
Electrocatalytic Reduction of CO2 to CO by Molecular Cobalt–Polypyridine Diamine Complexes

 , ,
, ,
Abstract
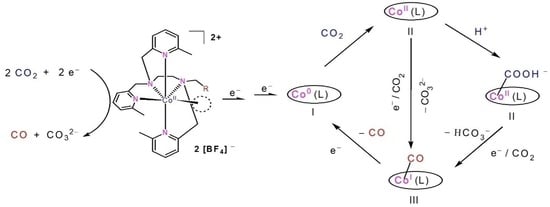
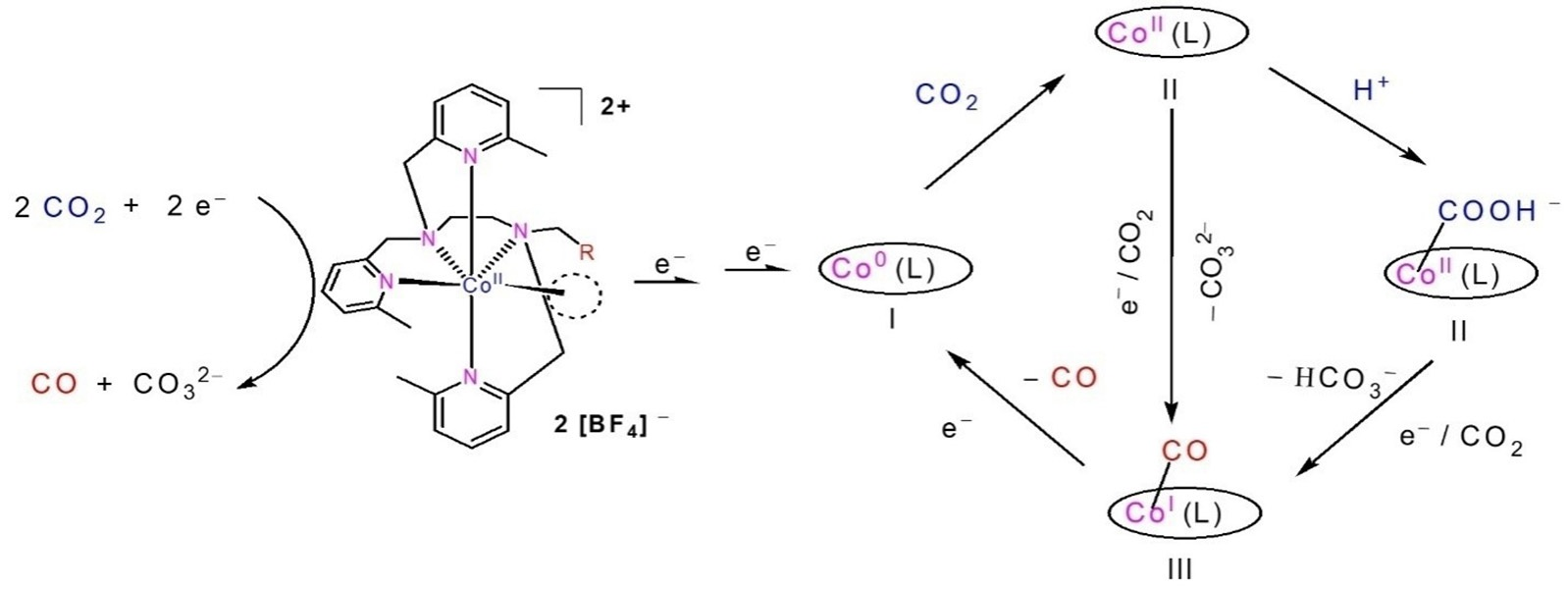{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
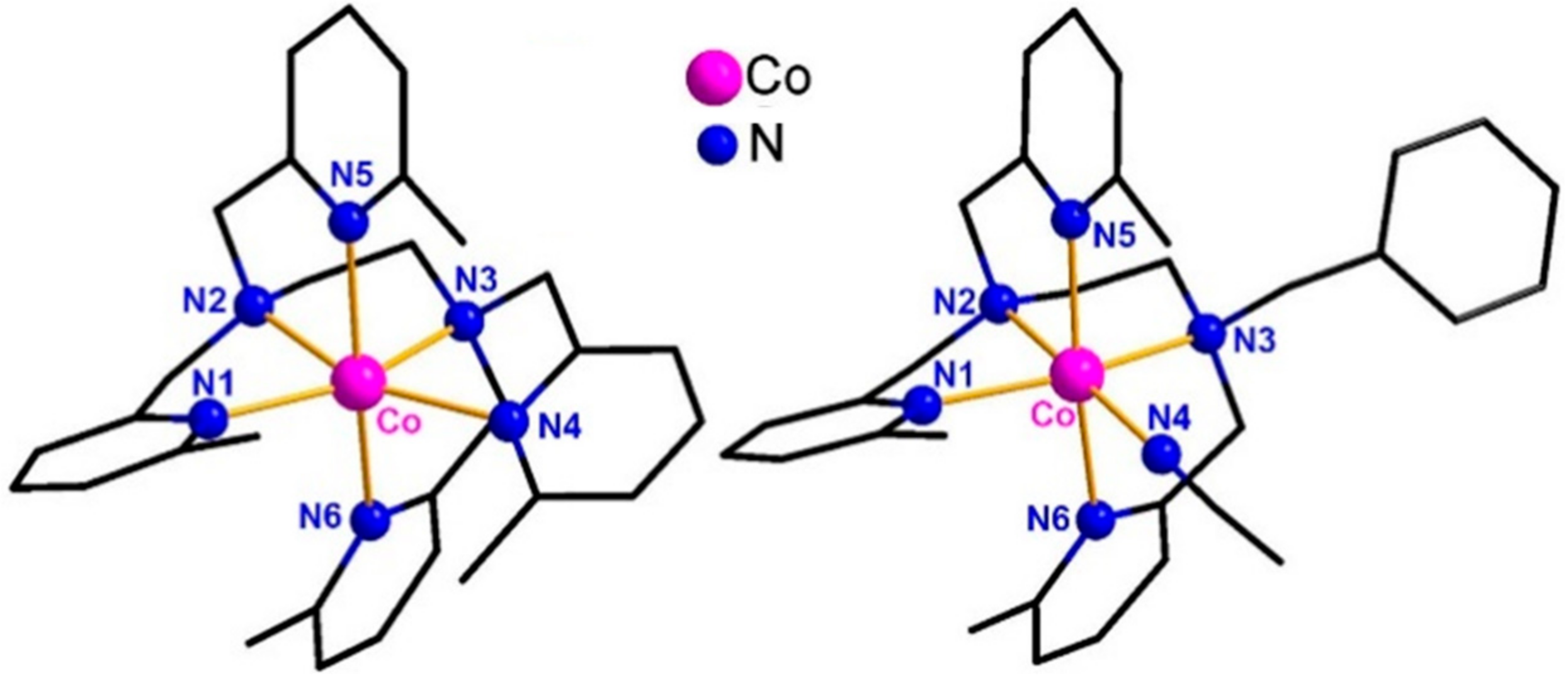
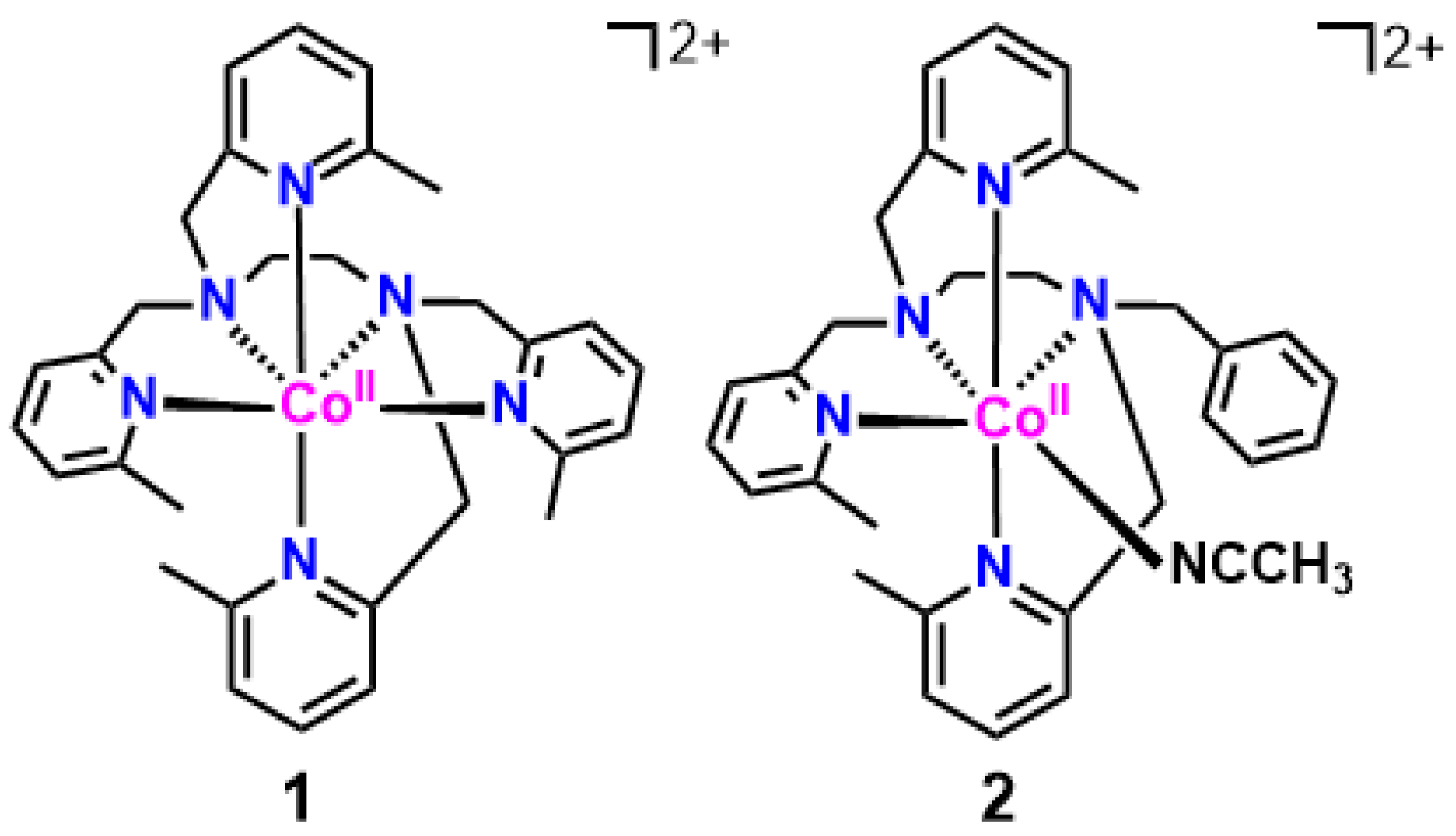
2.1. Synthesis, Characterization, and X-ray Structures
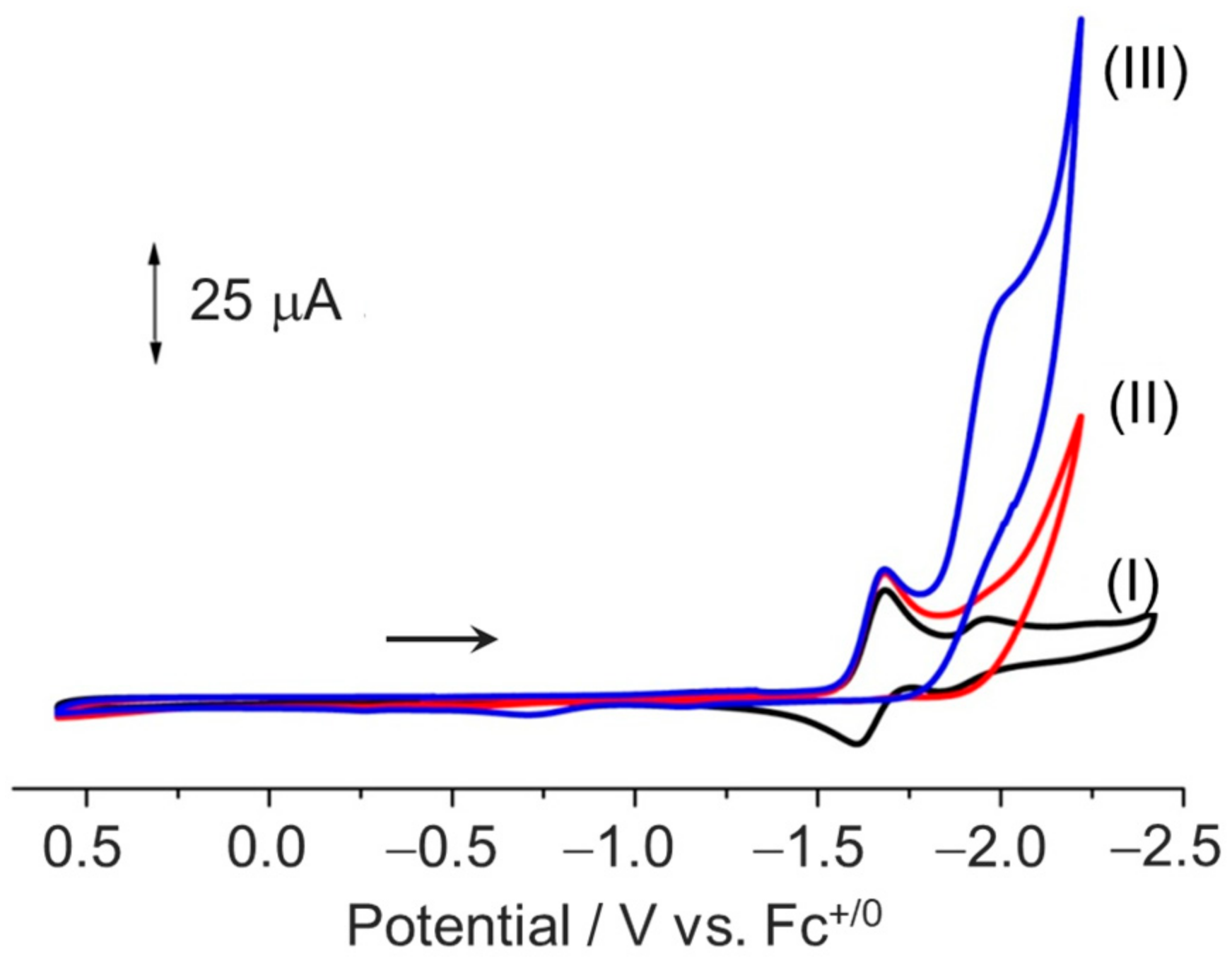
2.2. Electrochemistry
2.3. The Catalytic Rate and Stability of Cobalt Complexes
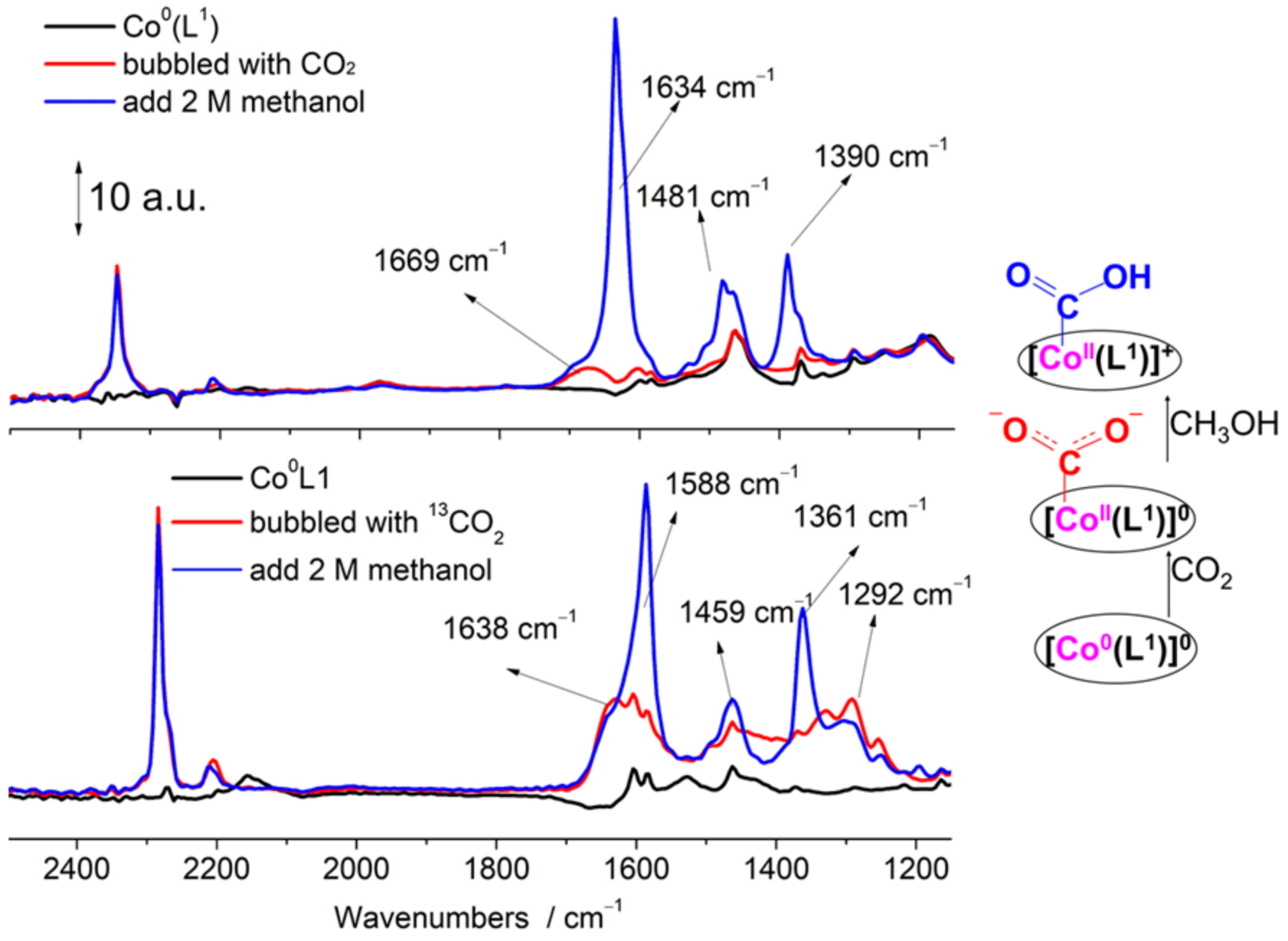
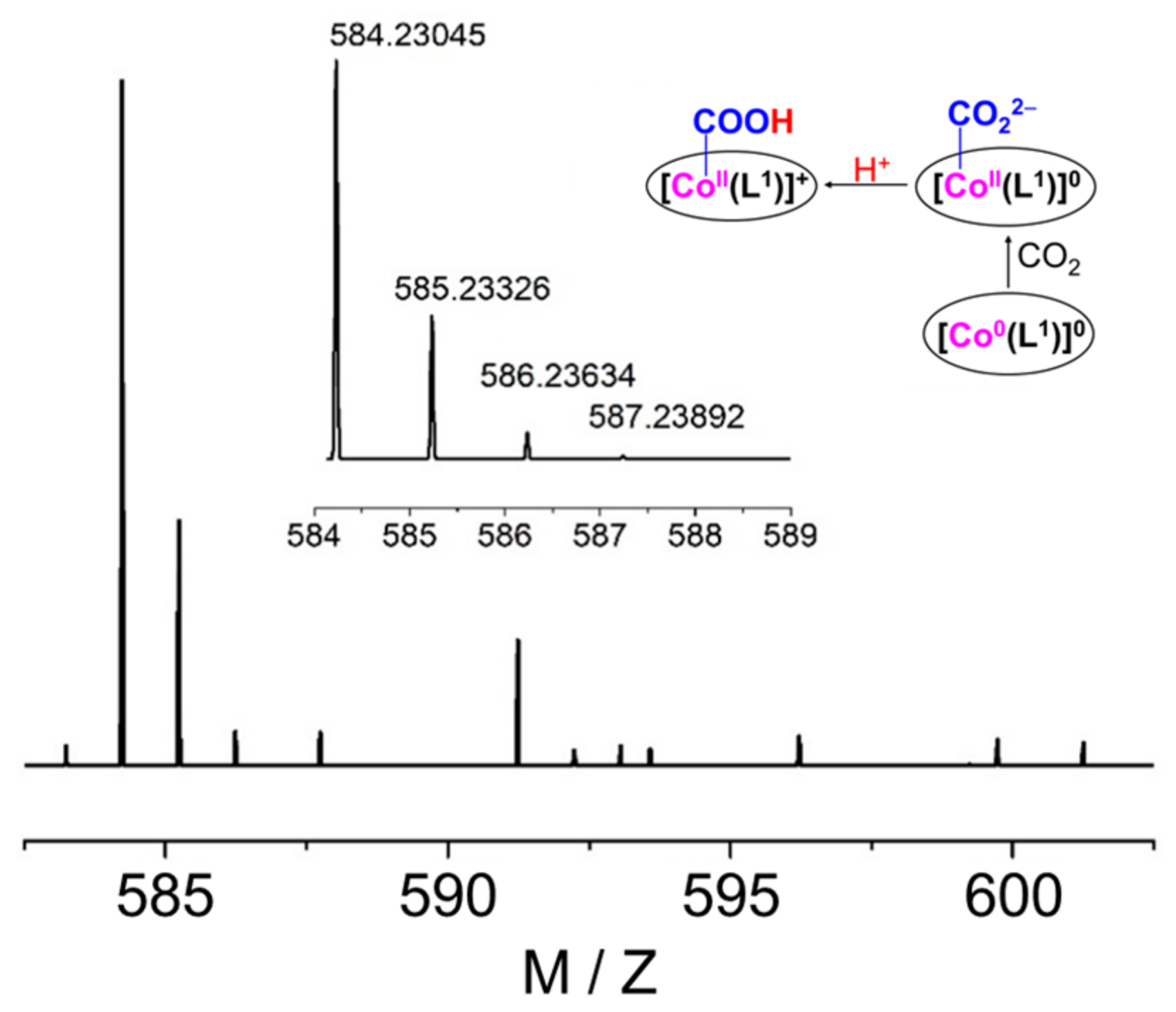
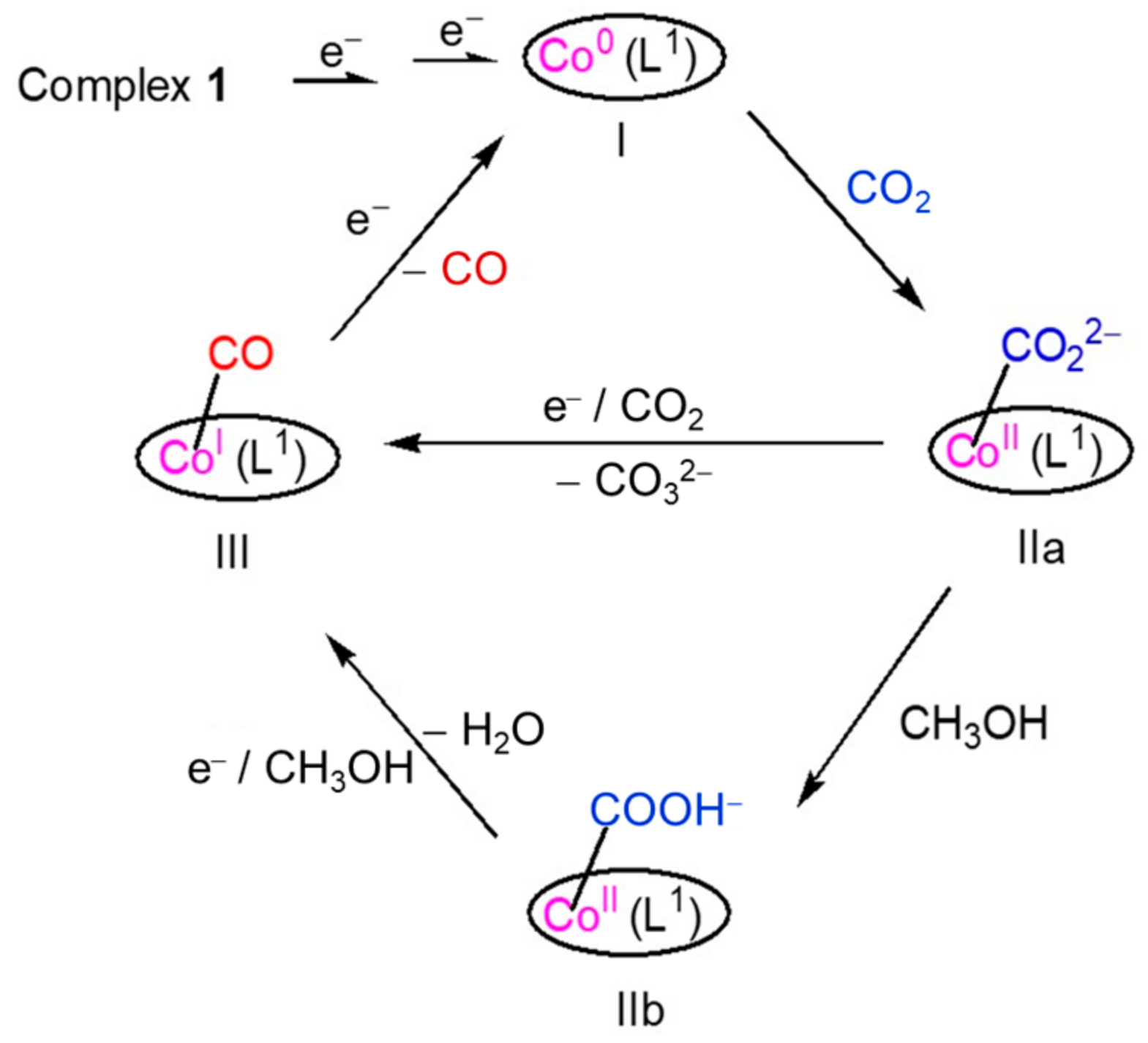
2.4. The Process and Intermediates Analysis
2.5. Density Functional Theory (DFT) Calculation and Mechanistic Consideration
3. Materials and Methods
3.1. Materials and Instruments
3.2. Synthesis
3.3. IR-SEC Test Details
3.4. Computational Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Davis, S.J.; Caldeira, K.; Matthews, H.D. Future CO2 Emissions and Climate Change from Existing Energy Infrastructure. Science 2010, 329, 1330–1333. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Khosrowabadi Kotyk, J.F.; Sheehan, S.W. Progress toward Commercial Application of Electrochemical Carbon Dioxide Reduction. Chem 2018, 4, 2571–2586. [Google Scholar] [CrossRef]
- Benson, E.E.; Kubiak, C.P.; Sathrum, A.J.; Smieja, J.M. Electrocatalytic and Homogeneous Approaches to conversion of CO2 to Liquid Fuels. Chem. Soc. Rev. 2009, 38, 89–99. [Google Scholar] [CrossRef] [PubMed]
- Boutin, E.; Merakeb, L.; Ma, B.; Boudy, B.; Wang, M.; Bonin, J.; Anxolabéhère-Mallart, E.; Robert, M. Molecular Catalysis of CO2 Reduction: Recent Advances and Perspectives in Electrochemical and Light-driven Processes with Selected Fe, Ni and Co Aza macrocyclic and Polypyridine Complexes. Chem. Soc. Rev. 2020, 49, 5772–5809. [Google Scholar] [CrossRef] [PubMed]
- Kinzel, N.W.; Werlé, C.; Leitner, W. Transition Metal Complexes as Catalysts for the Electroconversion of CO2: An Organometallic Perspective. Angew. Chem. Int. Ed. 2021, 60, 11628–11686. [Google Scholar] [CrossRef] [PubMed]
- Droghetti, F.; Amati, A.; Ruggi, A.; Natali, M. Bioinspired Motifs in Proton and CO2 Reduction with 3d-metal Polypyridine Complexes. Chem. Commun. 2024, 60, 658–673. [Google Scholar] [CrossRef] [PubMed]
- Costentin, C.; Drouet, S.; Robert, M.; Savéant, J.-M. Turnover Numbers, Turnover Frequencies, and Overpotential in Molecular Catalysis of Electrochemical Reactions. Cyclic Voltammetry and Preparative-Scale Electrolysis. J. Am. Chem. Soc. 2012, 134, 11235–11242. [Google Scholar] [CrossRef] [PubMed]
- Azcarate, I.; Costentin, C.; Robert, M.; Savéant, J.-M. Through-Space Charge Interaction Substituent Effects in Molecular Catalysis Leading to the Design of the Most Efficient Catalyst of CO2-to-CO Electrochemical Conversion. J. Am. Chem. Soc. 2016, 138, 16639–16644. [Google Scholar] [CrossRef]
- Jeoung, J.-H.; Dobbek, H. Carbon Dioxide Activation at the Ni,Fe-Cluster of Anaerobic Carbon Monoxide Dehydrogenase. Science 2007, 318, 1461–1464. [Google Scholar] [CrossRef]
- Can, M.; Armstrong, F.A.; Ragsdale, S.W. Structure, Function, and Mechanism of the Nickel Metalloenzymes, CO Dehydrogenase, and Acetyl-CoA Synthase. Chem. Rev. 2014, 114, 4149–4174. [Google Scholar] [CrossRef]
- Hawecker, J.; Lehn, J.-M.; Ziessel, R. Electrocatalytic Reduction of Carbon Dioxide Mediated by Re(bipy)(CO)3Cl (bipy = 2,2′-bipyridine). J. Chem. Soc. Chem. Commun. 1984, 6, 328–330. [Google Scholar] [CrossRef]
- Smieja, J.M.; Sampson, M.D.; Grice, K.A.; Benson, E.E.; Froehlich, J.D.; Kubiak, C.P. Manganese as a Substitute for Rhenium in CO2 Reduction Catalysts: The Importance of Acids. Inorg. Chem. 2013, 52, 2484–2491. [Google Scholar] [CrossRef] [PubMed]
- Machan, C.W.; Sampson, M.D.; Kubiak, C.P. A Molecular Ruthenium Electrocatalyst for the Reduction of Carbon Dioxide to CO and Formate. J. Am. Chem. Soc. 2015, 137, 8564–8571. [Google Scholar] [CrossRef]
- Johnson, B.A.; Maji, S.; Agarwala, H.; White, T.A.; Mijangos, E.; Ott, S. Activating a Low Overpotential CO2 Reduction Mechanism by a Strategic Ligand Modification on a Ruthenium Polypyridyl Catalyst. Angew. Chem. Int. Ed. 2016, 55, 1825–1829. [Google Scholar] [CrossRef]
- Kang, P.; Cheng, C.; Chen, Z.; Schauer, C.K.; Meyer, T.J.; Brookhart, M. Selective Electrocatalytic Reduction of CO2 to Formate by Water-Stable Iridium Dihydride Pincer Complexes. J. Am. Chem. Soc. 2012, 134, 5500–5503. [Google Scholar] [CrossRef] [PubMed]
- Todorova, T.K.; Huan, T.N.; Wang, X.; Agarwala, H.; Fontecave, M. Controlling Hydrogen Evolution during Photoreduction of CO2 to Formic Acid Using [Rh(R-bpy)(Cp*)Cl]+ Catalysts: A Structure–Activity Study. Inorg. Chem. 2019, 58, 6893–6903. [Google Scholar] [CrossRef] [PubMed]
- Caix, C.; Chardon-Noblat, S.; Deronzier, A. Electrocatalytic Reduction of CO2 into Formate with [(η5 Me5C5)M(L)Cl]+ Complexes (L = 2,2′-bipyridine ligands; M Rh(III) and Ir(III)). J. Electroanal. Chem. 1997, 434, 163–170. [Google Scholar] [CrossRef]
- Bourrez, M.; Molton, F.; Chardon-Noblat, S.; Deronzier, A. [Mn(bipyridyl)(CO)3Br]: An Abundant Metal Carbonyl Complex as Efficient Electrocatalyst for CO2 Reduction. Angew. Chem. Int. Ed. 2011, 50, 9903–9906. [Google Scholar] [CrossRef] [PubMed]
- Sampson, M.D.; Nguyen, A.D.; Grice, K.A.; Moore, C.E.; Rheingold, A.L.; Kubiak, C.P. Manganese Catalysts with Bulky Bipyridine Ligands for the Electrocatalytic Reduction of Carbon Dioxide: Eliminating Dimerization and Altering Catalysis. J. Am. Chem. Soc. 2014, 136, 5460–5471. [Google Scholar] [CrossRef]
- Sung, S.; Li, X.; Wolf, L.M.; Meeder, J.R.; Bhuvanesh, N.S.; Grice, K.A.; Panetier, J.A.; Nippe, M. Synergistic Effects of Imidazolium-Functionalization on fac-Mn(CO)3 Bipyridine Catalyst Platforms for Electrocatalytic Carbon Dioxide Reduction. J. Am. Chem. Soc. 2019, 141, 6569–6582. [Google Scholar] [CrossRef]
- Yang, Y.; Ertem, M.Z.; Duan, L. An Amide-based Second Coordination Sphere Promotes the Dimer Pathway of Mn-catalyzed CO2-to-CO Reduction at Low Overpotential. Chem. Sci. 2021, 12, 4779–4788. [Google Scholar] [CrossRef] [PubMed]
- He, S.; Qing, Y.; Zhang, P.; Xiong, Y.; Wu, Q.; Zhang, Y.; Chen, L.; Huang, F.; Li, F. Proton Tunneling Distances for Metal Hydrides Formation Manage the Selectivity of Electrochemical CO2 Reduction Reaction. Angew. Chem. Int. Ed. 2023, 62, e202216082. [Google Scholar] [CrossRef] [PubMed]
- Costentin, C.; Drouet, S.; Robert, M.; Savéant, J.-M. A Local Proton Source Enhances CO2 Electroreduction to CO by a Molecular Fe Catalyst. Science 2012, 338, 90–94. [Google Scholar] [CrossRef] [PubMed]
- Costentin, C.; Robert, M.; Savéant, J.-M.; Tatin, A. Efficient and Selective Molecular Catalyst for the CO2-to-CO Electrochemical Conversion in Water. Proc. Natl. Acad. Sci. USA 2015, 112, 6882–6886. [Google Scholar] [CrossRef] [PubMed]
- Nichols, A.W.; Chatterjee, S.; Sabat, M.; Machan, C.W. Electrocatalytic Reduction of CO2 to Formate by an Iron Schiff Base Complex. Inorg. Chem. 2018, 57, 2111–2121. [Google Scholar] [CrossRef] [PubMed]
- Froehlich, J.D.; Kubiak, C.P. The Homogeneous Reduction of CO2 by [Ni(cyclam)]+: Increased Catalytic Rates with the Addition of a CO Scavenger. J. Am. Chem. Soc. 2015, 137, 3565–3573. [Google Scholar] [CrossRef] [PubMed]
- Hong, D.; Tsukakoshi, Y.; Kotani, H.; Ishizuka, T.; Kojima, T. Visible-Light-Driven Photocatalytic CO2 Reduction by a Ni(II) Complex Bearing a Bioinspired Tetradentate Ligand for Selective CO Production. J. Am. Chem. Soc. 2017, 139, 6538–6541. [Google Scholar] [CrossRef]
- Enoki, O.; Imaoka, T.; Yamamoto, K. Electrochemical Reduction of Carbon Dioxide Catalyzed by Cofacial Dinuclear Metalloporphyrin. Macromol. Symp. 2003, 204, 151–158. [Google Scholar] [CrossRef]
- Lacy, D.C.; McCrory, C.C.L.; Peters, J.C. Studies of Cobalt-Mediated Electrocatalytic CO2 Reduction Using a Redox-Active Ligand. Inorg. Chem. 2014, 53, 4980–4988. [Google Scholar] [CrossRef]
- Su, X.; McCardle, K.M.; Chen, L.; Panetier, J.A.; Jurss, J.W. Robust and Selective Cobalt Catalysts Bearing Redox-Active Bipyridyl-N-heterocyclic Carbene Frameworks for Electrochemical CO2 Reduction in Aqueous Solutions. ACS Catal. 2019, 9, 7398–7408. [Google Scholar] [CrossRef]
- Johnson, E.M.; Liu, J.J.; Samuel, A.D.; Haiges, R.; Marinescu, S.C. Switching Catalyst Selectivity via the Introduction of a Pendant Nitrophenyl Group. Inorg. Chem. 2022, 61, 1316–1326. [Google Scholar] [CrossRef] [PubMed]
- Chapovetsky, A.; Liu, J.J.; Welborn, M.; Luna, J.M.; Do, T.; Haiges, R.; Miller Iii, T.F.; Marinescu, S.C. Electronically Modified Cobalt Aminopyridine Complexes Reveal an Orthogonal Axis for Catalytic Optimization for CO2 Reduction. Inorg. Chem. 2020, 59, 13709–13718. [Google Scholar] [CrossRef] [PubMed]
- Cometto, C.; Chen, L.; Lo, P.-K.; Guo, Z.; Lau, K.-C.; Anxolabéhère-Mallart, E.; Fave, C.; Lau, T.-C.; Robert, M. Highly Selective Molecular Catalysts for the CO2-to-CO Electrochemical Conversion at Very Low Overpotential. Contrasting Fe vs. Co Quaterpyridine Complexes upon Mechanistic Studies. ACS Catal. 2018, 8, 3411–3417. [Google Scholar] [CrossRef]
- Chen, L.; Chen, G.; Leung, C.-F.; Cometto, C.; Robert, M.; Lau, T.-C. Molecular Quaterpyridine-based Metal Complexes for Small Molecule Activation: Water Splitting and CO2 Reduction. Chem. Soc. Rev. 2020, 49, 7271–7283. [Google Scholar] [CrossRef] [PubMed]
- Hiratsuka, K.; Takahashi, K.; Sasaki, H.; Toshima, S. Electrocatalytic Behavior of Tetrasulfonated Metal Phthalocyanines in the Reduction of Carbon Dioxide. Chem. Lett. 2006, 6, 1137–1140. [Google Scholar] [CrossRef]
- Takahashi, K.; Hiratsuka, K.; Sasaki, H.; Toshima, S. Electrocatalytic Behavior of Metal Porphyrins in the Reduction of Carbon Dioxide. Chem. Lett. 2006, 8, 305–308. [Google Scholar] [CrossRef]
- Behar, D.; Dhanasekaran, T.; Neta, P.; Hosten, C.M.; Ejeh, D.; Hambright, P.; Fujita, E. Cobalt Porphyrin Catalyzed Reduction of CO2. Radiation Chemical, Photochemical, and Electrochemical Studies. J. Phys. Chem. A 1998, 102, 2870–2877. [Google Scholar] [CrossRef]
- Gangi, D.A.; Durand, R.R. Binding of Carbon Dioxide to Cobalt and Nickel Tetra-aza Macrocycles. J. Chem. Soc. Chem. Commun. 1986, 9, 697–699. [Google Scholar] [CrossRef]
- Chapovetsky, A.; Do, T.H.; Haiges, R.; Takase, M.K.; Marinescu, S.C. Proton-Assisted Reduction of CO2 by Cobalt Aminopyridine Macrocycles. J. Am. Chem. Soc. 2016, 138, 5765–5768. [Google Scholar] [CrossRef]
- Chapovetsky, A.; Welborn, M.; Luna, J.M.; Haiges, R.; Miller, T.F., III; Marinescu, S.C. Pendant Hydrogen-Bond Donors in Cobalt Catalysts Independently Enhance CO2 Reduction. ACS Cent. Sci. 2018, 4, 397–404. [Google Scholar] [CrossRef]
- Cometto, C.; Chen, L.; Anxolabéhère-Mallart, E.; Fave, C.; Lau, T.-C.; Robert, M. Molecular Electrochemical Catalysis of the CO2-to-CO Conversion with a Co Complex: A Cyclic Voltammetry Mechanistic Investigation. Organometallics 2019, 38, 1280–1285. [Google Scholar] [CrossRef]
- Costentin, C.; Passard, G.; Robert, M.; Savéant, J.-M. Ultraefficient Homogeneous Catalyst for the CO2-to-CO Electrochemical Conversion. Proc. Natl. Acad. Sci. USA 2014, 111, 14990–14994. [Google Scholar] [CrossRef] [PubMed]
- Nichols, E.M.; Derrick, J.S.; Nistanaki, S.K.; Smith, P.T.; Chang, C.J. Positional Effects of Second-sphere Amide Pendants on Electrochemical CO2 Eeduction Catalyzed by Iron Porphyrins. Chem. Sci. 2018, 9, 2952–2960. [Google Scholar] [CrossRef] [PubMed]
- Anxolabéhère-Mallart, E.; Costentin, C.; Fournier, M.; Nowak, S.; Robert, M.; Savéant, J.-M. Boron-Capped Tris(glyoximato) Cobalt Clathrochelate as a Precursor for the Electrodeposition of Nanoparticles Catalyzing H2 Evolution in Water. J. Am. Chem. Soc. 2012, 134, 6104–6107. [Google Scholar] [CrossRef]
- Lassalle-Kaiser, B.; Zitolo, A.; Fonda, E.; Robert, M.; Anxolabéhère-Mallart, E. In Situ Observation of the Formation and Structure of Hydrogen-Evolving Amorphous Cobalt Electrocatalysts. ACS Energy Lett. 2017, 2, 2545–2551. [Google Scholar] [CrossRef]
- Nie, W.; Wang, Y.; Zheng, T.; Ibrahim, A.; Xu, Z.; McCrory, C.C.L. Electrocatalytic CO2 Reduction by Cobalt Bis(pyridylmonoimine) Complexes: Effect of Ligand Flexibility on Catalytic Activity. ACS Catal. 2020, 10, 4942–4959. [Google Scholar] [CrossRef]
- Sheng, H.; Frei, H. Direct Observation by Rapid-Scan FT-IR Spectroscopy of Two-Electron-Reduced Intermediate of Tetraaza Catalyst [CoIIN4H(MeCN)]2+ Converting CO2 to CO. J. Am. Chem. Soc. 2016, 138, 9959–9967. [Google Scholar] [CrossRef]
- Kou, Y.; Nabetani, Y.; Masui, D.; Shimada, T.; Takagi, S.; Tachibana, H.; Inoue, H. Direct Detection of Key Reaction Intermediates in Photochemical CO2 Reduction Sensitized by a Rhenium Bipyridine Complex. J. Am. Chem. Soc. 2014, 136, 6021–6030. [Google Scholar] [CrossRef] [PubMed]
- Szalda, D.J.; Fujita, E.; Creutz, C. Cobalt(I), -(II), and -(III) Complexes of a Tetraaza 14-membered Macrocycle, 5,7,7,12,14,14-hexamethyl-1,4,8,11-tetraazacyclotetradeca-4,11-diene (L). Crystal and Molecular Structures of [CoL(CO)]ClO4, trans-CoLCl2, and cis-[CoL(CO3)]ClO4. Inorg. Chem. 1989, 28, 1446–1450. [Google Scholar] [CrossRef]
- Schneider, J.; Jia, H.; Muckerman, J.T.; Fujita, E. Thermodynamics and Kinetics of CO2, CO, and H+ Binding to the Metal Centre of CO2 Reduction catalysts. Chem. Soc. Rev. 2012, 41, 2036–2051. [Google Scholar] [CrossRef]
- Smieja, J.M.; Benson, E.E.; Kumar, B.; Grice, K.A.; Seu, C.S.; Miller, A.J.M.; Mayer, J.M.; Kubiak, C.P. Kinetic and Structural Studies, Origins of Selectivity, and Interfacial Charge Transfer in the Artificial Photosynthesis of CO. Proc. Natl. Acad. Sci. USA 2012, 109, 15646–15650. [Google Scholar] [CrossRef]
- Mei, F.; Ou, C.; Wu, G.; Cao, L.; Han, F.; Meng, X.; Li, J.; Li, D.; Liao, Z. Non-heme Iron(ii/iii) Complexes that Model the Reactivity of Lipoxygenase with a Redox Switch. Dalton Trans. 2010, 39, 4267–4269. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Wang, M.; Gloaguen, F.; Chen, L.; Quentel, F.; Sun, L. Electrocatalytic Hydrogen Evolution from Neutral Water by Molecular Cobalt Tripyridine–diamine Complexes. Chem. Commun. 2013, 49, 9455–9457. [Google Scholar] [CrossRef] [PubMed]
- Becke, A.D. A New Mixing of Hartree-Fock and Local Density Functional Theories. J. Chem. Phys. 1993, 98, 1372–1377. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti Correlation-energy Formula into a Functional of the Electron Density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Chen, L.; Guo, Z.; Wei, X.-G.; Gallenkamp, C.; Bonin, J.; Anxolabéhère-Mallart, E.; Lau, K.-C.; Lau, T.-C.; Robert, M. Molecular Catalysis of the Electrochemical and Photochemical Reduction of CO2 with Earth-Abundant Metal Complexes. Selective Production of CO vs. HCOOH by Switching of the Metal Center. J. Am. Chem. Soc. 2015, 137, 10918–10921. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, Y.; Xie, F.; Chen, J.; Qiu, S.; Qiang, N.; Lu, M.; Peng, Z.; Yang, J.; Liu, G. Electrocatalytic Reduction of CO2 to CO by Molecular Cobalt–Polypyridine Diamine Complexes. Molecules 2024, 29, 1694. https://doi.org/10.3390/molecules29081694
Yang Y, Xie F, Chen J, Qiu S, Qiang N, Lu M, Peng Z, Yang J, Liu G. Electrocatalytic Reduction of CO2 to CO by Molecular Cobalt–Polypyridine Diamine Complexes. Molecules. 2024; 29(8):1694. https://doi.org/10.3390/molecules29081694
Chicago/Turabian StyleYang, Yong, Fang Xie, Jiahui Chen, Si Qiu, Na Qiang, Ming Lu, Zhongli Peng, Jing Yang, and Guocong Liu. 2024. "Electrocatalytic Reduction of CO2 to CO by Molecular Cobalt–Polypyridine Diamine Complexes" Molecules 29, no. 8: 1694. https://doi.org/10.3390/molecules29081694
APA StyleYang, Y., Xie, F., Chen, J., Qiu, S., Qiang, N., Lu, M., Peng, Z., Yang, J., & Liu, G. (2024). Electrocatalytic Reduction of CO2 to CO by Molecular Cobalt–Polypyridine Diamine Complexes. Molecules, 29(8), 1694. https://doi.org/10.3390/molecules29081694