triacylglycerol structure on the accumulation of docosahexaenoic acid [22:6(n-3)] in organs in a short-term feeding trial with mildly omega-3 deficient rats. Mol. Nutr. Food Res. 2024, 68, 2300635. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Kalpio, M.; Tao, L.; Haraldsson, G.G.; Gudmundsson, H.G.; Fang, X.; Linderborg, K.M.; Zhang, Y.; Yang, B. Metabolic fate of DHA from regio- and stereospecific positions of triacylglycerols in a long-term feeding trial in rats. Food Res. Intern. 2023, 174, 113626. [Google Scholar] [CrossRef] [PubMed]
- Beltrame, G.; Ahonen, E.; Damerau, A.; Gudmundsson, H.G.; Haraldsson, G.G.; Linderborg, K.M. Lipid structure influences the digestion and oxidation behavior of docosahexaenoic and eicosapentaenoic acids in simulated digestion system. J. Agric. Food Chem. 2023, 71, 10087–10096. [Google Scholar] [CrossRef]
- Damerau, A.; Ahonen, E.; Kortesniemi, M.; Gudmundsson, H.G.; Yang, B.; Haraldsson, G.G.; Linderborg, K.M. Docosahexaenoic acid in regio- and enantiopure triacylglycerols: Oxidative stability and influence of chiral antioxidant. Food Chem. 2023, 402, 134271. [Google Scholar] [CrossRef] [PubMed]
- Ahonen, E.; Damerau, A.; Linderborg, K.M. Antioxidative effect of dihydrosphingosine (d18:0) and α-tocopherol on tridocosahexaenoin (DHA-TAG). J. Agric. Food Chem. 2023, 71, 14769–14781. [Google Scholar] [CrossRef]
- Greene, T.W.; Wuts, P.G.M. Protection for the hydroxyl group, including 1,2- and 1,3-diols. In Greene’s Protective Groups in Organic Synthesis, 4th ed.; Greene, T.W., Wuts, P.G.M., Eds.; John Wiley and sons: Hoboken, NJ, USA, 2006; pp. 16–366. [Google Scholar]
- Kociensky, P.J. Protecting Groups, 3rd ed.; Thieme Verlag: Stuttgart, Germany, 2005; 279p. [Google Scholar]
- Kodali, D.R.; Tercyak, A.; Fahey, D.A.; Small, D.M. Acyl migration in 1,2-dipalmitoyl-sn-glycerol. Chem. Phys. Lipids 1990, 52, 163–170. [Google Scholar] [CrossRef]
- Laszlo, J.A.; Compton, D.L.; Vermillion, K.E. Acyl migration kinetics of vegetable oil 1,2-diacylglycerols. J. Am. Oil Chem. Soc. 2008, 85, 307–312. [Google Scholar] [CrossRef]
- Compton, D.L.; Laszlo, J.A.; Appell, M.; Vermillion, K.E.; Evans, K.O. Influence of fatty acid desaturation on spontaneous acyl migration in 2-monoacylglycerols. J. Am. Oil Chem. Soc. 2012, 89, 2259–2267. [Google Scholar] [CrossRef]
- Yadav, J.S.; Satyanarayana, M.; Raghavendra, S.; Balanarsaiah, E. Chemoselective hydrolysis of terminal isopropylidene acetals in acetonitrile using molecular iodine as a mild and efficient catalyst. Tetrahedron Lett. 2005, 46, 8745–8748. [Google Scholar] [CrossRef]
- Sun, J.; Dong, Y.; Cao, L.; Wang, X.; Wang, S.; Hu, Y. Highly efficient chemoselective deprotection of O,O-acetals and O,O-ketals catalyzed by molecular iodine in acetone. J. Org. Chem. 2004, 69, 8932–8934. [Google Scholar] [CrossRef] [PubMed]
- Halldorsson, A.; Magnusson, C.D.; Haraldsson, G.G. Chemoenzymatic synthesis of structured triacylglycerols by highly regioselective acylation. Tetrahedron 2003, 59, 9101–9109. [Google Scholar] [CrossRef]
- Magnusson, C.D.; Haraldsson, G.G. Chemoenzymatic synthesis of symmetrically structured triacylglycerols possessing short-chain fatty acids. Tetrahedron 2010, 66, 2728–2731. [Google Scholar] [CrossRef]
- Christie, W.W. Lipid Analysis. Isolation, Separation, Identification and Structural Analysis of Lpids, 3rd ed.; The Oily Press: High Wycombe, Buckinghamshire, UK, 2003; Chapter 4; pp. 105–135. [Google Scholar]
- Thomas, A.E.; Scharoun, J.E.; Ralston, H. Quantitative estimation of isomeric monoglycerides by thin-layer chromatography. J. Am. Oil Chem. Soc. 1965, 42, 789–792. [Google Scholar] [CrossRef]
- Oikawa, Y.; Yoshioka, T.; Yonemitsu, O. Specific removal of o-methoxybenzyl protection by DDQ oxidation. Tetrahedron Lett. 1982, 23, 885–888. [Google Scholar] [CrossRef]
- Frankel, E.N. Lipid oxidation. Progr. Lipid Res. 1980, 19, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Frankel, E.N. Volatile lipid oxidation products. Progr. Lipid Res. 1982, 22, 1–33. [Google Scholar] [CrossRef]
- Frankel, E.N. Lipid oxidation: Mechanisms, products and biological significance. J. Am. Oil Chem. Soc. 1984, 61, 1908–1917. [Google Scholar] [CrossRef]
- Porter, N.A.; Caldwell, S.E.; Mills, K.A. Mechanisms of free radical oxidation of unsaturated lipids. Lipids 1995, 30, 277–290. [Google Scholar] [CrossRef] [PubMed]
- Schlenk, W., Jr. Synthesis and analysis of optically active triglycerides. J. Am. Oil Chem. Soc. 1965, 42, 945–957. [Google Scholar] [CrossRef] [PubMed]
- Mislow, K.; Bickart, P. An epistemological note on chirality. Isr. J. Chem. 1976/1977, 15, 1–6. [Google Scholar] [CrossRef]
- Foss, B.J.; Sliwka, H.-R.; Partali, V.; Köpsel, C.; Mayer, B.; Martin, H.-D.; Zsila, F.; Bikadi, Z.; Simonyi, M. Optically active oligomer units in aggregates of a highly unsaturated optically inactive carotenid phospholipid. Chem. Eur. J. 2005, 11, 4103–4108. [Google Scholar] [CrossRef] [PubMed]
- Mislow, K. Molecular chirality. In Topics in Stereochemistry; Denmark, S.E., Ed.; Wiley & Sons: New York, NY, USA, 1999; Volume 22, pp. 1–82. [Google Scholar]
- Magnusson, C.D.; Haraldsson, G.G. Activation of n-3 polyunsaturated fatty acids as oxime esters: A novel approach for their exclusive incorporation into the primary alcoholic positions of the glycerol moiety by lipase. Chem. Phys. Lipids 2012, 165, 712–720. [Google Scholar] [CrossRef]
- Gudmundsdottir, A.V.; Hansen, K.-A.; Magnusson, C.D.; Haraldsson, G.G. Synthesis of reversed structured triacylglycerols possessing EPA and DHA at their terminal positions. Tetrahedron 2015, 71, 8544–8550. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | sn-1 | sn-2 | sn-3 | Yield | [α]20D |
|---|---|---|---|---|---|
| (R)-9a | PMB | OH | 12:0 | >99% | −1.28 |
| (R)-9b | PMB | OH | 14:0 | 91% | −1.93 |
| (R)-9c | PMB | OH | 16:0 | 94% | −1.18 |
| (R)-14 | PMB | OH | 18:1 | 87% | −0.72 |
| Compound | sn-1 | sn-2 | sn-3 | Yield | [α]20D |
|---|---|---|---|---|---|
| (R)-10a | PMB | 18:1 | 12:0 | 99% | −6.77 |
| (R)-10b | PMB | 18:1 | 14:0 | 99% | −6.61 |
| (R)-10c | PMB | 18:1 | 16:0 | 88% | −6.24 |
| (R)-10d | PMB | 18:2 | 12:0 | 88% | −6.05 |
| (R)-10e | PMB | 18:2 | 16:0 | 89% | −6.80 |
| (R)-15a | PMB | 14:0 | 18:1 | 94% | −6.86 |
| (R)-15b | PMB | 16:0 | 18:1 | 91% | −6.75 |
| Compound | sn-1 | sn-2 | sn-3 | Yield | [α]20D |
|---|---|---|---|---|---|
| (R)-11a | OH | 18:1 | 12:0 | 91% | +2.43 |
| (R)-11b | OH | 18:1 | 14:0 | 92% | +2.09 |
| (R)-11c | OH | 18:1 | 16:0 | 85% | +2.10 |
| (R)-16a | OH | 14:0 | 18:1 | 95% | +2.41 |
| (R)-16b | OH | 16:0 | 18:1 | 91% | +2.39 |
| Compound | sn-1 | sn-2 | sn-3 | Yield | [α]20D |
|---|---|---|---|---|---|
| (S)-1 | 18:2 | 18:1 | 12:0 | 85% | +0.03 |
| (S)-2 | 18:2 | 18:1 | 16:0 | 92% | −0.01 |
| (S)-5 | 18:2 | 14:0 | 18:1 | 94% | +0.05 |
| (S)-6 | 18:2 | 16:0 | 18:1 | 95% | −0.02 |
| (R)-12a | 12:0 | 18:1 | 14:0 | 90% | +0.04 |
| (R)-12b | 10:0 | 18:1 | 16:0 | 98% | +0.05 |
| (R)-12c | 12:0 | 18:1 | 16:0 | 99% | +0.02 |
| (R)-12d | 14:0 | 18:1 | 16:0 | 96% | −0.02 |
| (S)-12e | 20:0 | 18:1 | 16:0 | 97% | +0.01 |
| (R)-12f | 12:0 | 18:2 | 16:0 | 86% | +0.09 |
| Compound | sn-1 | sn-2 | sn-3 | Yield | [α]20D |
|---|---|---|---|---|---|
| (S)-7 | isopropylidene | PMB | 76% | +0.03 | |
| (R)-8 | OH | OH | PMB | 92% | −1.84 |
| (S)-14 | 18:1 | OH | PMB | 74% | +0.79 |
| (S)-19 | 18:1 | OH | OH | 70% | +1.05 |
| (S)-20a | 18:1 | OH | 12:0 | 83% | −1.28 |
| (S)-20b | 18:1 | OH | 16:0 | 87% | −0.76 |
| (S)-3 | 18:1 | 18:2 | 12:0 | 97% | +0.08 |
| (S)-4 | 18:1 | 18:2 | 16:0 | 96% | +0.09 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Haraldsdottir, H.; Gudmundsson, H.G.; Linderborg, K.M.; Yang, B.; Haraldsson, G.G. Chemoenzymatic Synthesis of ABC-Type Enantiostructured Triacylglycerols by the Use of the p-Methoxybenzyl Protective Group. Molecules 2024, 29, 1633. https://doi.org/10.3390/molecules29071633
Haraldsdottir H, Gudmundsson HG, Linderborg KM, Yang B, Haraldsson GG. Chemoenzymatic Synthesis of ABC-Type Enantiostructured Triacylglycerols by the Use of the p-Methoxybenzyl Protective Group. Molecules. 2024; 29(7):1633. https://doi.org/10.3390/molecules29071633
Chicago/Turabian StyleHaraldsdottir, Hafdis, Haraldur G. Gudmundsson, Kaisa M. Linderborg, Baoru Yang, and Gudmundur G. Haraldsson. 2024. "Chemoenzymatic Synthesis of ABC-Type Enantiostructured Triacylglycerols by the Use of the p-Methoxybenzyl Protective Group" Molecules 29, no. 7: 1633. https://doi.org/10.3390/molecules29071633
APA StyleHaraldsdottir, H., Gudmundsson, H. G., Linderborg, K. M., Yang, B., & Haraldsson, G. G. (2024). Chemoenzymatic Synthesis of ABC-Type Enantiostructured Triacylglycerols by the Use of the p-Methoxybenzyl Protective Group. Molecules, 29(7), 1633. https://doi.org/10.3390/molecules29071633