
Investigation of Structures, Stabilities, and Electronic and Magnetic Properties of Niobium Carbon Clusters Nb7Cn (n = 1–7)

 ,
,
Abstract
1. Introduction
2. Results and Discussion
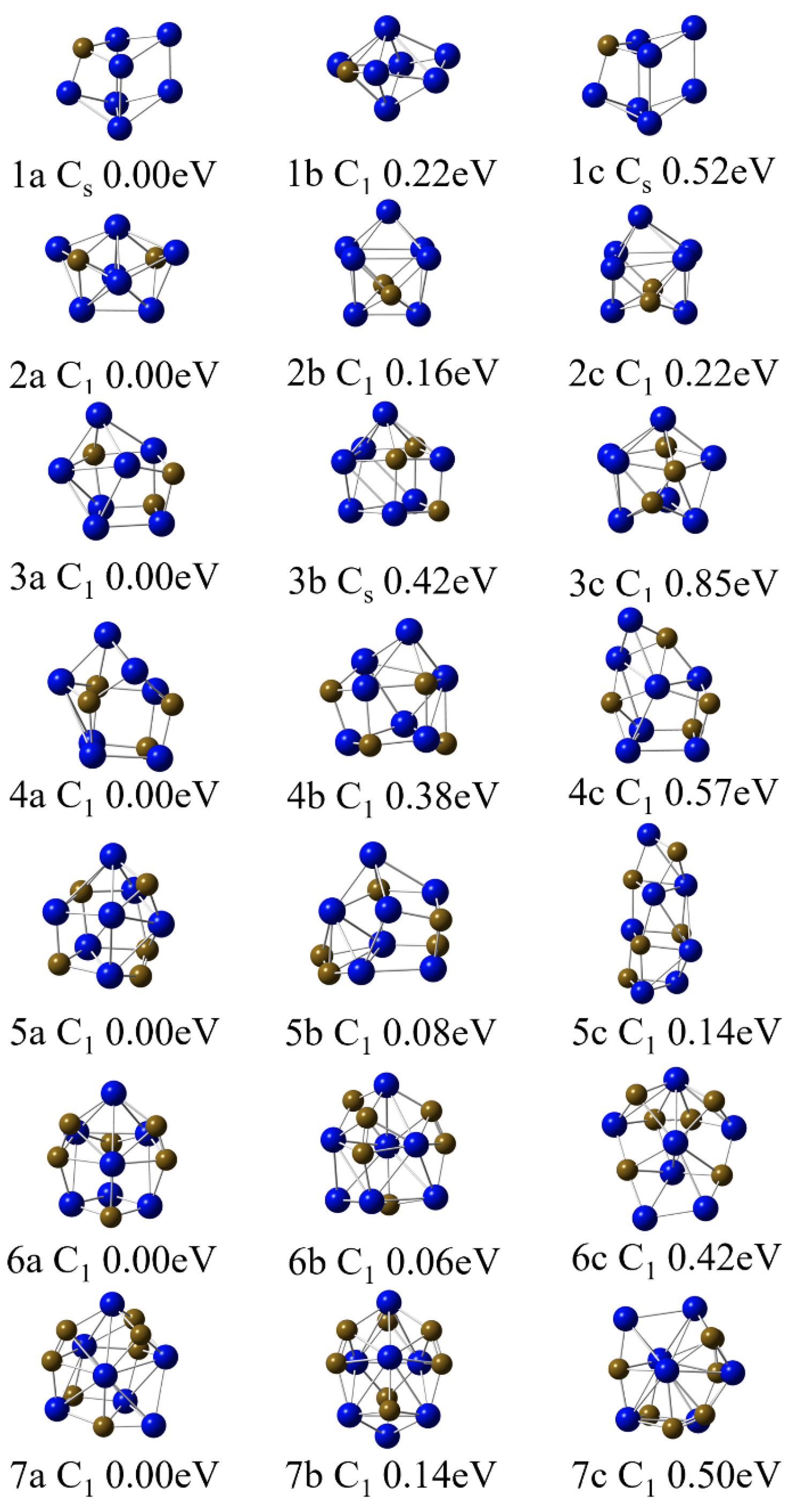
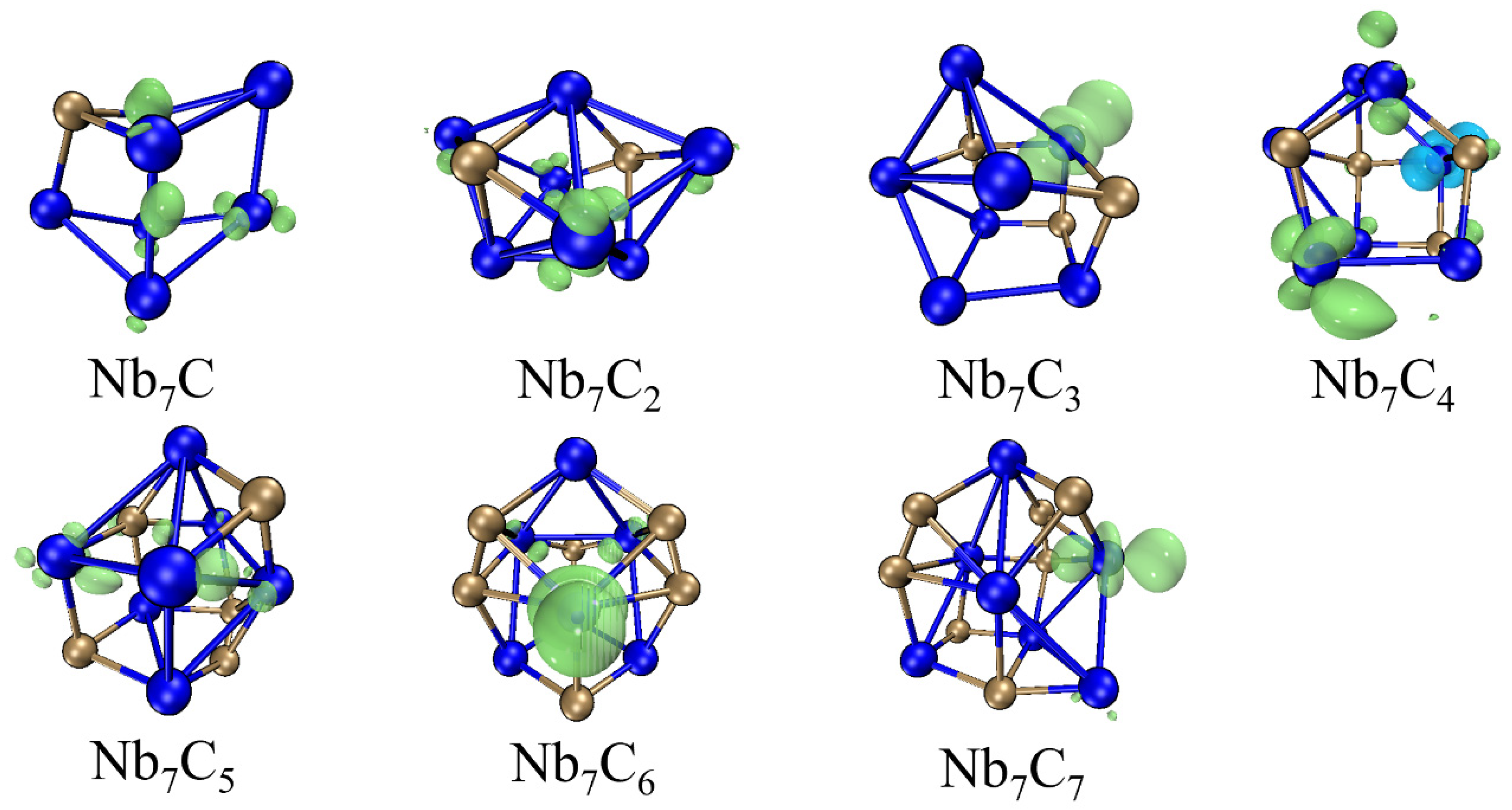
2.1. Structure of Nb7Cn (n = 1–7)
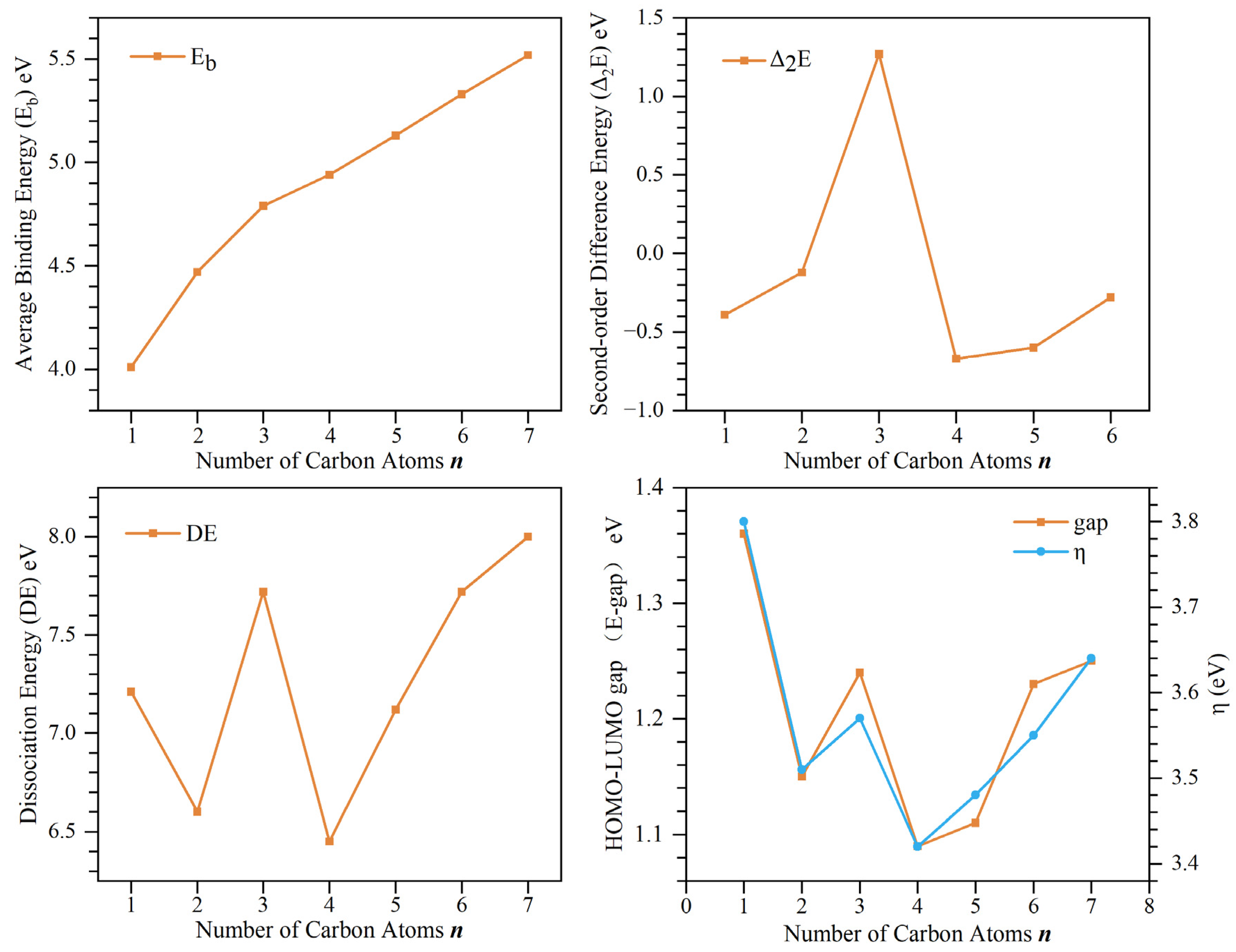
2.2. Stability
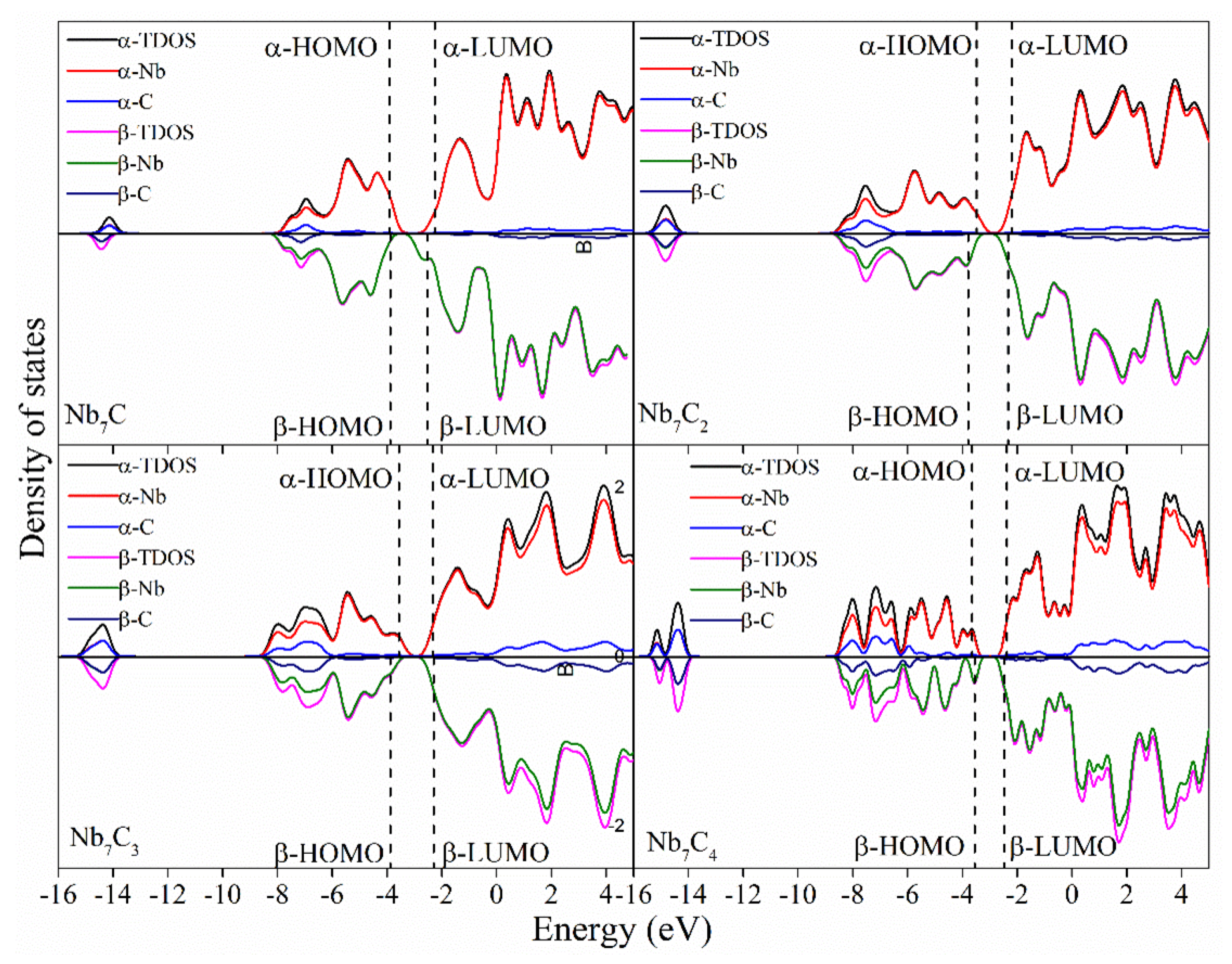
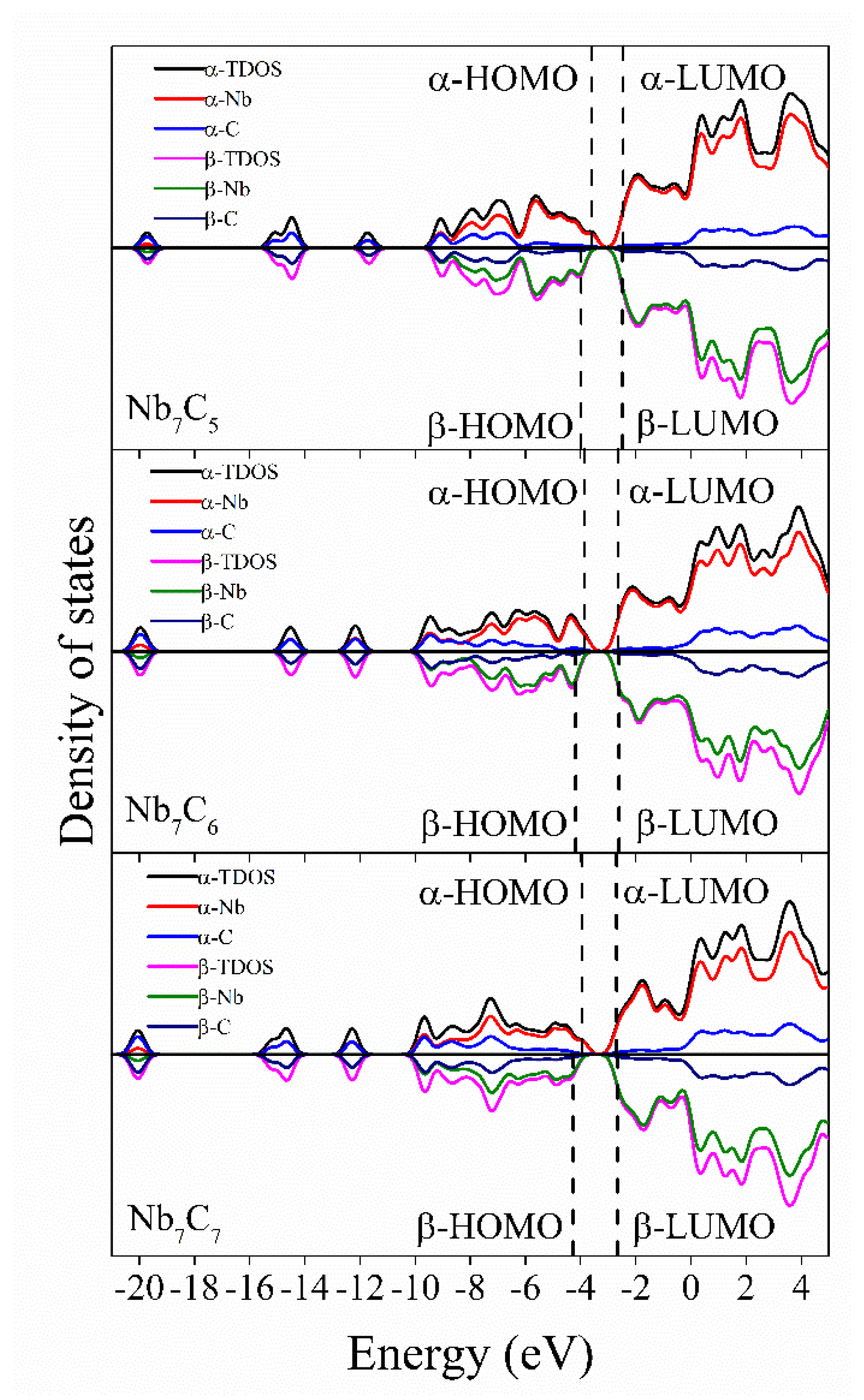
2.3. Density of States
2.4. Magnetic Properties
3. Computational Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lu, C.; Gong, W.G.; Li, Q.; Chen, C.F. Elucidating stress-strain relations of ZrB12 from first-principles studies. J. Phys. Chem. Lett. 2020, 11, 9165–9170. [Google Scholar] [CrossRef] [PubMed]
- Zhao, R.; Chen, R.; Zhao, H.; Lin, F.; Han, J.-G. Exploration on electronic properties of self-assembled Indium nitrogen nanosheets and nanowires by a density functional method. Molecules 2023, 28, 7358. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Zhang, M.; Lu, X.-Q.; Yan, Q.-Q.; Zhao, X.-N.; Li, S.-D. Sc@B28−, Ti@B28, V@B28+, and V@B292−: Spherically aromatic endohedral seashell-like metallo-borospherenes. Molecules 2023, 28, 3892. [Google Scholar] [CrossRef] [PubMed]
- Uchiyama, T.; Nakamura, T.; Hiyama, M.; Kudo, T. Theoretical study of Si/C equally mixed dodecahedrane analogues. Molecules 2023, 28, 2769. [Google Scholar] [CrossRef] [PubMed]
- Khanna, V.; McGrady, J.E. Mn2 dimers encapsulated in silicon cages: A complex challenge to MC-SCF theory. Molecules 2022, 27, 7544. [Google Scholar] [CrossRef] [PubMed]
- Duan, X.F.F.; Burggraf, L.W.; Huang, L.Y. Searching for stable SinCn clusters: Combination of stochastic potential surface search and pseudopotential Plane-Wave Car-Parinello simulated annealing simulations. Molecules 2013, 18, 8591–8606. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Deng, J.-J.; Yao, W.-W.; Gurti, J.I.; Li, W.; Wang, W.-J.; Yao, J.-X.; Ding, X.-L. Non-stoichiometric molybdenum sulfide clusters and their reactions with the hydrogen molecule. Phys. Chem. Chem. Phys. 2021, 23, 347–355. [Google Scholar] [CrossRef]
- Valeeva, A.A.; Gusev, A.I. Effect of nonstoichiometry on elastic properties of niobium carbide NbCy. Int. J. Refract. Met. Hard Mater. 2021, 95, 105435. [Google Scholar] [CrossRef]
- Jalalya, M.; Gotorb, F.J.; Sayagués, M.J. Mechanochemical combustion synthesis of vanadium carbide (VC), niobium carbide (NbC) and tantalum carbide (TaC) nanoparticles. Int. J. Refract. Met. Hard Mater. 2019, 79, 177–184. [Google Scholar] [CrossRef]
- Laskoski, M.; Prestigiacomo, J.; Dyatkin, B.; Keller, T.M.; Mahzabeen, W.; Shepherd, A.R.; Daftary, M.N.; Clarke, J.S.; Neal, A.; Qadri, S.B.; et al. Synthesis and material properties of polymer-derived niobium carbide and niobium nitride nanocrystalline ceramics. Ceram. Int. 2021, 47, 1163–1168. [Google Scholar] [CrossRef]
- Yeh, C.S.; Byun, Y.G.; Afzaal, S.; Kan, S.Z.; Lee, S.; Freiser, B.S.; Hay, P.J. Experimental and theoretical studies on Nb4C40/+: Reactivity and structure of the smallest cubic niobium-carbon cluster. J. Am. Chem. Soc. 1995, 117, 4042–4048. [Google Scholar] [CrossRef]
- Byun, Y.G.; Lee, S.A.; Kan, S.Z.; Freiser, B.S. Reactivities of metallocarbohedrenes: Nb8C12+. J. Phys. Chem. 1996, 100, 14281–14288. [Google Scholar]
- Li, S.; Wu, H.; Wang, L. Probing the electronic structure of metallocarbohedrenes: M8C12 (M = Ti, V, Cr, Zr, Nb). J. Am. Chem. Soc. 1997, 119, 7417–7422. [Google Scholar] [CrossRef]
- Yang, D.S.; Zgierski, M.Z.; Berces, A.; Hackett, P.A.; Roy, P.N.; Martinez, A.; Carrington, T.; Salahub, D.R.; Fournier, R.; Pang, T.; et al. Vibrational and geometric structures of Nb3C2 and Nb3C2+ from pulsed field ionization-zero electron kinetic energy photoelectron spectra and density functional calculations. J. Chem. Phys. 1996, 105, 10663–10671. [Google Scholar] [CrossRef]
- Knappenberger, K.L., Jr.; Clayborne, P.A.; Reveles, J.U.; Sobhy, M.A.; Jones, C.E.; Gupta, U.U.; Khanna, S.N.; Iordanov, I.; Sofo, J.; Castleman, A.W., Jr. Anion photoelectron spectroscopy and density functional investigation of diniobium-carbon clusters. ACS Nano 2007, 1, 319–326. [Google Scholar] [CrossRef] [PubMed]
- Zhai, H.J.; Liu, S.R.; Wang, L.S. Photoelectron spectroscopy of mono-niobium carbide clusters NbCn− (n = 2–7): Evidence for a cyclic to linear structural transition. J. Chem. Phys. 2001, 115, 5170–5178. [Google Scholar] [CrossRef]
- Chebanenko, M.I.; Danilovich, D.P.; Lobinsky, A.A.; Popkov, V.I.; Rempel, A.A.; Valeeva, A.A. Novel high stable electrocatalyst based on non-stoichiometric nanocrystalline niobium carbide toward effective hydrogen evolution. Int. J. Hydrogen Energy 2021, 46, 16907–16916. [Google Scholar] [CrossRef]
- Il’in, E.G.; Parshakov, A.S.; Teterin, Y.A.; Maslakov, K.I.; Teterin, A.Y. Surface morphology and composition of a NbC/C composite studied by scanning electron microscopy and X-ray photoelectron spectroscopy. Inorg. Mater. 2020, 56, 443–450. [Google Scholar] [CrossRef]
- Pilgrim, J.S.; Brock, L.R.; Duncan, M.A. Photodissociation of niobium-carbon clusters and nanocrystals. J. Phys. Chem. 1995, 99, 544–550. [Google Scholar] [CrossRef]
- Wei, S.; Guo, B.C.; Deng, H.T.; Kerns, K.; Purnell, J.; Buzza, S.A.; Castleman, A.W., Jr. Formation of met-cars and face-centered cubic structures: Thermodynamically or kinetically controlled? J. Am. Chem. Soc. 1994, 116, 4475–4476. [Google Scholar] [CrossRef]
- Fukushima, N.; Miyajima, K.; Mafune, F. Ionization energies of niobium carbide clusters NbnCm (n = 3–10, m = 0–7). J. Phys. Chem. A 2009, 113, 2309–2315. [Google Scholar] [CrossRef] [PubMed]
- Dryza, V.; Addicoat, M.A.; Gascooke, J.R.; Buntine, M.A.; Metha, G.F. Threshold photoionization and density functional theory studies of the niobium carbide clusters Nb3Cn (n = 1–4) and Nb4Cn (n = 1–6). J. Phys. Chem. A 2008, 112, 5582–5592. [Google Scholar] [CrossRef] [PubMed]
- Gusev, A.I. Anisotropy of microstructure and elastic properties of niobium carbide nanopowders. Solid State Sci. 2020, 100, 106092. [Google Scholar] [CrossRef]
- Heijnsbergen, D.V.; Fielicke, A.; Meijer, G.; Helden, G.V. Structure determination of gas-phase niobium and tantalum carbide nanocrystals via infrared spectroscopy. Phys. Rev. Lett. 2002, 89, 013401. [Google Scholar] [CrossRef] [PubMed]
- Clemmer, D.E.; Hunter, J.M.; Shelimov, K.B.; Jarrold, M.F. Physical and chemical evidence for metallofullerenes with metal atoms as part of the cage. Lett. Nat. 1994, 372, 230–248. [Google Scholar] [CrossRef]
- Pham, V.N.; Minh, T.N. Structures, spectra, and energies of Niobium Clusters from Nb13 to Nb20. J. Phys. Chem. A 2012, 116, 7405–7418. [Google Scholar]
- Harris, H.; Dance, I. The geometric and electronic structures of niobium carbon clusters. J. Phys. Chem. A 2001, 105, 3340–3358. [Google Scholar] [CrossRef]
- Dai, D.; Roszak, S.; Balasubramanian, K. Electronic structures of niobium carbides: NbCn (n = 3–8). J. Phys. Chem. A 2000, 104, 9760–9769. [Google Scholar] [CrossRef]
- Jeffrey, P.M.; Damian, M.; Leo, R. An evaluation of harmonic vibrational frequency scale factors. J. Phys. Chem. A 2007, 111, 11683–11700. [Google Scholar]
- Roy, D.; Corminboeuf, C.; Wannere, C.S.; King, R.B.; Schleyer, P.V.R. Planar tetracoordinate carbon atoms centered in bare four-membered rings of late transition metals. Inorg. Chem. 2006, 45, 8902–8906. [Google Scholar] [CrossRef]
- Bera, P.P.; Sattelmeyer, K.W.; Saunders, M.; Schaefer, H.F.; Schleyer, P.R. Mindless chemistry. J. Phys. Chem. A 2006, 110, 4287–4290. [Google Scholar] [CrossRef] [PubMed]
- Saunders, M. Stochastic search for isomers on a quantum mechanical surface. J. Comput. Chem. 2004, 25, 621–626. [Google Scholar] [CrossRef] [PubMed]
- Xie, B.; Wang, H.Q.; Li, H.F.; Zhang, J.M.; Zeng, J.K.; Mei, X.J.; Zhang, Y.H.; Zheng, H.; Qin, L.X. Making sense of the growth behavior of ultra-high magnetic Gd2-doped silicon clusters. Molecules 2023, 28, 5071. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.M.; Wang, H.Q.; Li, H.F.; Mei, X.J.; Zeng, J.K.; Qin, L.X.; Zheng, H.; Zhang, Y.H.; Jiang, K.L.; Zhang, B.; et al. Aromatic and magnetic properties in a series of heavy rare earth-doped Ge6 cluster anions. J. Comput. Chem. 2024, in press. [Google Scholar] [CrossRef]
- Li, H.F.; Wang, H.Q. Stabilization of golden cages by encapsulation of a single transition metal atom. R. Soc. Open Sci. 2018, 5, 171019. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.W.; Wang, H.Q.; Li, H.F. Structural and electronic properties of exohedrally doped neutral silicon clusters LnSin (n = 5, 10; Ln = Sm, Eu, Yb). Phys. Chem. Chem. Phys. 2020, 22, 20545–20552. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.W.; Kong, X.Y.; Zhao, L.J.; Wang, H.Q.; Li, H.F.; Zhan, Q.; Xie, B.; Xu, H.G.; Zheng, W.J. A joint experimental and theoretical study on structural, electronic, and magnetic properties of MnGen− (n = 3–14) clusters. J. Chem. Phys. 2021, 154, 204302. [Google Scholar] [CrossRef]
- Xie, B.; Wang, H.Q.; Li, H.F.; Zeng, J.K. Structural and electronic properties of Ln2Si6q: (Sm, Eu, Yb; q = 0, −1) clusters. Chem. Phys. 2023, 566, 111782. [Google Scholar] [CrossRef]
- Fan, Y.W.; Wang, H.Q.; Li, H.F. Probing the structural and electronic properties of anionic europium-doped silicon clusters by density functional theory and comparison of experimental photoelectron spectroscopy. Chem. Phys. 2020, 538, 110918. [Google Scholar] [CrossRef]
- Wang, H.Q.; Li, H.F. Structure identification of endohedral golden cage nanoclusters. RSC Adv. 2015, 5, 94685–94693. [Google Scholar] [CrossRef]
- Wang, H.Q.; Li, H.F. Probing the structural and electronic properties of al-doped small niobium clusters. Chem. Phys. Lett. 2012, 554, 231–235. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [PubMed]
- Igel-Mann, G.; Stoll, H.; Preuss, H. Pseudopotentials for main group elements (IIIa through VIIa). Mol. Phys. 1988, 65, 1321–1328. [Google Scholar] [CrossRef]
- Mclean, A.D.; Chandler, G.S. Contracted gaussian basis sets for molecular calculations. I. Second row atoms, Z = 11–18. J. Chem. Phys. 1980, 72, 5639–5648. [Google Scholar] [CrossRef]
- Ganesh, M.; Gamidi, R.K.; Sashi, D.; Anjan, B. Origin of optoelectronic contradicions in 3,4-cycloalkyl[c]-chalcogenophenes: A computational study. Polymers 2020, 15, 4240. [Google Scholar]
- Shida, N.; Nishiyama, H.; Zheng, F.; Ye, S.; Seferos, D.S.; Tomita, I.; Inagi, S. Redox chemistry of π-extended tellurophenes. Commun. Chem. 2019, 2, 124–132. [Google Scholar] [CrossRef]
- Dishi, O.; Gidron, O. Macrocyclic Oligofurans: A Computational Study. J. Org. Chem. 2018, 83, 3119–3125. [Google Scholar] [CrossRef]
- Wu, B.; Melvina; Wu, X.; Lee Yeow, E.K.; Yoshikai, N. Versatile telluracycle synthesis via the sequential electrophilic telluration of C(sp2)-Zn and C(sp2)-H bonds. Chem. Sci. 2017, 8, 4527–4532. [Google Scholar] [CrossRef]
- Zhang, R.Q.; Chu, T.S.; Lee, S.T. Computation of large systems with an economic basis set: Ab initio calculations of silicon oxide clusters. J. Chem. Phys. 2001, 114, 5531–5536. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision C.01; Gaussian Inc.: Wallingford, CT, USA, 2010. [Google Scholar]
- Lu, T.; Chen, F.W. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Isomers | VIP | Eb | Δ2E | DE | E-Gap | η | |
|---|---|---|---|---|---|---|---|
| Calc. | Expt. [21] | ||||||
| Nb7C | 5.16 | 5.20 ± 0.08 | 4.01 | −0.39 | 7.21 | 1.36 | 3.80 |
| Nb7C2 | 4.72 | 4.7 ± 0.1 | 4.47 | −0.12 | 6.60 | 1.15 | 3.51 |
| Nb7C3 | 4.78 | 4.7 ± 0.08 | 4.79 | 1.27 | 7.72 | 1.24 | 3.57 |
| Nb7C4 | 4.76 | 4.75 ± 0.07 | 4.94 | −0.67 | 6.45 | 1.09 | 3.42 |
| Nb7C5 | 4.86 | 4.7 ± 0.1 | 5.13 | −0.60 | 7.12 | 1.11 | 3.48 |
| Nb7C6 | 5.08 | 4.91 ± 0.07 | 5.33 | −0.28 | 7.72 | 1.23 | 3.55 |
| Nb7C7 | 5.19 | 5.1 ± 0.1 | 5.52 | 8.00 | 1.25 | 3.64 | |
| Isomers | Atoms | α-HOMO | α-LUMO | β-HOMO | β-LUMO |
|---|---|---|---|---|---|
| Nb7C | Nb | 98.72% | 98.80% | 98.68% | 99.87% |
| C | 1.28% | 1.20% | 1.32% | 0.13% | |
| Nb7C2 | Nb | 99.45% | 97.73% | 98.88% | 99.47% |
| C | 0.55% | 2.27% | 1.12% | 0.53% | |
| Nb7C3 | Nb | 94.32% | 96.29% | 92.69% | 96.63% |
| C | 5.68% | 3.71% | 7.31% | 3.37% | |
| Nb7C4 | Nb | 91.37% | 95.49% | 96.44% | 92.42% |
| C | 8.63% | 4.51% | 3.56% | 7.58% | |
| Nb7C5 | Nb | 88.95% | 92.15% | 88.60% | 92.54% |
| C | 11.05% | 7.85% | 11.40% | 7.46% | |
| Nb7C6 | Nb | 93.80% | 94.57% | 84.35% | 94.29% |
| C | 6.20% | 5.43% | 15.65% | 5.71% | |
| Nb7C7 | Nb | 94.49% | 91.86% | 84.66% | 92.29% |
| C | 5.51% | 8.14% | 15.34% | 7.71% |
| Isomers | Nb | C | Total | ||||
|---|---|---|---|---|---|---|---|
| 5s | 4d | 5p | 6s | 6p | |||
| Nb7C | 0.15 | 0.74 | 0.12 | 0.00 | −0.01 | −0.04 | 1 |
| Nb7C2 | 0.12 | 0.63 | 0.20 | 0.00 | 0.00 | −0.02 | 1 |
| Nb7C3 | 0.01 | 0.75 | 0.14 | 0.12 | 0.00 | −0.01 | 1 |
| Nb7C4 | 0.03 | 0.47 | 0.24 | 0.08 | 0.00 | 0.12 | 1 |
| Nb7C5 | 0.02 | 0.36 | 0.18 | 0.14 | 0.15 | 0.16 | 1 |
| Nb7C6 | 0.05 | 0.74 | 0.08 | 0.07 | 0.02 | 0.02 | 1 |
| Nb7C7 | 0.00 | 0.66 | 0.13 | 0.14 | 0.05 | 0.02 | 1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, H.-F.; Wang, H.-Q.; Zhang, J.-M.; Qin, L.-X.; Zheng, H.; Zhang, Y.-H. Investigation of Structures, Stabilities, and Electronic and Magnetic Properties of Niobium Carbon Clusters Nb7Cn (n = 1–7). Molecules 2024, 29, 1692. https://doi.org/10.3390/molecules29081692
Li H-F, Wang H-Q, Zhang J-M, Qin L-X, Zheng H, Zhang Y-H. Investigation of Structures, Stabilities, and Electronic and Magnetic Properties of Niobium Carbon Clusters Nb7Cn (n = 1–7). Molecules. 2024; 29(8):1692. https://doi.org/10.3390/molecules29081692
Chicago/Turabian StyleLi, Hui-Fang, Huai-Qian Wang, Jia-Ming Zhang, Lan-Xin Qin, Hao Zheng, and Yong-Hang Zhang. 2024. "Investigation of Structures, Stabilities, and Electronic and Magnetic Properties of Niobium Carbon Clusters Nb7Cn (n = 1–7)" Molecules 29, no. 8: 1692. https://doi.org/10.3390/molecules29081692
APA StyleLi, H.-F., Wang, H.-Q., Zhang, J.-M., Qin, L.-X., Zheng, H., & Zhang, Y.-H. (2024). Investigation of Structures, Stabilities, and Electronic and Magnetic Properties of Niobium Carbon Clusters Nb7Cn (n = 1–7). Molecules, 29(8), 1692. https://doi.org/10.3390/molecules29081692