

Metalloid–Organic Intermolecular Complexes with Charge State-Controlled Conformations


Abstract
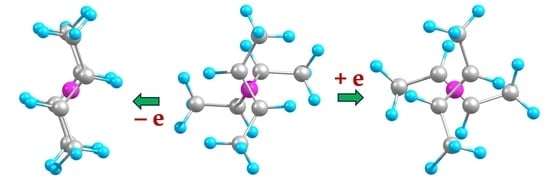
1. Introduction
2. Results and Discussion
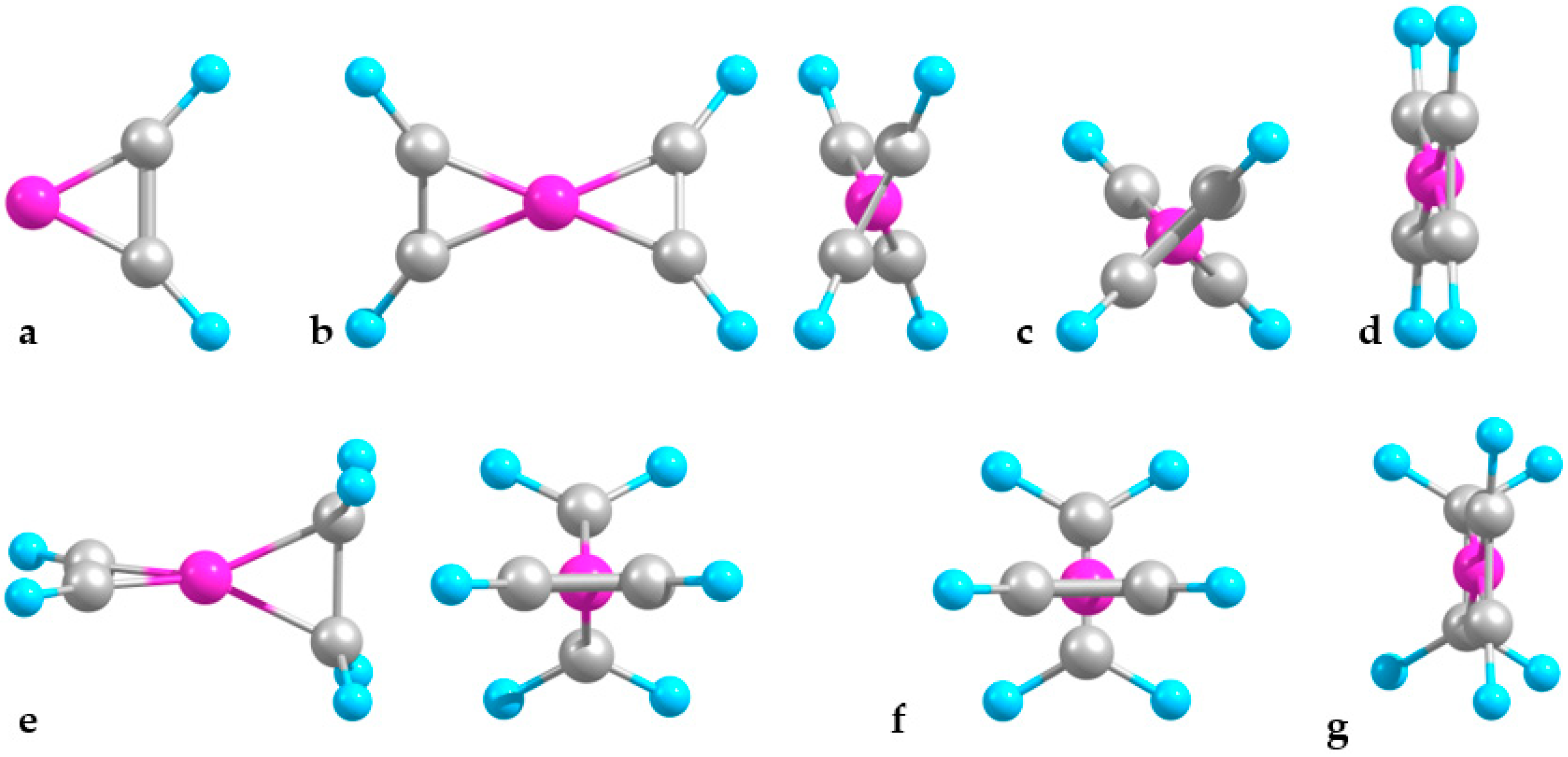
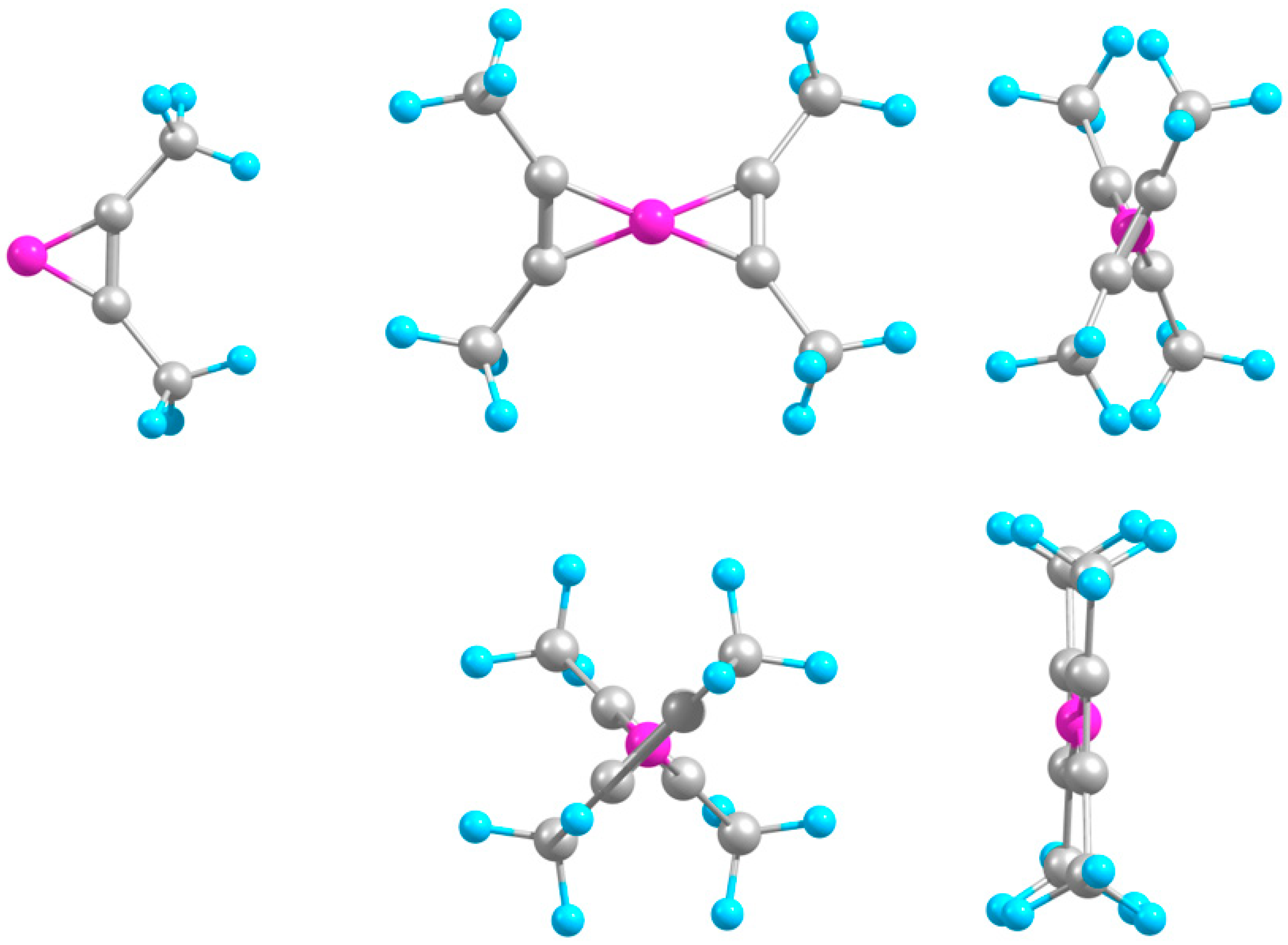
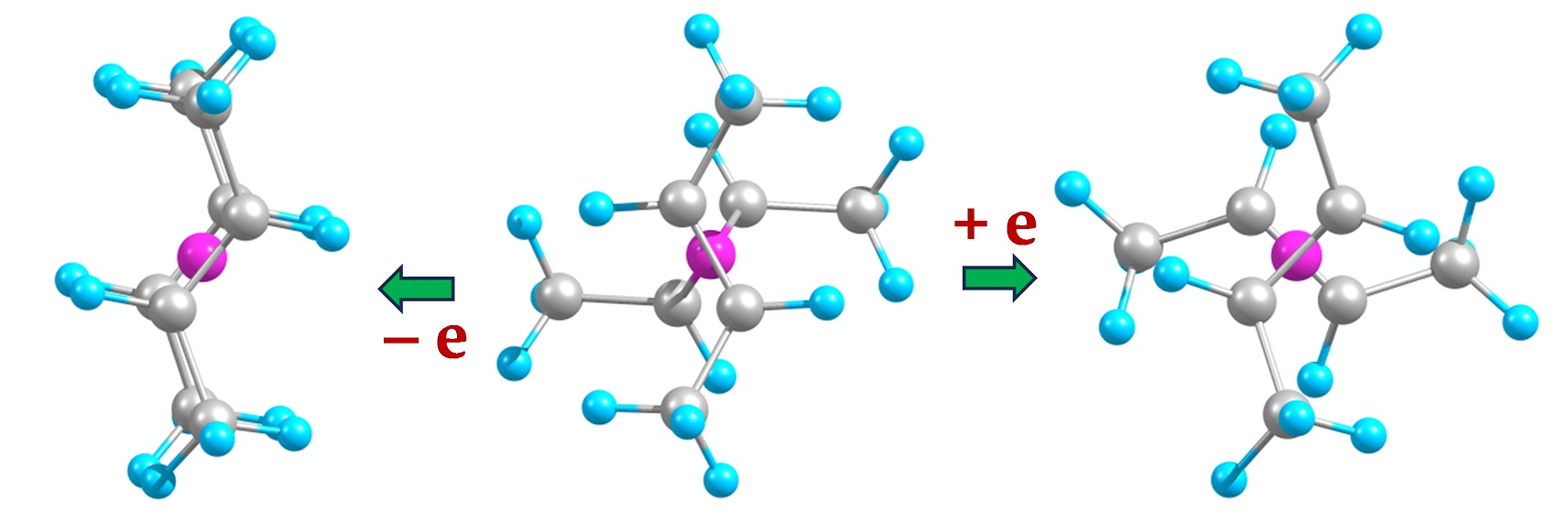
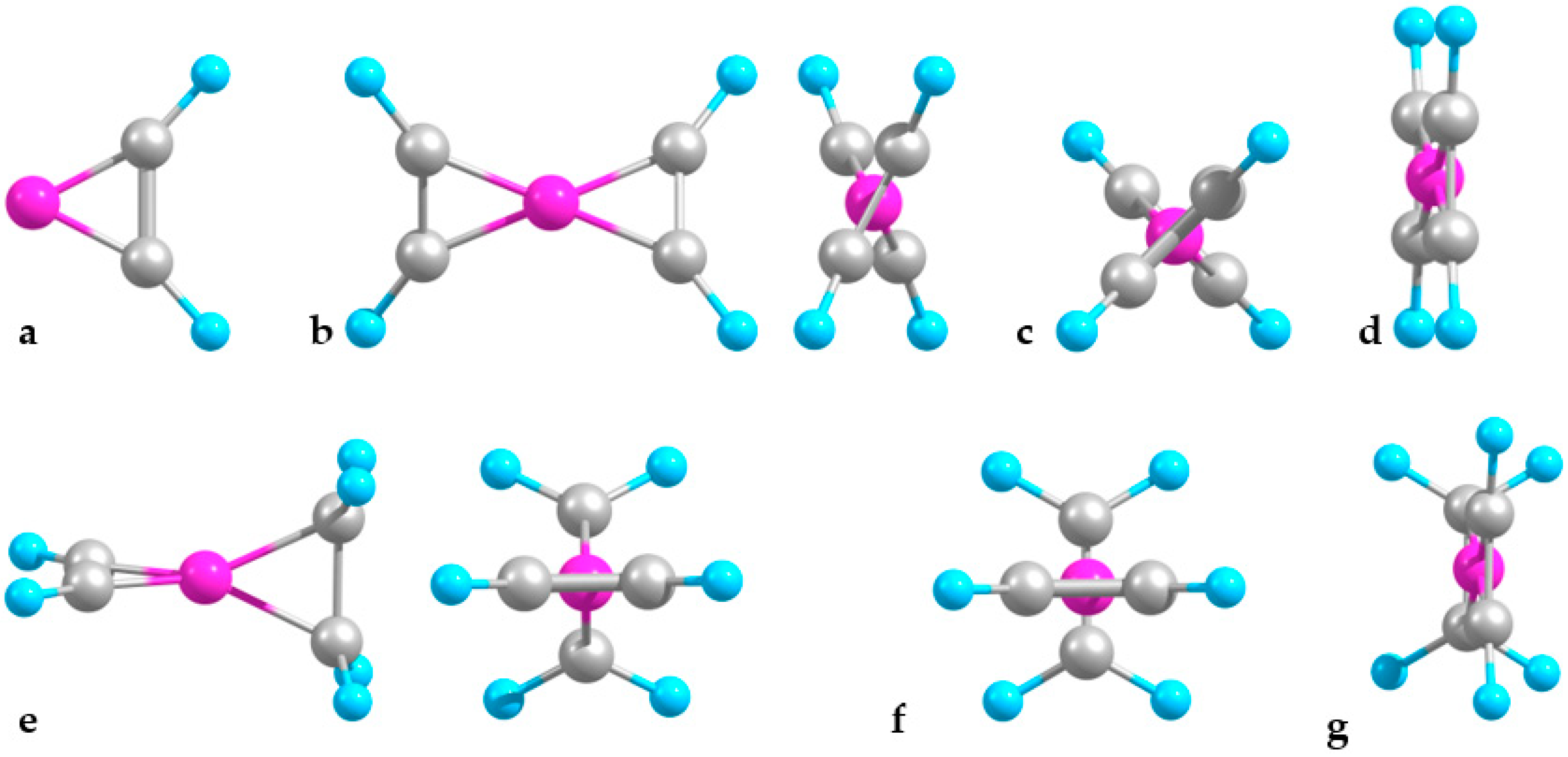
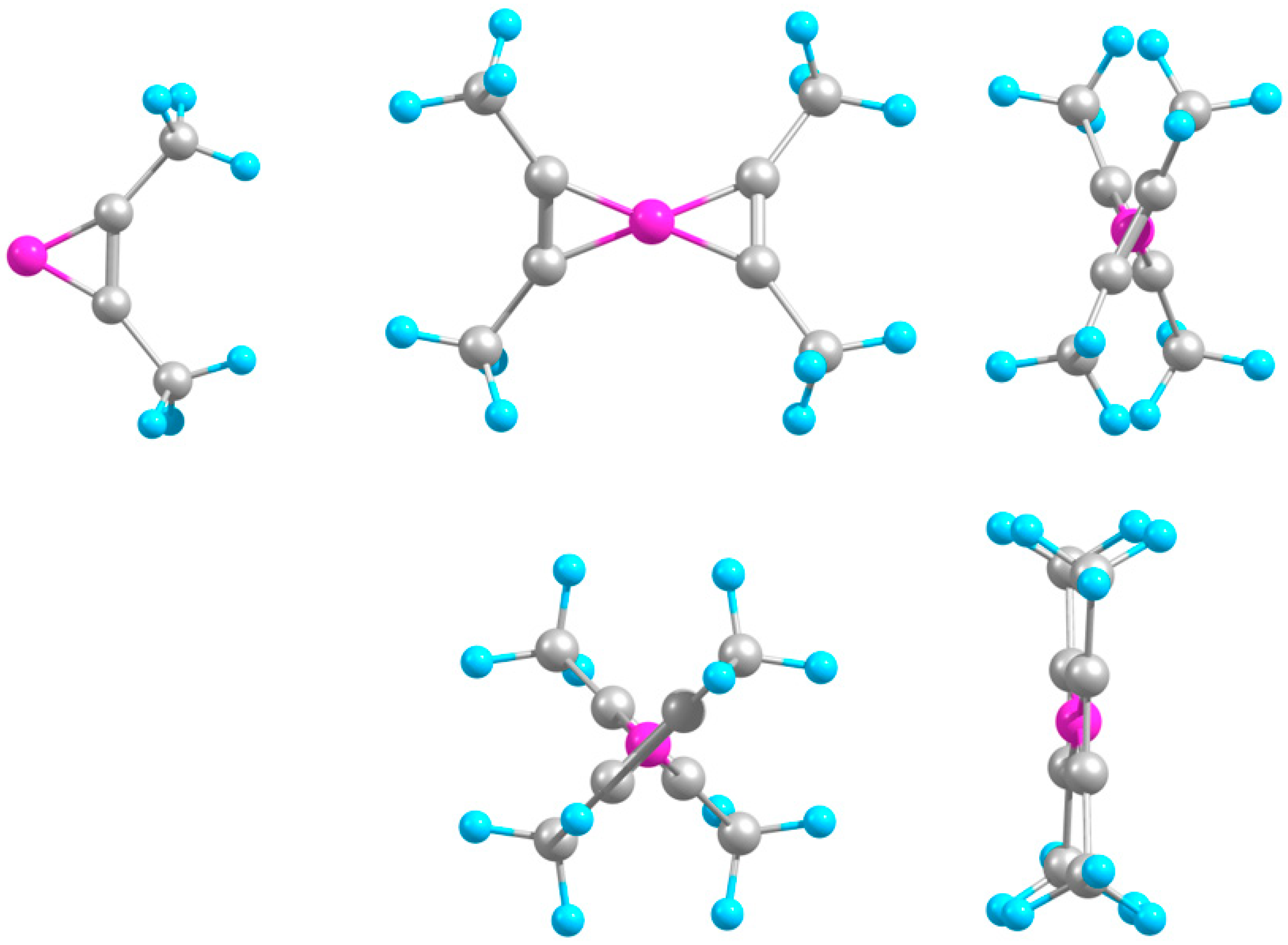
2.1. Minimal Systems
2.1.1. Structures and Stabilities
2.1.2. Charge Distributions
2.1.3. Ionization Energies and Electron Affinities
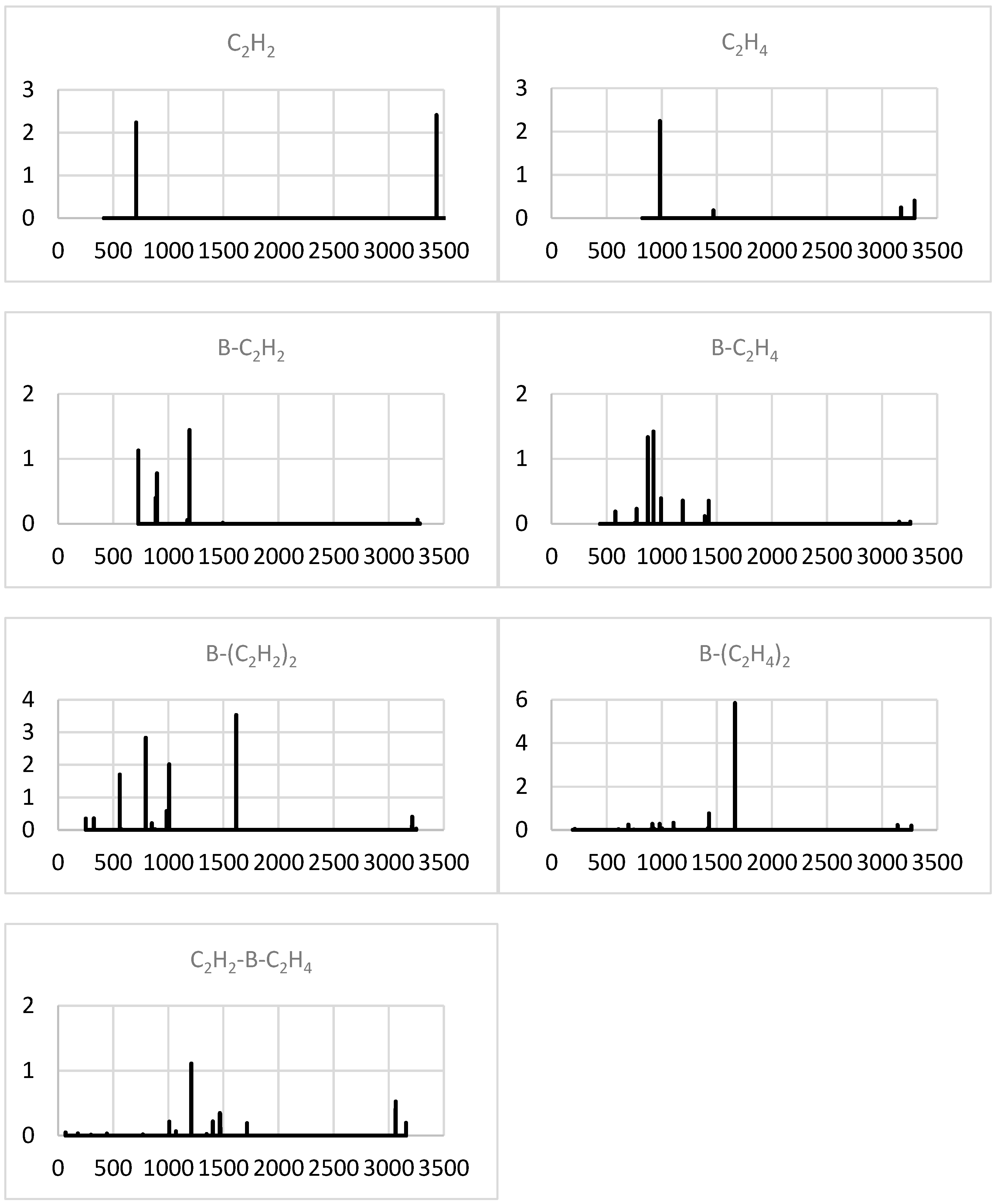
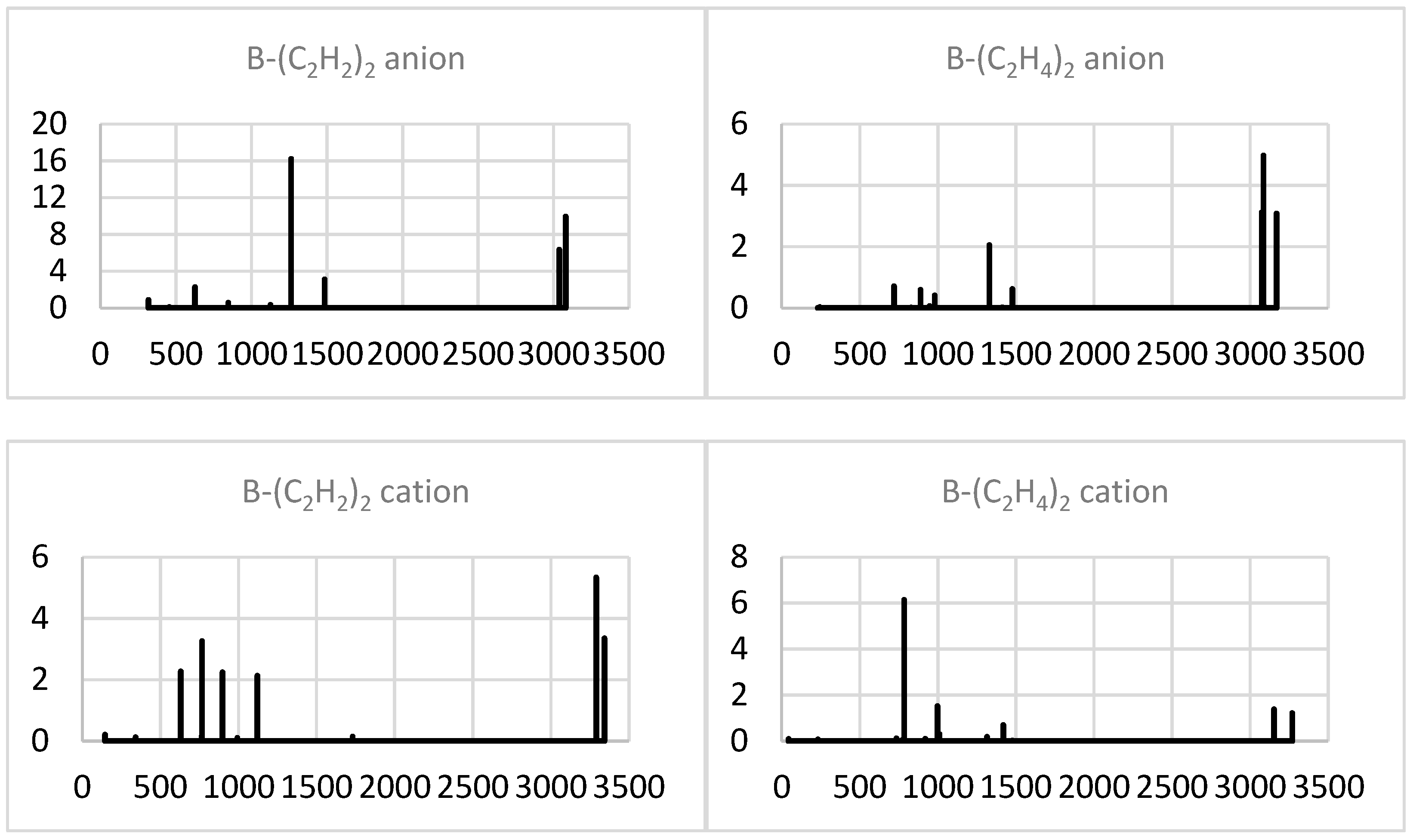
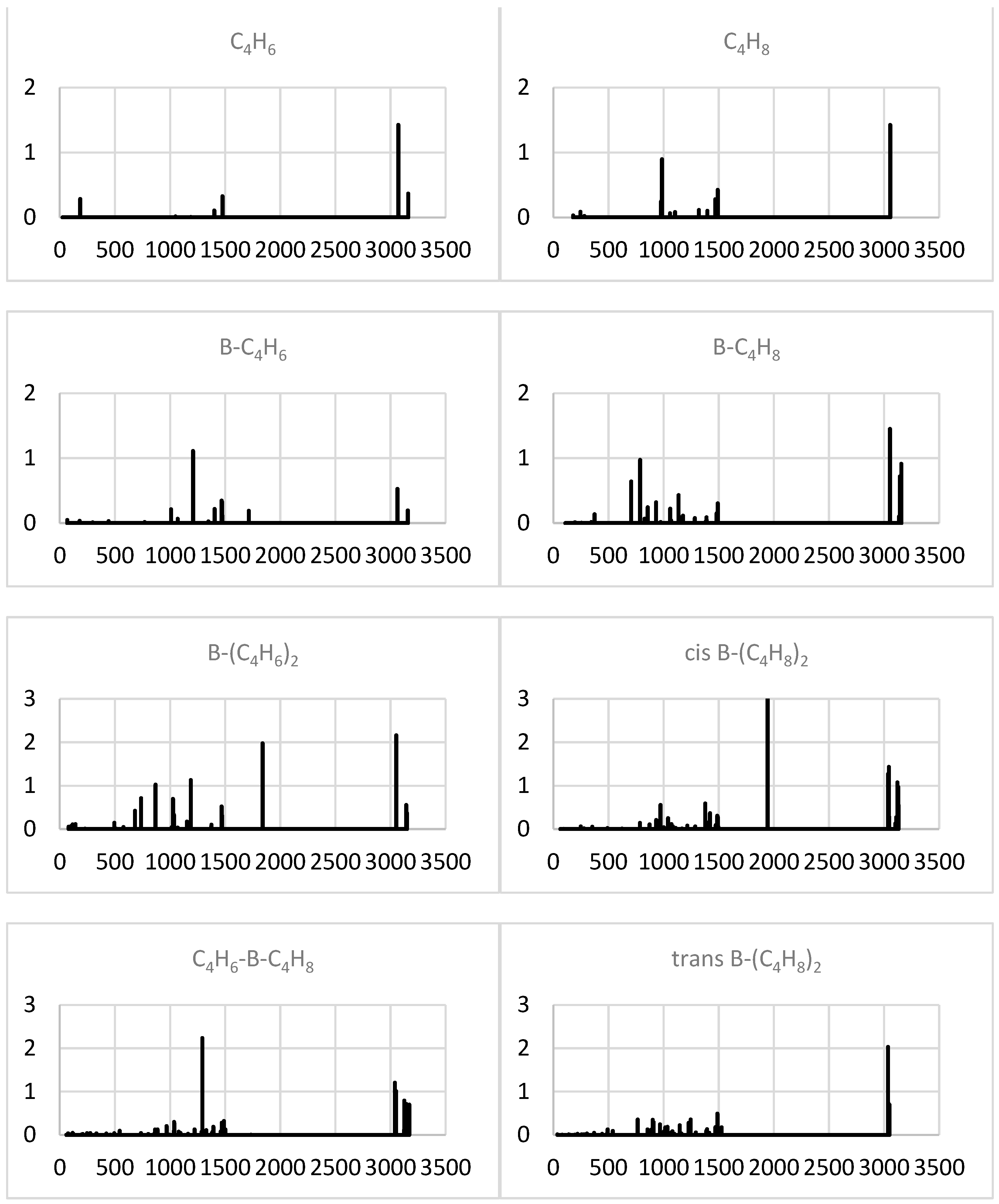
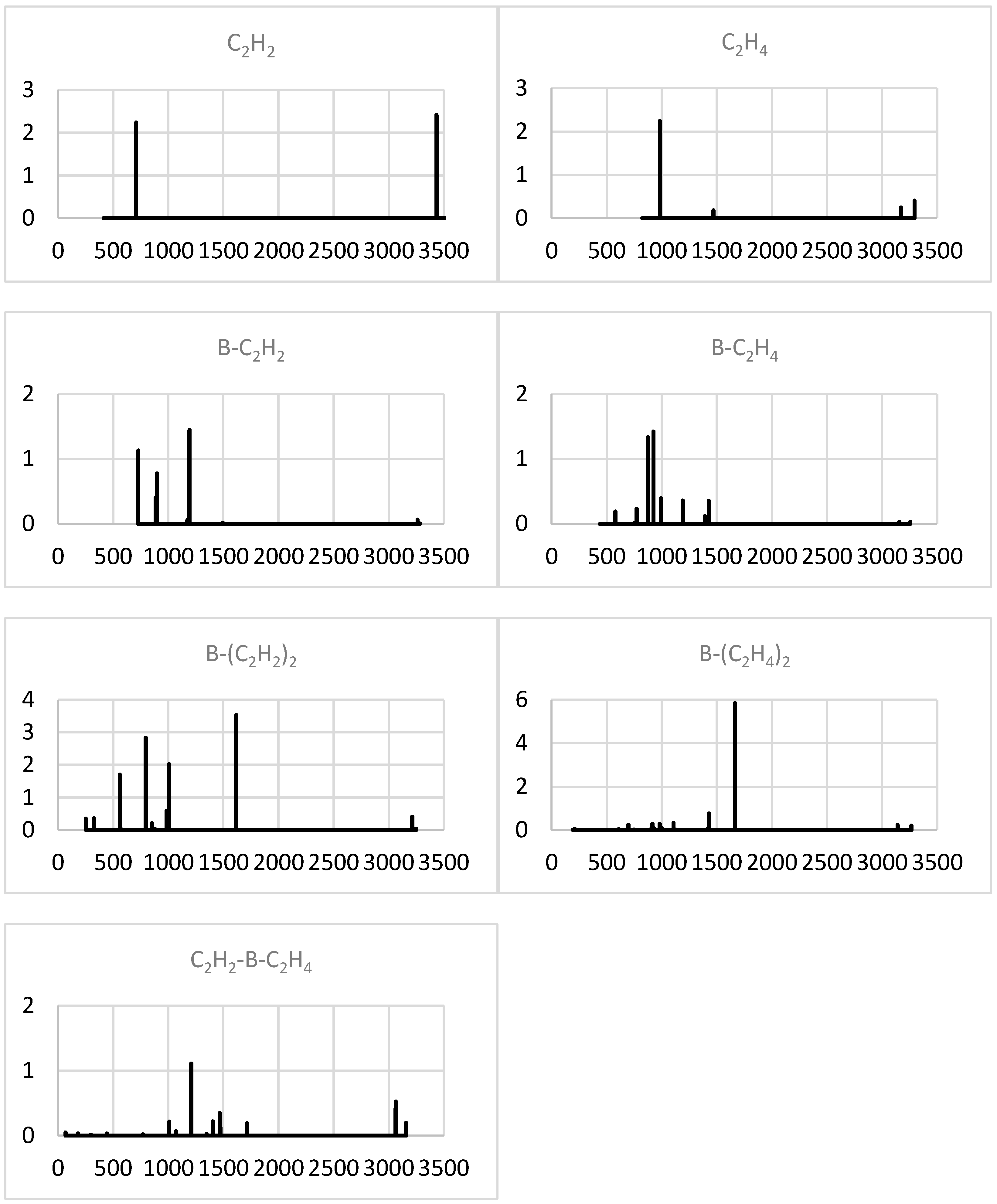
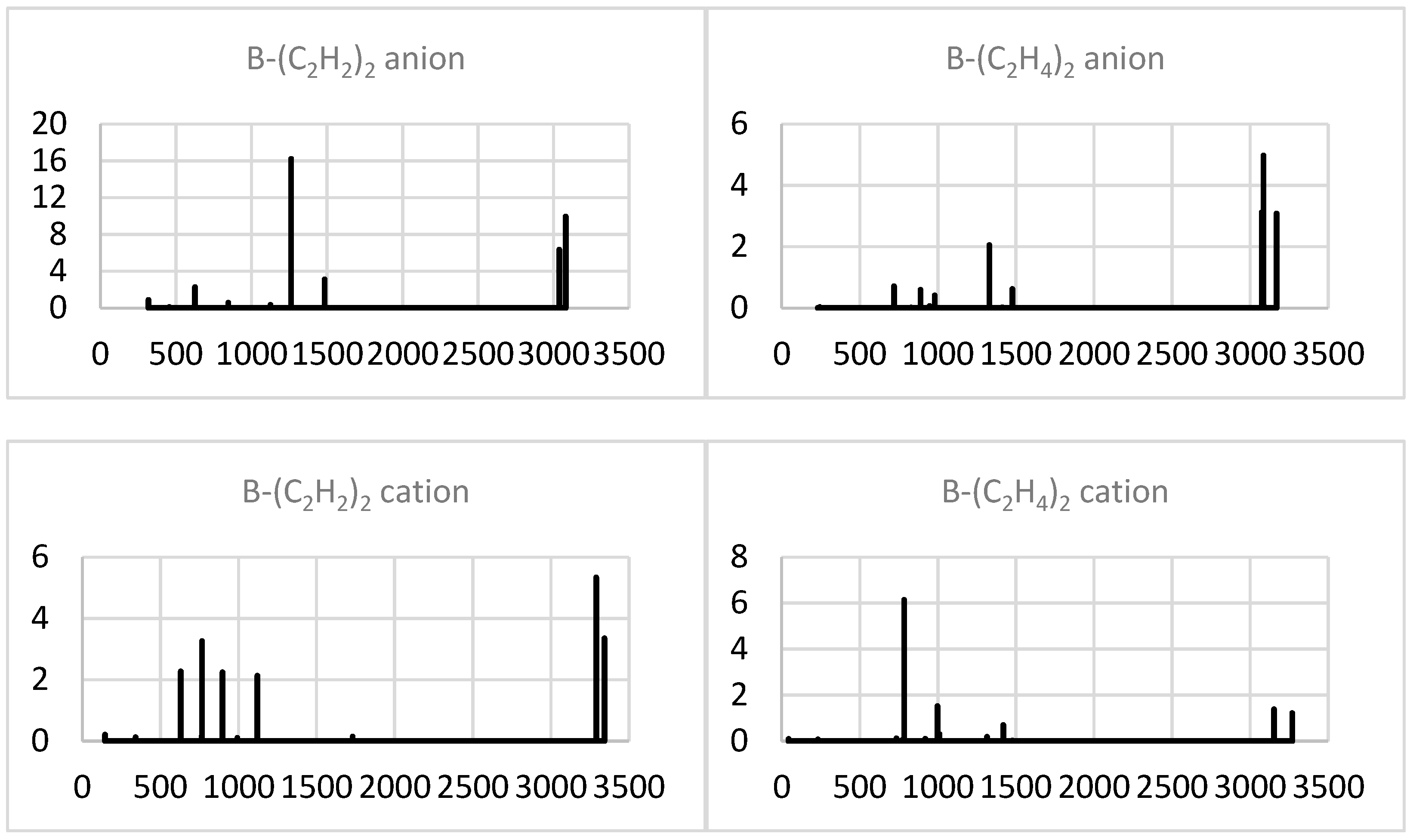
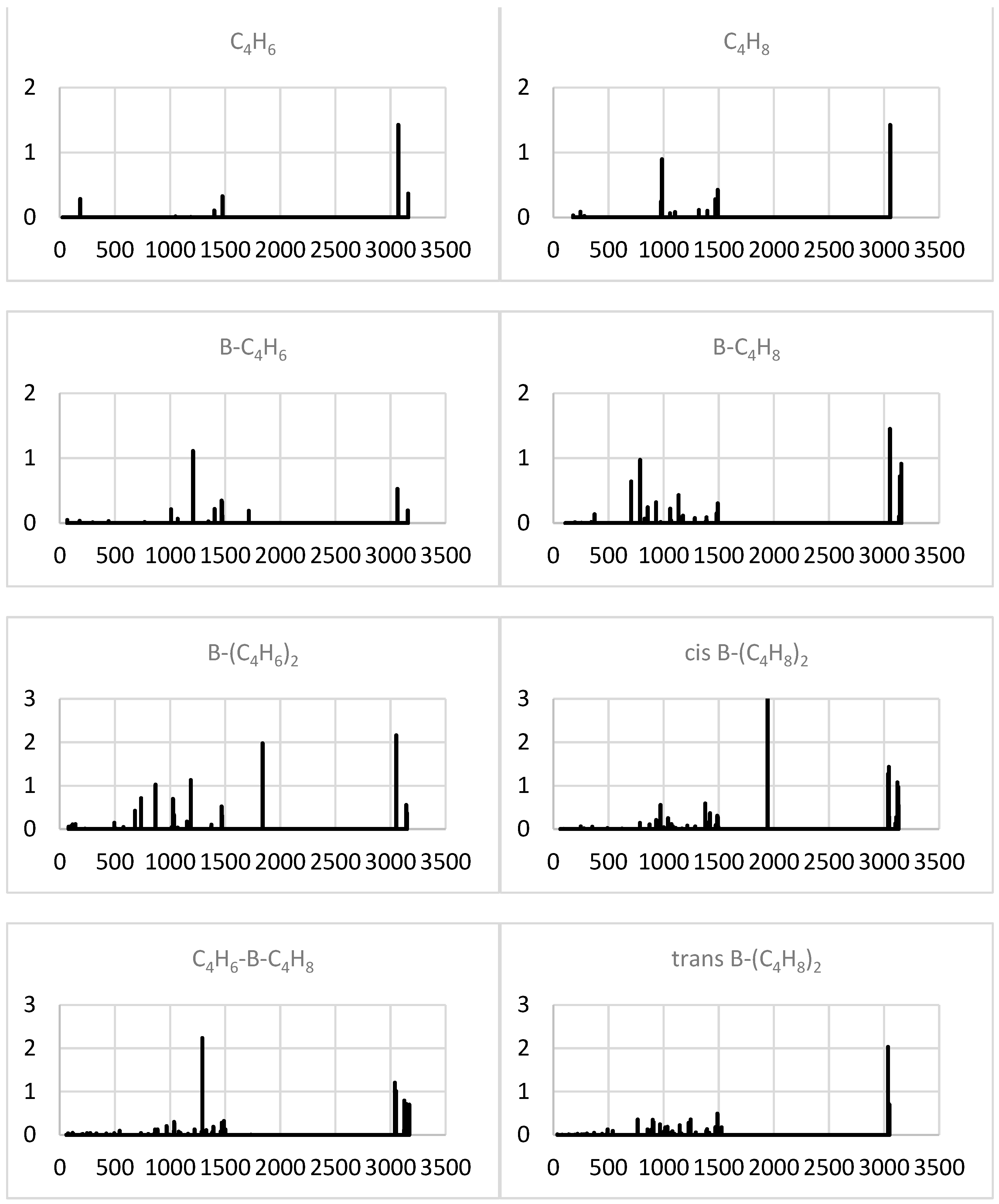
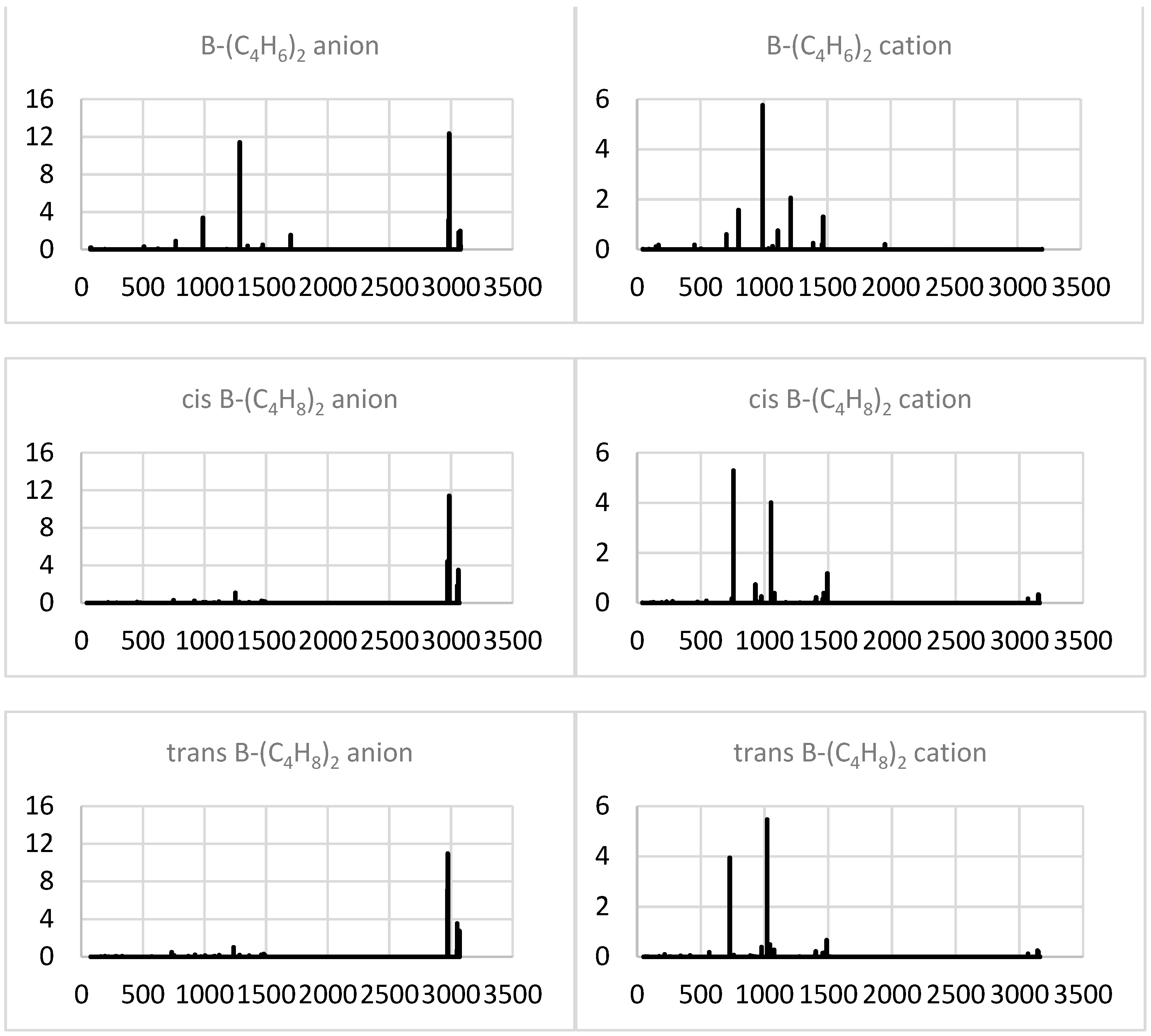
2.1.4. Simulated IR Spectra
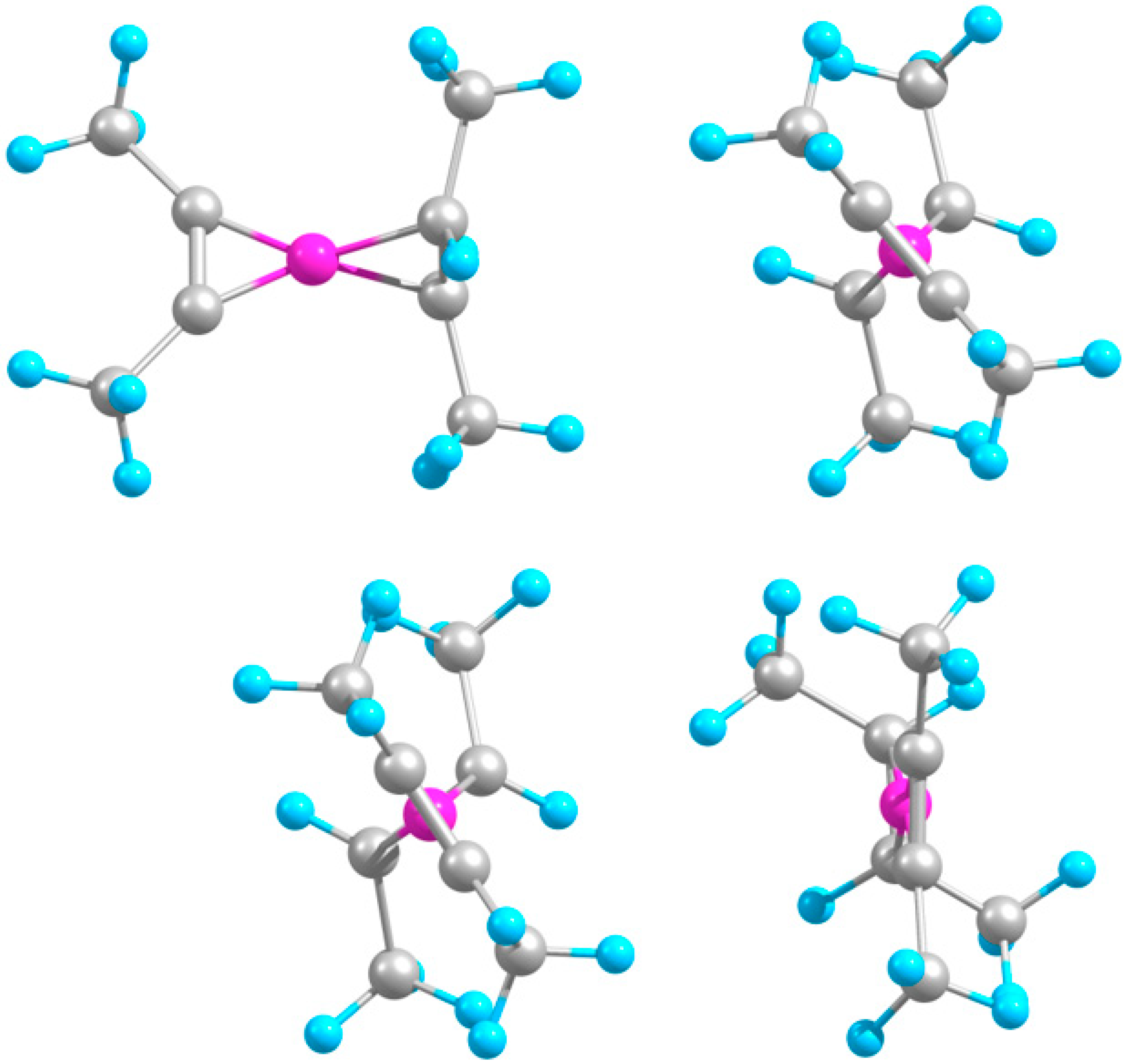
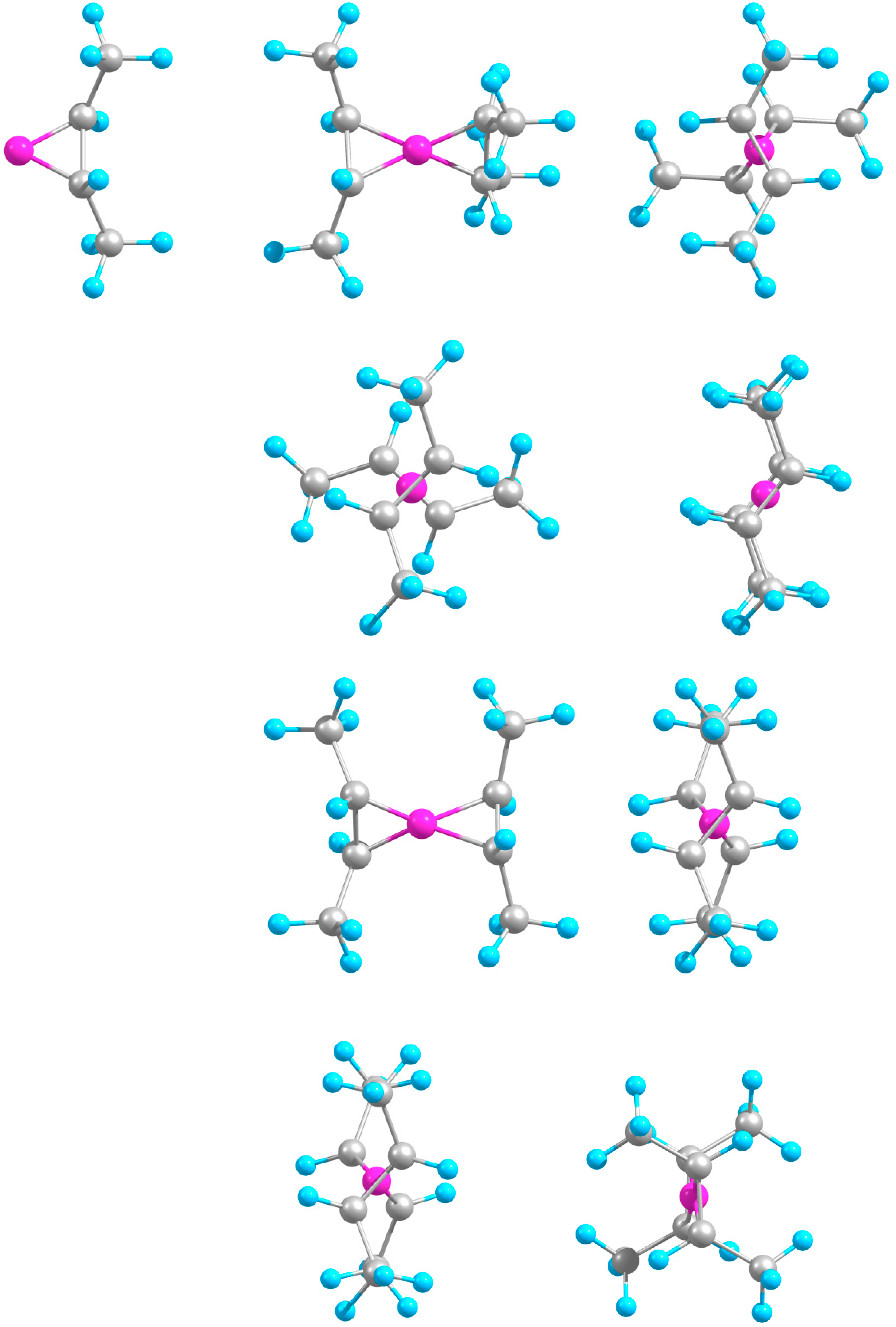
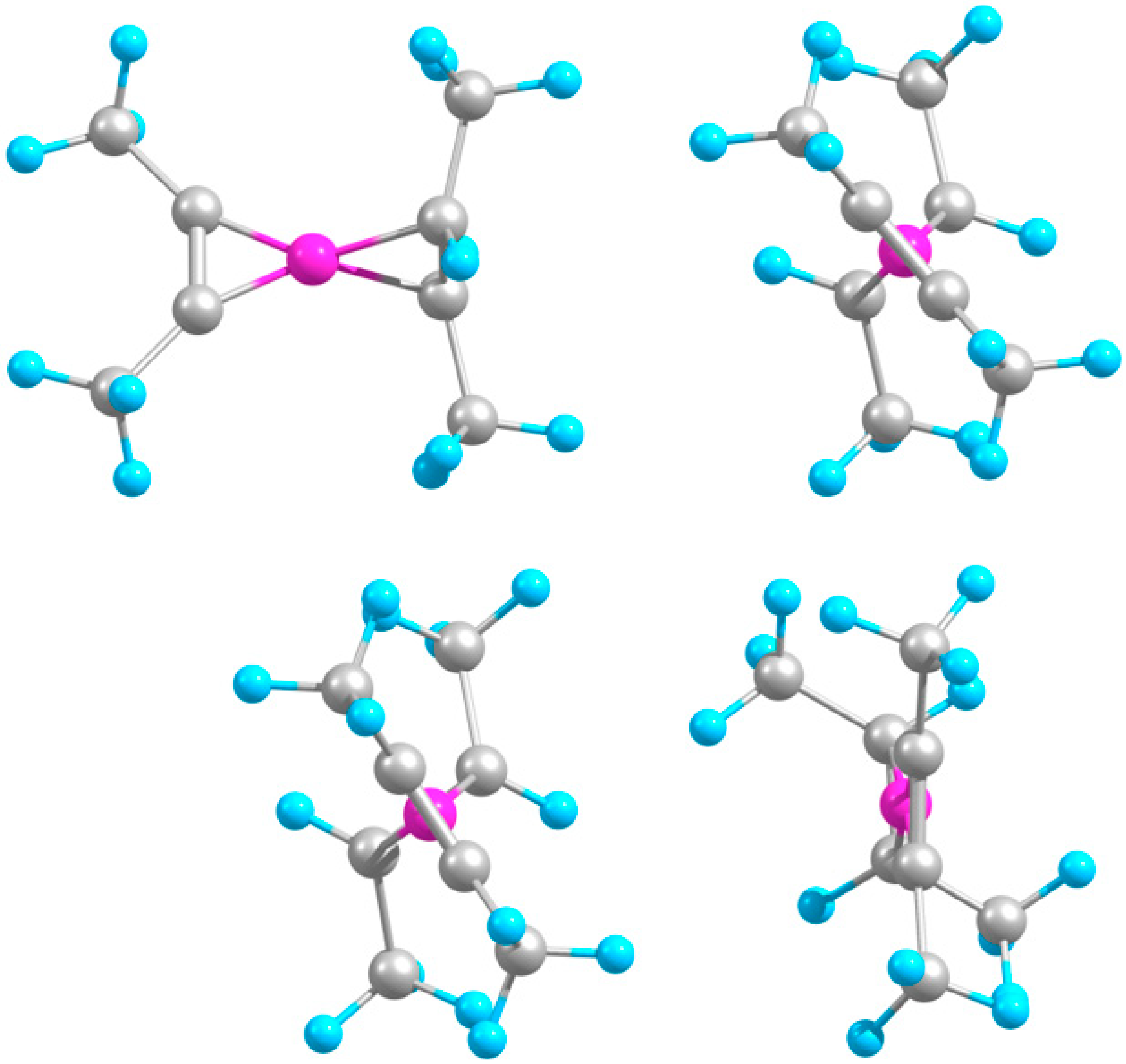
2.2. Extended Systems
2.2.1. Structures and Stabilities
2.2.2. Charge Distributions
2.2.3. Ionization Energies and Electron Affinities
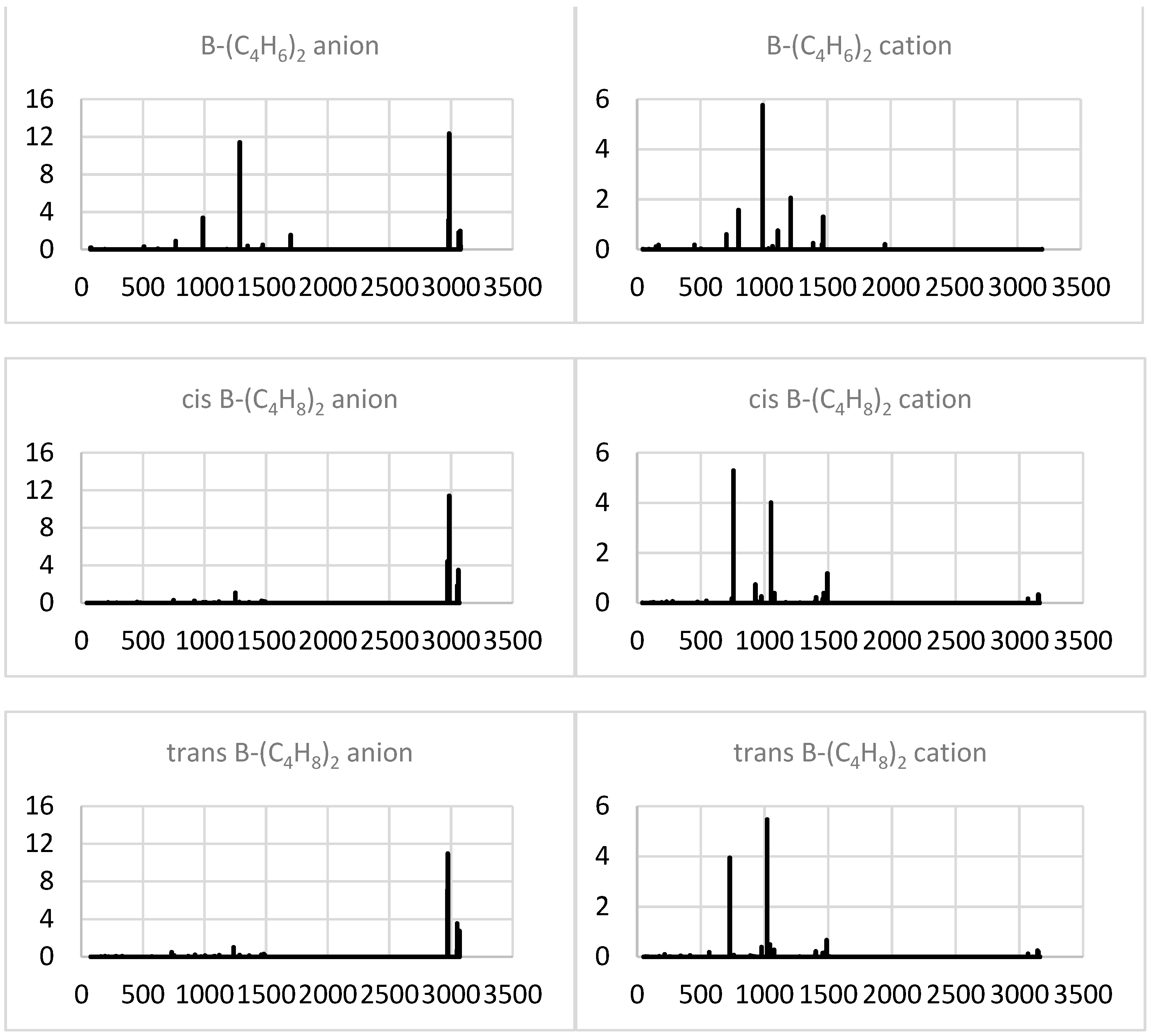
2.2.4. Simulated IR Spectra
3. Computational Methods
4. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- DeFrancesco, H.; Dudley, J.; Coca, A. Boron Chemistry: An Overview. In Boron Reagents in Synthesis; Coca, A., Ed.; ACS Symposium Series 1236; U.S. American Chemical Society: Washington, DC, USA, 2016; Chapter 1; pp. 1–25. [Google Scholar]
- Fyfe, J.W.B.; Watson, A.J.B. Recent Developments in Organoboron Chemistry: Old Dogs, New Tricks. Chem 2017, 3, 31–55. [Google Scholar] [CrossRef]
- Ali, F.; Hosmane, N.S.; Zhu, Y. Boron Chemistry for Medical Applications. Molecules 2020, 25, 828–852. [Google Scholar] [CrossRef] [PubMed]
- Grams, R.J.; Santos, W.L.; Scorei, I.R.; Abad-García, A.; Rosenblum, C.A.; Bita, A.; Cerecetto, H.; Viñas, C.; Soriano-Ursúa, M.A. The Rise of Boron-Containing Compounds: Advancements in Synthesis, Medicinal Chemistry, and Emerging Pharmacology. Chem. Rev. 2024, 124, 2441–2511. [Google Scholar] [CrossRef] [PubMed]
- Naumkin, F.Y.; Fisher, K. Small metal–organic molecular sandwiches: Versatile units for induced structure manipulation. Chem. Phys. Lett. 2013, 590, 52–57. [Google Scholar] [CrossRef]
- Dong Xiang, D.; Wang, X.; Jia, C.; Lee, T.; Guo, X. Molecular-Scale Electronics: From Concept to Function. Chem. Rev. 2016, 116, 4318–4440. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Stoddart, J.F. From molecular to supramolecular electronics. Nat. Rev. Mater. 2021, 6, 804–828. [Google Scholar] [CrossRef]
- Aprahamian, I. The Future of Molecular Machines. ACS Cent. Sci. 2020, 6, 347–358. [Google Scholar] [CrossRef]
- Kassem, S.; van Leeuwen, T.; Lubbe, A.S.; Wilson, M.R.; Feringa, B.L.; Leigh, D.A. Artificial molecular motors. Chem. Soc. Rev. 2017, 46, 2592–2621. [Google Scholar] [CrossRef] [PubMed]
- Balucani, N.; Zhang, F.; Kaiser, R.I. Elementary Reactions of Boron Atoms with Hydrocarbons toward the Formation of Organo-Boron Compounds. Chem. Rev. 2010, 110, 5107–5127. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Gu, X.; Kaiser, R.I.; Bettinger, H. A reinvestigation of the gas phase reaction of boron atoms, 11B/10B with acetylene, C2H2. Chem. Phys. Lett. 2007, 450, 178–185. [Google Scholar] [CrossRef]
- Zhang, F.; Gu, X.; Kaiser, R.I.; Balucani, N.; Huang, C.H.; Kao, C.H.; Chang, A.H.H. A Crossed Beam and Ab Initio Study of the Reaction of Atomic Boron with Ethylene. J. Phys. Chem. A 2008, 112, 3837–3845. [Google Scholar] [CrossRef] [PubMed]
- Linstrom, P.J.; Mallard, W.G. (Eds.) NIST Chemistry WebBook, NIST Standard Ref. Database Number 69; NIST: Gaithersburg, MD, USA, 2023. [Google Scholar]
- Zhang, F.; Kao, C.H.; Chang, A.H.H.; Gu, X.; Guo, Y.; Kaiser, R.I. A Crossed Molecular Beam Study on the Reaction of Boron Atoms with Methylacetylene. ChemPhysChem 2008, 9, 95–105. [Google Scholar] [CrossRef] [PubMed]
- Sillars, D.; Kaiser, R.I.; Galland, N.; Hannachi, Y. Crossed-Beam Reaction of Boron Atoms, B (2Pj), with Dimethylacetylene, CH3CCCH3 (X 1A1g). J. Phys. Chem. A 2003, 107, 5149–5156. [Google Scholar] [CrossRef]
- Schuchardt, K.L.; Didier, B.T.; Elsethagen, T.; Sun, L.; Gurumoorthi, V.; Chase, J.; Li, J.; Windus, T.L. Basis Set Exchange: A Community Database for Computational Sciences. J. Chem. Inf. Model. 2007, 47, 1045–1052. [Google Scholar] [CrossRef] [PubMed]
- Boys, S.F.; Bernardi, F. The Calculation of Small Molecular Interactions by the Differences of Separate Total Energies. Some Procedures with Reduced Errors. Mol. Phys. 1970, 19, 553–566. [Google Scholar] [CrossRef]
- Naumkin, F.Y. Main-Group Metal Complexes of Benzene: Predicted Features of Stabilization and Isomerization. Molecules 2023, 28, 5985. [Google Scholar] [CrossRef]
- Valiev, M.; Bylaska, E.J.; Govind, N.; Kowalski, K.; Straatsma, T.P.; van Dam, H.J.J.; Wang, D.; Nieplocha, J.; Apra, E.; Windus, T.L.; et al. NWChem: A comprehensive and scalable open-source solution for large scale molecular simulations. Comput. Phys. Commun. 2010, 181, 1477–1489. [Google Scholar] [CrossRef]
- Reed, A.E.; Robert, B.; Weinstock, R.B.; Weinhold, F. Natural population analysis. J. Chem. Phys. 1985, 83, 735–746. [Google Scholar] [CrossRef]
- Nikolaienko, T.Y.; Bulavin, L.A.; Hovorun, D.M. JANPA: An open source cross-platform implementation of the Natural Population Analysis on the Java platform. Comput. Theor. Chem. 2014, 1050, 15–22. [Google Scholar] [CrossRef]
- Jian, J.; Li, W.; Wu, X.; Zhou, M. Double C-H bond activation of acetylene by atomic boron in forming aromatic cyclic-HBC2BH in solid neon. Chem Sci. 2017, 8, 4443–4449. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| System | Charge State | De/eV | Re (B-C)/Å | Re (C-C) | φe 1/° | θe (CC-CC) 2 |
|---|---|---|---|---|---|---|
| B-C2H2 | neutral | 3.41 | 1.467 | 1.360 | 138 | |
| anion | 3.75 | 1.539 | 1.336 | 139 | ||
| cation | 3.02 | 1.391 | 1.464 | 132 | ||
| B-(C2H2)2 | neutral | 4.48 | 1.558 | 1.309 | 144 | 53 |
| anion | 5.89 | 1.573 | 1.340 | 134 | 90 | |
| cation | 7.06 | 1.538 | 1.280 | 156 | 0 | |
| B-C2H4 * | neutral | 2.48 | 1.519 | 1.574 | 138 | |
| anion | 2.95 | 1.604 | 1.515 | 141 | ||
| B-(C2H4)2 * | neutral | 4.47 | 1.594 | 1.475 | 148 ± 2 | 60 |
| anion | 6.39 | 1.574 | 1.550 | 133 | 90 | |
| cation | 6.44 | 1.600 | 1.438 | 159 | 0 | |
| C2H2-B-C2H4 | neutral | 4.77 | 1.492, 1.680 | 1.347, 1.442 | 138; 156 | 90 |
| anion | 5.95 | 1.568, 1.581 | 1.331, 1.562 | 136; 132 | 90 | |
| cation | 6.91 | 1.487, 1.663 | 1.310, 1.406 | 149; 165 | 0 |
| System | Charge State | q(B)/e | System | Charge State | q(B)/e |
|---|---|---|---|---|---|
| B-C2H2 | neutral | 0.514 | B-C4H6 | neutral | 0.543 |
| anion | −0.221 | anion | −0.185 | ||
| cation | 0.999 | cation | 0.936 | ||
| B-(C2H2)2 | neutral | 0.447 | B-(C4H6)2 | neutral | 0.537 |
| anion | 0.455 | anion | 0.564 | ||
| cation | 0.423 | cation | 0.515 | ||
| B-C2H4 * | neutral | 0.69 | B-C4H8 | neutral | 0.762 |
| anion | −0.04 | anion | 0.029 | ||
| B-(C2H4)2 * | neutral | 0.59 | cis B-(C4H8)2 | neutral | 0.712 |
| anion | 0.37 | anion | 0.539 | ||
| cation | 0.92 | cation | 1.000 | ||
| C2H2-B-C2H4 | neutral | 0.541 | trans B-(C4H8)2 | neutral | 0.791 |
| anion | 0.463 | anion | 0.542 | ||
| cation | 0.553 | cation | 1.003 | ||
| C4H6-B-C4H8 | neutral | 0.609 | |||
| anion | 0.549 | ||||
| cation | 0.623 | ||||
| System | IE/eV | EA/eV | System | IE/eV | EA/eV |
|---|---|---|---|---|---|
| B-C2H2 | 8.65 | 0.58 | B-C4H6 | 7.60 | 0.51 |
| B-(C2H2)2 | 5.76 | 1.71 | B-(C4H6)2 | 4.62 | 1.33 |
| B-C2H4 * | 7.37 | 0.69 | B-C4H8 | 0.75 | |
| B-(C2H4)2 * | 6.42 | 2.21 | cis B-(C4H8)2 | 5.81 | 2.15 |
| C2H2-B-C2H4 | 6.17 | 1.46 | trans B-(C4H8)2 | 5.75 | 2.21 |
| B | 8.28 [8.30] † | 0.21 [0.28] † | C4H6-B-C4H8 | 5.30 | 1.26 |
| System | Charge State | De/eV | Re (B-C)/Å | Re (C-C) 1 | φe 2/° | θe (CCCC) 3 |
|---|---|---|---|---|---|---|
| B-C4H6 | neutral | 3.43 | 1.492 | 1.386 | 139 | |
| anion | 3.69 | 1.552 | 1.354 | 143 | ||
| cation | 4.06 | 1.416 | 1.532 | 133 | ||
| B-(C4H6)2 | neutral | 4.49 | 1.586 | 1.334 | 145 | 54 |
| anion | 5.58 | 1.599 | 1.357 | 136 | 90 | |
| cation | 8.18 | 1.563 | 1.311 | 139 | 0 | |
| B-C4H8 | neutral | 2.14 | 1.545 | 1.595 | 118 | |
| anion | 2.64 | 1.640 | 1.513 | 116 | ||
| cis B-(C4H8)2 | neutral | 4.12 | 1.612 | 1.494 | 145 ± 3 | 62 |
| anion | 5.95 | 1.589 | 1.553 | 136 | 90 | |
| cation | 6.61 | 1.623 | 1.462 | 153 | 0 | |
| trans B-(C4H8)2 | neutral | 4.07 | 1.553, 1.704 | 1.566, 1.446 | 136, 153 | 77 |
| anion | 5.95 | 1.590 | 1.553 | 137 | 83 | |
| cation | 6.61 | 1.620 | 1.463 | 154 | 6 | |
| C4H6-B-C4H8 | neutral | 4.72 | 1.517, 1.695 | 1.372, 1.463 | 140; 151 | 94 |
| anion | 5.70 | 1.588, 1.603 | 1.347, 1.565 | 139; 136 | 86 | |
| cation | 7.71 | 1.508, 1.690 | 1.345, 1.429 | 149; 158 | 4 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Naumkin, F.Y. Metalloid–Organic Intermolecular Complexes with Charge State-Controlled Conformations. Molecules 2024, 29, 1635. https://doi.org/10.3390/molecules29071635
Naumkin FY. Metalloid–Organic Intermolecular Complexes with Charge State-Controlled Conformations. Molecules. 2024; 29(7):1635. https://doi.org/10.3390/molecules29071635
Chicago/Turabian StyleNaumkin, Fedor Y. 2024. "Metalloid–Organic Intermolecular Complexes with Charge State-Controlled Conformations" Molecules 29, no. 7: 1635. https://doi.org/10.3390/molecules29071635
APA StyleNaumkin, F. Y. (2024). Metalloid–Organic Intermolecular Complexes with Charge State-Controlled Conformations. Molecules, 29(7), 1635. https://doi.org/10.3390/molecules29071635