1. Introduction
Diabetic patients suffer from chronic non-healing wounds [
1], mainly due to tissue loss and inadequate blood flow to the wound bed [
2,
3]. Therefore, the reconstruction of the micro-vascular network becomes a significant challenge for the tissue repair of diabetic wounds. A large number of experimental and clinical studies have shown that the healing of diabetic wounds does not follow the normal process of wound healing seen in acute wounds due to the complex physiological micro-environment in chronic wounds, and the repair effect achieved by using conventional wound dressings is very limited. Therefore, new technologies are being translated from the test bench to clinical practice using bio-active materials and synthetic bio-polymers, aiming to provide new strategies for diabetic wound healing.
At present, the dressings used for the treatment of diabetic wounds mainly include hydrogels, electrospun nanofibers, bio-macromolecule scaffolds/sponges and bio-active glass wound dressings [
4,
5], all of which have shown the potential to enhance diabetic wound healing. For example, Lv et al. reported a silicate-containing poly(caprolactone)/gelatin nanofibrous composite scaffold for diabetic wounds and suggested that the composite scaffold had a synergetic effect, improving the efficiency of diabetic wound healing [
6]. Pietramaggiori et al. fabricated a poly N-acetylglucosamine hydrogel matrix system for the treatment of diabetic foot ulcers [
7,
8,
9]. Yu and co-workers prepared a curcumin-loaded in situ injectable hydrogel based on sodium alginate and chitosan. Nano-curcumin in a hydrogel was able to leach slowly to promote fibroblast proliferation, collagen generation and capillary formation, leading to diabetic wound healing [
10,
11]. Yang et al. [
12] utilized electrospinning technology to prepare nanofiber pad silk fibroin dressings containing epidermal growth factor (EGF) and basic fibroblast growth factor (bFGF). The results indicated that reconstructed nanofiber pads could support and enhance cell migration to the wound bed in high-glucose environments, then promote wound healing [
13]. In addition, bio-macromolecules and their complexes based on carboxymethyl cellulose, silk fibroin, chitosan and collagen have also been used as wound dressings and show great promise in inducing tissue regeneration in diabetic wound healing [
6].
Silk fibroin is a natural amino acid polymer with a composition and structure similar to that of a natural extracellular matrix. It can penetrate the surface of wounds and maintain a gentle, moist environment to promote wound healing [
14]. Collagen-based skin dressings have also been reported to have a rapid and effective therapeutic effect on diabetic wounds [
15] because collagen and its degradation products and metabolic derivatives can attract fibroblasts to wounds and enhance skin repair [
16,
17]. For example, collagen dressings combined with silver and sodium alginate have achieved great efficacy in the treatment of diabetes wounds [
18]. Han et al. constructed a new type of collagen-based dermal scaffold coated with silver nanoparticles (NAg). The results demonstrated that NAg-containing scaffolds exerted bactericidal and anti-inflammatory effects and promoted wound healing by regulating fibroblast migration and macrophage activation [
19]. Moreover, collagen can be used in the form of a hydrogel, freeze-dried sponge, membrane, inner membrane or nanofibers generated by electrospinning [
20], showing advantages in various wound-repair scenarios [
15].
In recent years, with the continuous growth and evolution of the number of tissue repair models used in research, our understanding of tissue repair and our ability to combine this knowledge with the pathogenesis of related diseases have also been expanded. Nowadays, it is widely believed that the immune system plays a crucial role in tissue healing. Immune cells directly affect the host response at the site of injury, as well as the activity of tissue-specific cell populations and recruited and tissue-resident stem cells. In fact, the immune system has been shown to have positive and negative regulatory effects on tissue repair processes, leading to effective tissue regeneration or fibrosis and scar formation, respectively [
21,
22]. In the early stage of inflammation [
23], primitive and inactive M0 macrophages (Mφs) are primarily activated to the pro-inflammatory M1 phenotype, which initiates an immune response and engulfs micro-organisms, pathogens, abnormal cells and cell fragments. Additional white blood cells are also recruited and produce chemokines [
24]. During the later healing process, M0 macrophages mainly transform into the M2 phenotype with anti-inflammatory effects and can release various anti-inflammatory cytokines to assist in wound healing [
25]. The biological functions of M2 macrophages also include promoting fibroblast proliferation; collagen remodeling; neovascularization; and the expression of anti-inflammatory mediators such as
IL-1R antagonists,
IL-10, transforming growth factor β (
TGF-β) and vascular endothelial growth factor (
VEGF) [
26]. However, unlike normal acute wounds, there is a large number of M1 phenotype macrophages in chronic wounds of diabetes, and these macrophages excessively produce related pro-inflammatory cytokines such as tumor necrosis factor α (
TNF-α), interleukin-6 and
IL-1β. The increase in these key pro-inflammatory cytokines leads to a prolonged inflammatory phase and delayed wound healing [
27]. Therefore, in view of the special micro-environment of diabetes wounds, transforming macrophages from the M1 to M2 phenotype and promoting the secretion of anti-inflammatory cytokines may be effective ways to regulate the inflammatory process of diabetic wounds. In addition, in the process of immune regulation, the characteristics of implanted materials inducing the aggregation of immune cells in vivo [
28] have been shown to play a direct role in angiogenesis [
29,
30], which can improve the reconstruction of the micro-vascular network during ischemia of diabetes symptoms and promote the repair of wound tissue.
Heparin (Hep) is a highly sulfated glycosaminoglycan (GAG) that interacts with many proteins to regulate physiological and pathological processes [
31]. The combination of heparin and angiogenic growth factor has been shown to have significant therapeutic effects in the treatment of ischemic injury [
32,
33]. However, heparin has limitations in clinical application due to its anticoagulant properties [
34,
35]. Sulfated polysaccharides (such as sulfated chitosan (SCS)) are a kind of heparin-like anionic polysaccharide with a higher degree of sulfation and a similar structure to that of heparin. SCS has shown promise for a wide range of applications because of its capacity to bind to protein growth factors [
36,
37] and regulate the biological activity of growth factors [
37]. Liu et al. developed a high-affinity SCS for vascular endothelial growth factor (
VEGF) and found that it could effectively promote angiogenesis in the presence of extremely low concentrations of exogenous
VEGF [
38]. The release of SCS from an SCS-coated gelatin sponge (SCS GS) was able to effectively stimulate M1-M2 polarization of Mφs and induce endothelial cell migration and capillary formation, thus promoting angiogenesis with the help of very low doses of
VEGF [
38].
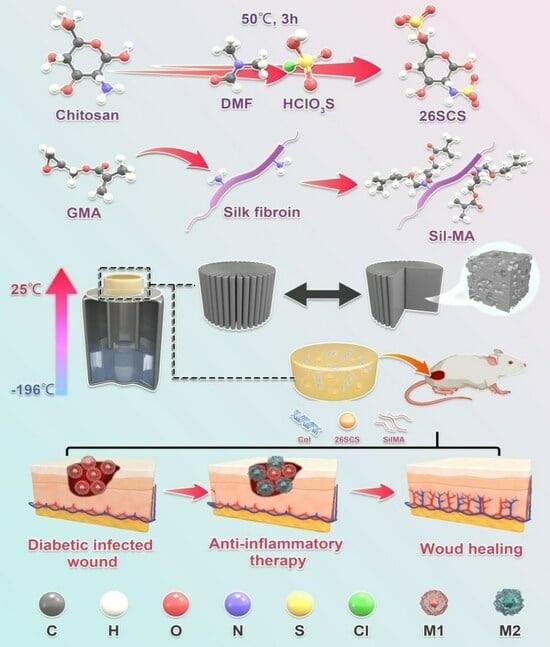
In this work, we develop a lamellar-oriented collagen/silk fibroin porous sponge loaded with SCS, aiming at guiding the polarization of Mφs from M1 to M2 and inducing efficient angiogenesis in diabetic wounds. The excellent bio-compatibility and skin affinity of collagen/silk fibroin sponges can provide a wetting environment for wounds and reduce wound infiltration and bleeding. Importantly, by regulating the release of SCS in collagen/silk fibroin sponges, Mφs polarization towards the M2 phenotype can be regulated, thereby increasing the secretion of endogenous VEGF, mediating neovascularization and ultimately promoting late vascular maturation, granulation tissue growth and wound repair function. Our research can provide a new strategy for the clinical treatment of diabetes dressings.
2. Materials and Methods
2.1. Materials
Chitosan (deacetylation degree ≥95%, viscosity from 100 to 200 mpa.s.), sodium-hydroxide, chlorosulfonic acid, lithium bromide, glycidyl methacrylate, acetic acid and anhydrous ethanol were obtained from Aladdin (Wuhan, China). N-N dimethylformamide, anhydrous sodium carbonate and glutaraldehyde were purchased from Macklin (Shanghai, China). Dulbecco’s modified Eagle medium (DMEM), penicillin–streptomycin, fetal bovine serum (FBS) and phosphate buffer saline (PBS) were supplied by Gibco (Grand Island, NY, USA). A live/dead cell double-staining kit was obtained from Proteintech (Rosemont, IL, USA), and a cell counting kit-8 was purchased from KeyGen Biotech (Nanjing, China). Enzyme-linked immunosorbent assay kits were obtained from 4A Biotech (Beijing, China). Streptozotocin was obtained from Sigma Aldrich (Burlington, MA, USA). Citric acid was purchased from Solarbio (Beijing, China). Sodium citrate was supplied by YuanYe Biotech (Shanghai, China). A hematoxylin and eosin dying kit was purchased from Beyotime (Shanghai, China), and Masson’s Trichrome dying kit was purchased from Solabio (Beijing, China), dying according to the procedure in the instructions.
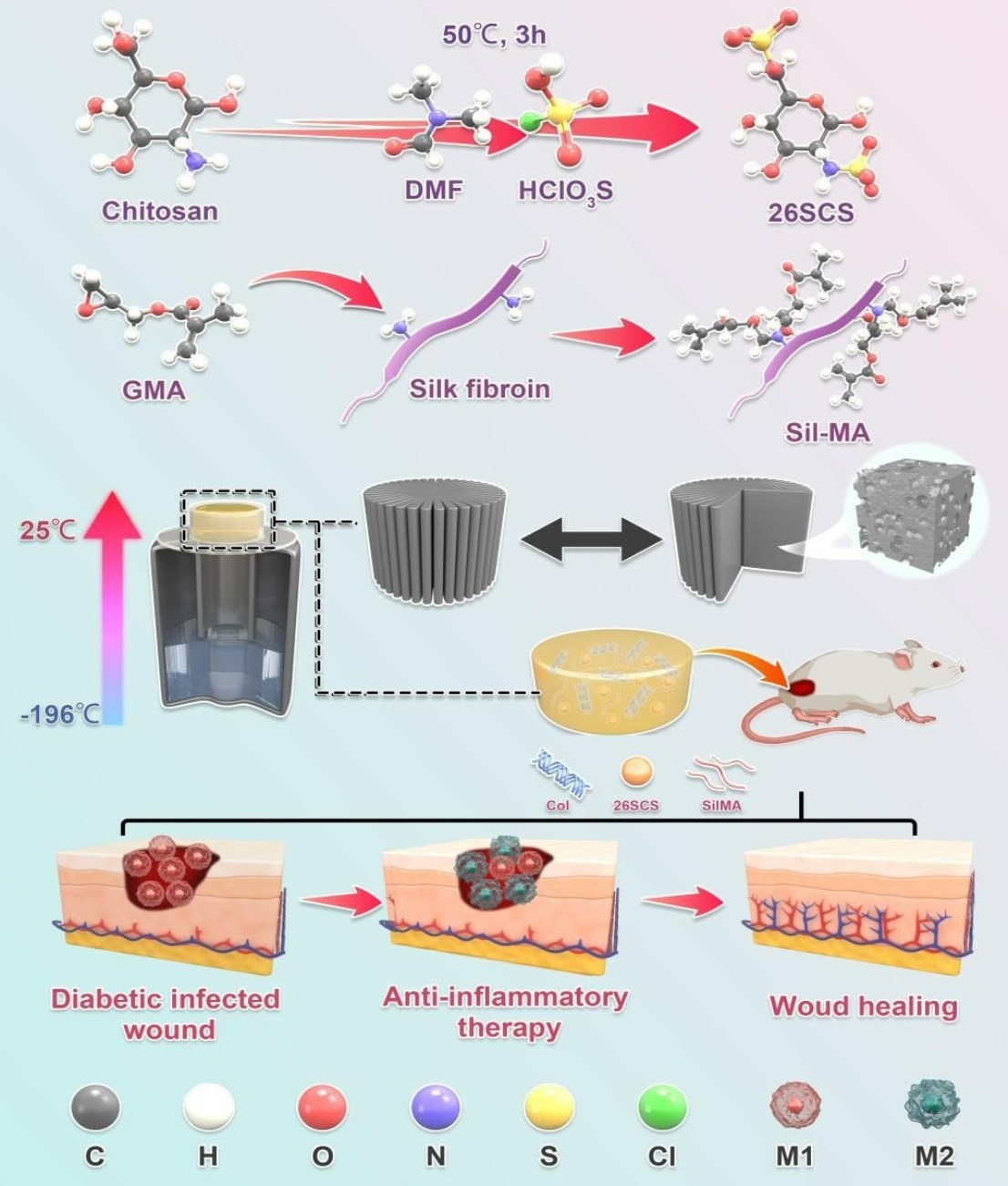
2.2. Synthesis of 2-N,6-O-Sulfonated Chitosan (26SCS)
The synthesis of 26SCS was performed with a slight modification of a previously described protocol. First, the sulfonation reagent was prepared by adding 10 mL HClSO3 into 30 mL N-N dimethylformamide (DMF), which was cooled at 4 °C in advance and stirred for 15 min. Secondly, the sulfonation reagent was transferred to a three-necked, flat-bottomed flask containing chitosan solution (2 g chitosan dispersed in 50 mL N-N dimethylformamide and 10 mL formic acid) and reacted at 50 °C for 3 h to obtain a light-yellow solution.
After the reaction was completed, the following post processing was performed. First, 100 mL deionized water was added to the above light-yellow solution to stop the reaction, and 1 M NaOH was used to adjust the pH to 7–8; then, 1500 mL anhydrous ethanol was added to precipitate product. The extracted product was dissolved in deionized water; then, the solution was dialyzed against water for 3–7 d in a 2000 Da cut-off dialysis membrane. Finally, the 26SCS was obtained after freeze-drying.
2.3. Preparation of SilMA
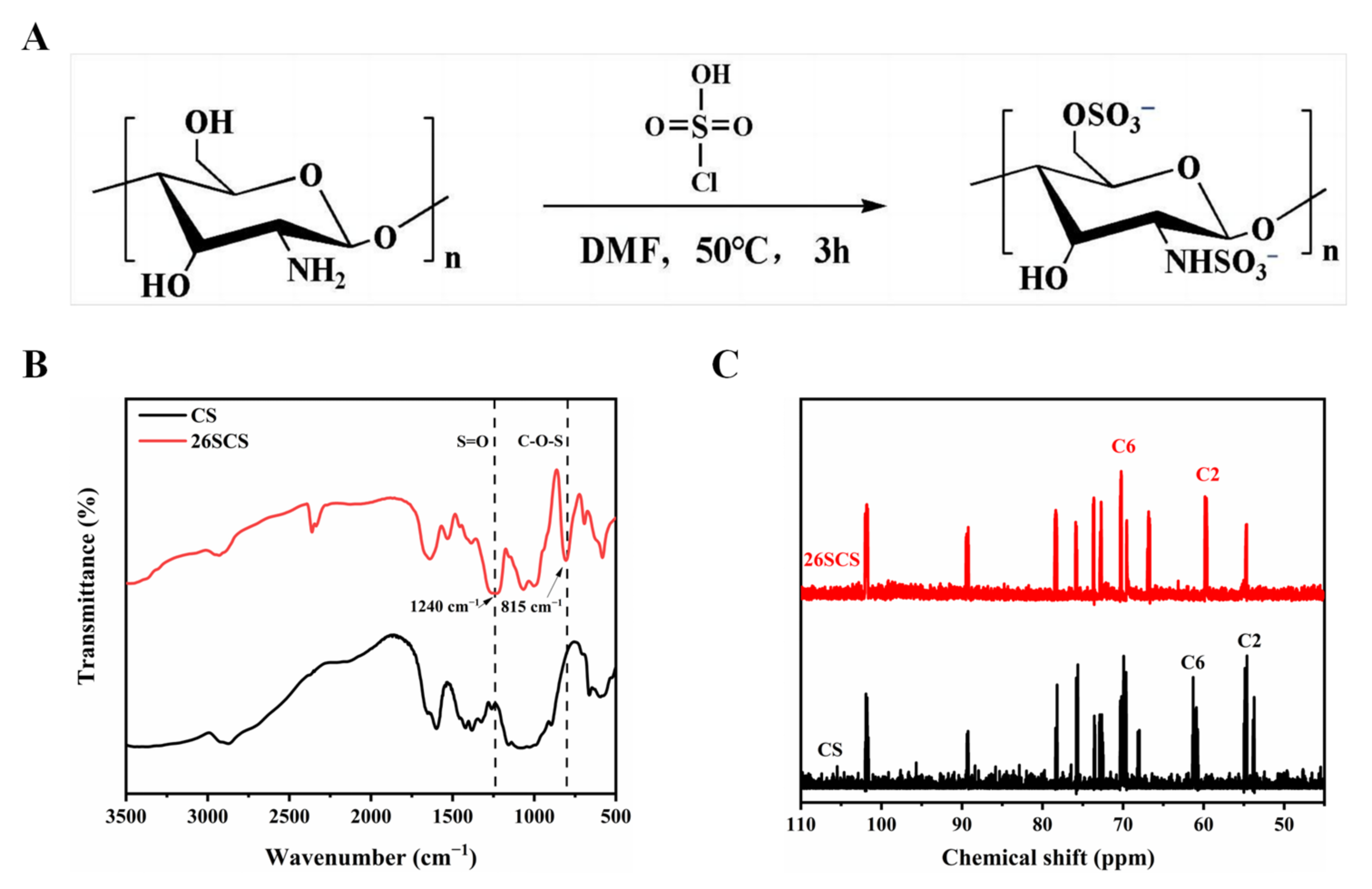
First, the natural silk was boiled with a 0.25% Na2CO3 aqueous solution for 30 min to remove sericin, and the step was repeated 3 times; then, the silk was dried in a 60 °C oven. Subsequently, the 10 g dried silk was dissolved in 50 mL lithium bromide solution (9.2 M); then, 424 mM glycidyl methacrylate (GMA) solution was added to the solution and reacted at 60 °C for 5 h. Finally, impurities were filtered from the solution, and the filtrate was dialyzed against water for 3–7 d in a 8000–14,000 Da cut-off dialysis membrane, and the water-soluble silk (SilMA) was obtained after lyophilization.
2.4. Preparation of 26SCS-Loaded Composite Sponges
First, 0.05 g collagen and 0.05 g SilMA were dissolved in 5 mL acetic acid (0.5 M) and mixed evenly; then different concentrations of 26SCS (2.5 mg/mL, 5 mg/mL, 7.5 mg/mL and 10 mg/mL) were added, and a uniform mixture was obtained after full stirring. The mixed solution was injected into the polytetrafluoroethylene mold, and a one-way temperature gradient with bottom-up temperature difference was set to form directional crystallization of the mixed solution, then freeze-dried. The obtained composite sponges were cross-linked in a glutaraldehyde atmosphere for 5 h. Finally, the composite sponges were immersed in anhydrous ethanol for 1 h and soaked in deionized water for 3 h. The 26SCS-loaded composite sponges were obtained after lyophilization ((2.5/5.0/7.5/10.0) 26SCS-loaded composite sponges).
2.5. Characterization Methods
2.5.1. Fourier Transform Infrared Spectroscopy (FTIR)
The chemical structure changes of 26SCS and SilMA were recorded using a Fourier transform infrared spectrometer (Spectrum Two, PerkinElmer, Shelton, CT, USA) in the wavenumber range of 3500–500 cm−1 with a resolution of 4 cm−1.
2.5.2. Nuclear Magnetic Resonance Spectroscopy (NMR)
The changes in functional groups before and after modification of chitosan and silk were recorded by nuclear magnetic resonance (AVANCE III 300, BRUKER, Ettlingen, Germany).
2.5.3. Scanning Electron Microscopy (SEM)
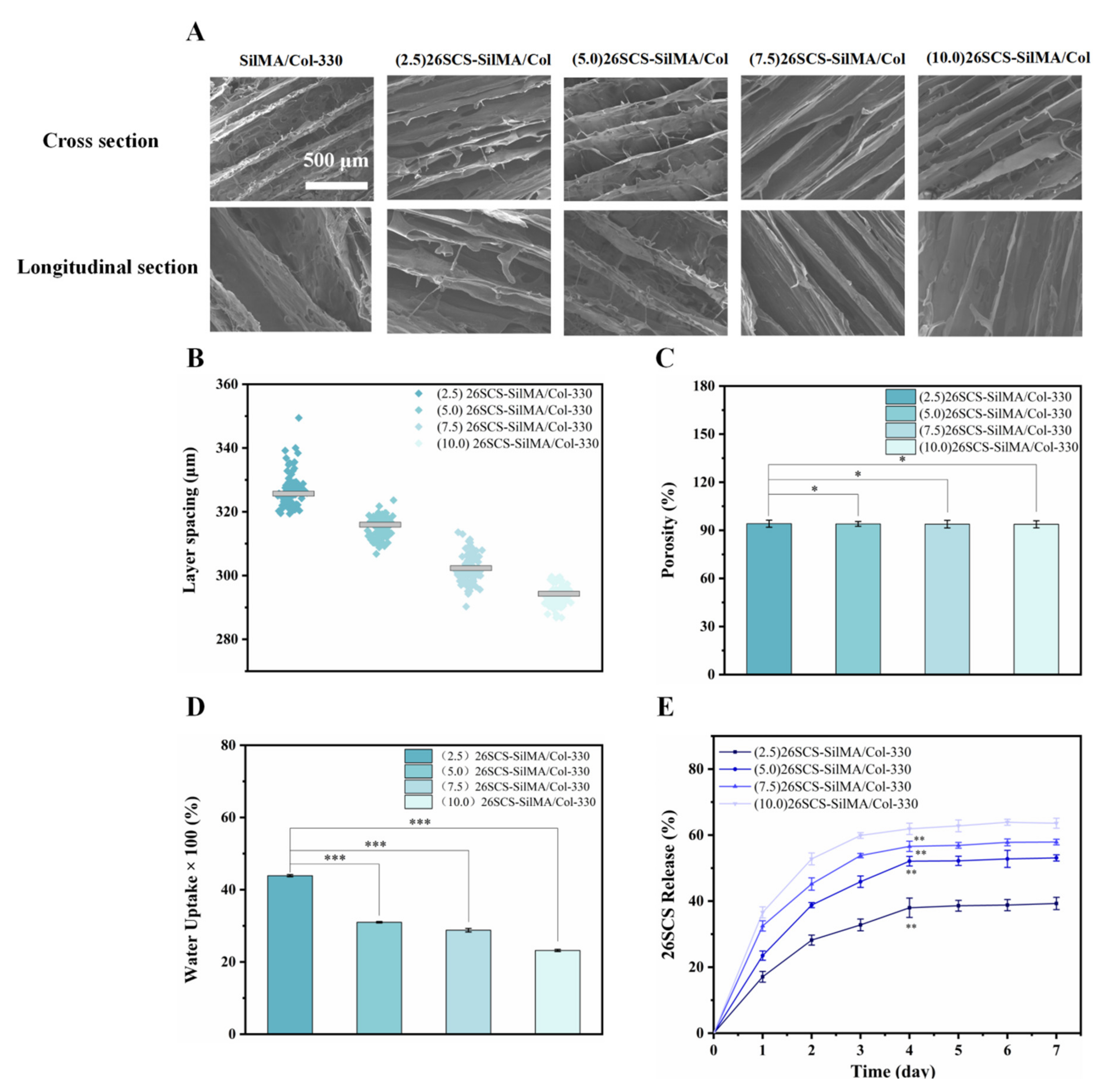
The morphology and the layer spacing of composite sponges were characterized by SEM (LEO153VP, Carl Zeiss, Jena, Germany). The samples were pasted to the copper platform and gilded and sputtered; then, their cross-sectional/longitudinal morphologies were observed. SEM images of composite sponges with a magnification of 500× were captured; then, 150 holes were randomly selected to calculate the layer spacing using ImageJ (x64) 1.8.0. (National Institute of Health, Bethesda, MD, USA).
2.5.4. Measurement of Porosity
The porosity of the composite sponges was determined by the ethanol immersion method. Briefly, the sponges were immersed in anhydrous ethanol under a vacuum atmosphere to ensure that the anhydrous ethanol fully permeated into the sponges. The wet weight (
Ww) of the composite sponges were measured after 24 h, and the porosity (
P(%)) was defined as follows:
where
ρ is the density of anhydrous ethanol, and
Wd is the dry weight of the composite sponges.
2.5.5. Measurement of Water Absorption
The fully dried composite sponges were weighed (
M0), then immersed in DI water. Filter paper was used to remove the water adsorbed on the surface of the sponges, and the wet weight (
M1) was recorded. The water absorption (
W(%)) was defined as follows:
2.5.6. Measurement of the Release of 26SCS
The 26SCS-loaded composite sponges were immersed in centrifuge tubes containing PBS solution and incubated at 37 °C in a shaker with a speed of 90 r/min. The supernatant was withdrawn at different time points (1–7 days) for detection [
37]. The absorbance of each sample was measured at a specific wavelength; then, the cumulative release of 26SCS was calculated according to the standard curve. Three parallel experiments were performed on each sample.
2.6. In Vitro Cell Response to 26SCS-SilMA/Col Sponges
2.6.1. Cell Culture
The fibroblasts (L929), macrophages (RAW264.7) and HUVECs were purchased from the American Type Culture Collection (ATCC). All cells were cultured in Dulbecco’s modified Eagle medium containing 1% penicillin and 10% fetal bovine serum at 37 °C in a 5% CO
2 cell incubator [
37]. The medium was updated every other day. In order to evaluate the effect of the 26SCS concentration on cell behaviors, we set six SCS concentrations, namely 0 mg/mL, 1 mg/mL, 2 mg/mL, 3 mg/mL, 4 mg/mL and 5 mg/mL (named (0) 26SCS, (1) 26SCS, (2) 26SCS, (3) 26SCS, (4) 26SCS and (5) 26SCS, respectively), to determine the appropriate concentration range for SCS that could have a positive effect on cell function.
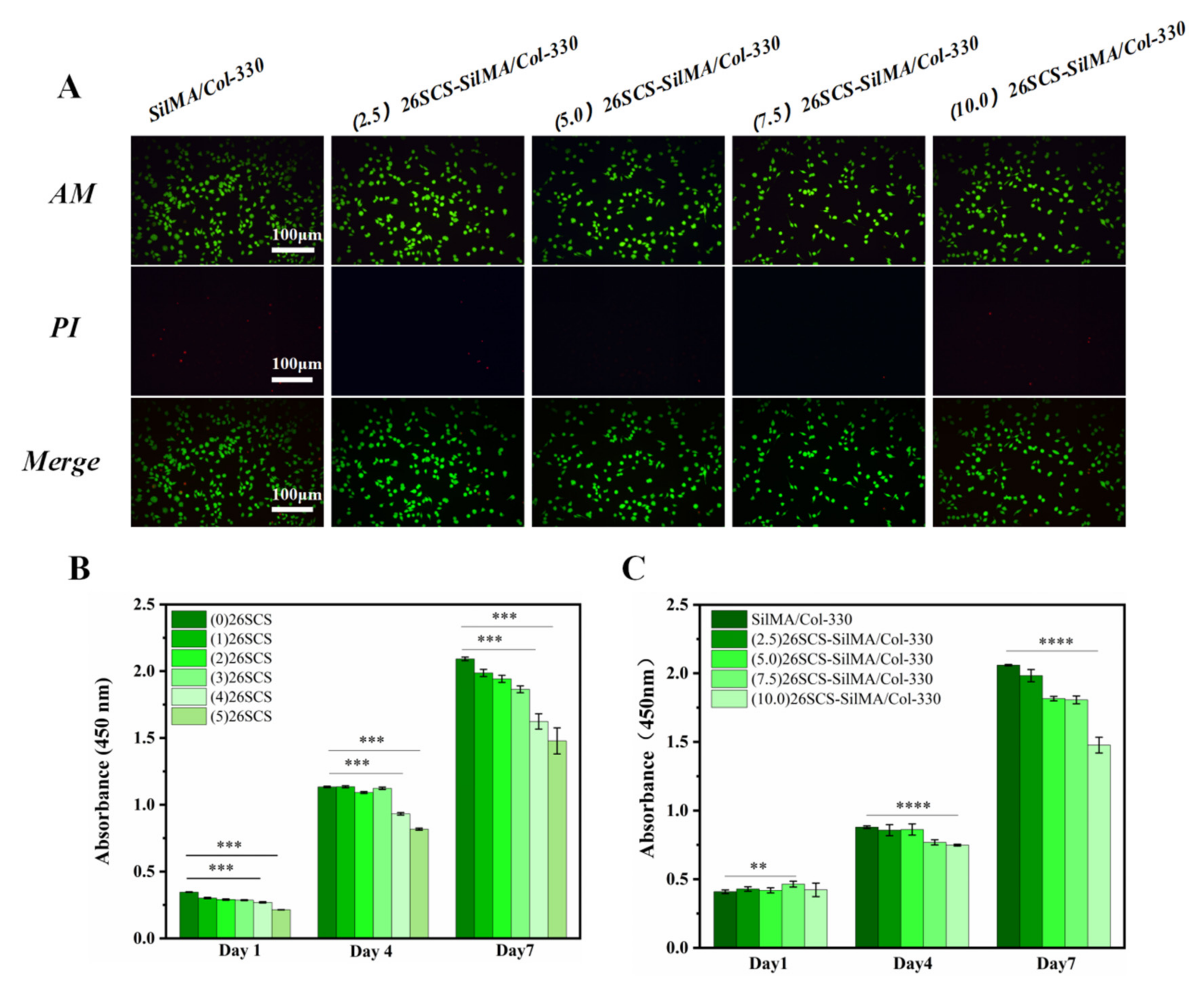
2.6.2. Cytotoxicity Assay
The fibroblasts (L929) were seeded in 48-well plates containing different concentrations of 26SCS or covered with (2.5/5.0/7.5/10.0) 26SCS-loaded sponges at a density of 2 × 104 cells per well. After 1, 4 and 7 days of incubation, the medium containing 10% CCK-8 was substituted for the original medium; then, the absorbance at 450 nm was measured using an enzyme-linked immunosorbent assay plate reader (MK3, Thermo Fisher Scientific Ltd., Waltham, MA, USA).
2.6.3. Cell Viability Assay
Similarly, the fibroblasts (L929) were seeded at a density of 2 × 104 cells per well in 48-well plates covered with 26SCS-loaded composite sponges. After 48 h of incubation, the original medium was replaced with reagent for live/dead cell staining. A fluorescence microscope (XDY-2, Guangzhou Liss Optical Instrument Ltd., Guangzhou, China) was used to observe the distribution of live/dead cells.
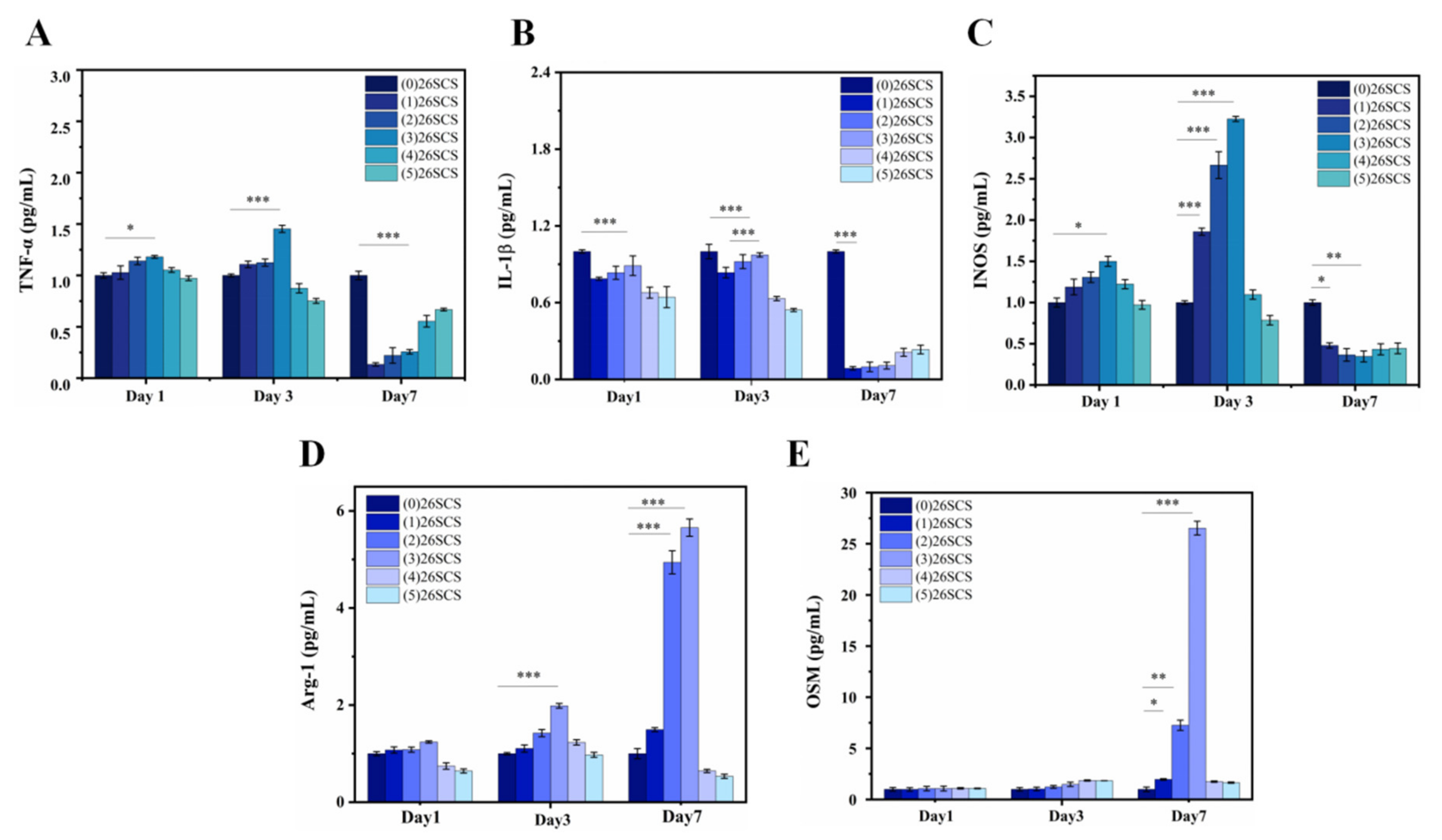
2.6.4. Quantitative Real-Time RT-PCR (qRT-PCR)
The macrophages (RAW264.7) were co-cultured with the (0/1/2/3/4/5) 26SCS in 6-well plates at a density of 5 × 10
4 cells/well. After 1, 3 and 7 days of incubation, the expression of polarization-related genes in Mφs was evaluated by qRT-PCR. In brief, total RNA was extracted from Mφs using a TrizolReagent Kit (Invitrogen, Carlsbad, CA, USA). The extracted RNA was reverse-transcribed into DNA using a PrimeScript RT reagent kit (Takara, Maebashi, Japan) according to the manufacturer’s instructions. The mRNA expressions of
IL-1β,
TNF-α,
INOS,
OSM and
Arg-1 were quantized by a SYBR Green PCR Master Mix real-time PCR system, and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was used as the normalized steward gene. The forward and reverse sequences of each gene primer are shown in
Table 1.
2.6.5. Enzyme-Linked Immunosorbent Assay (ELISA)
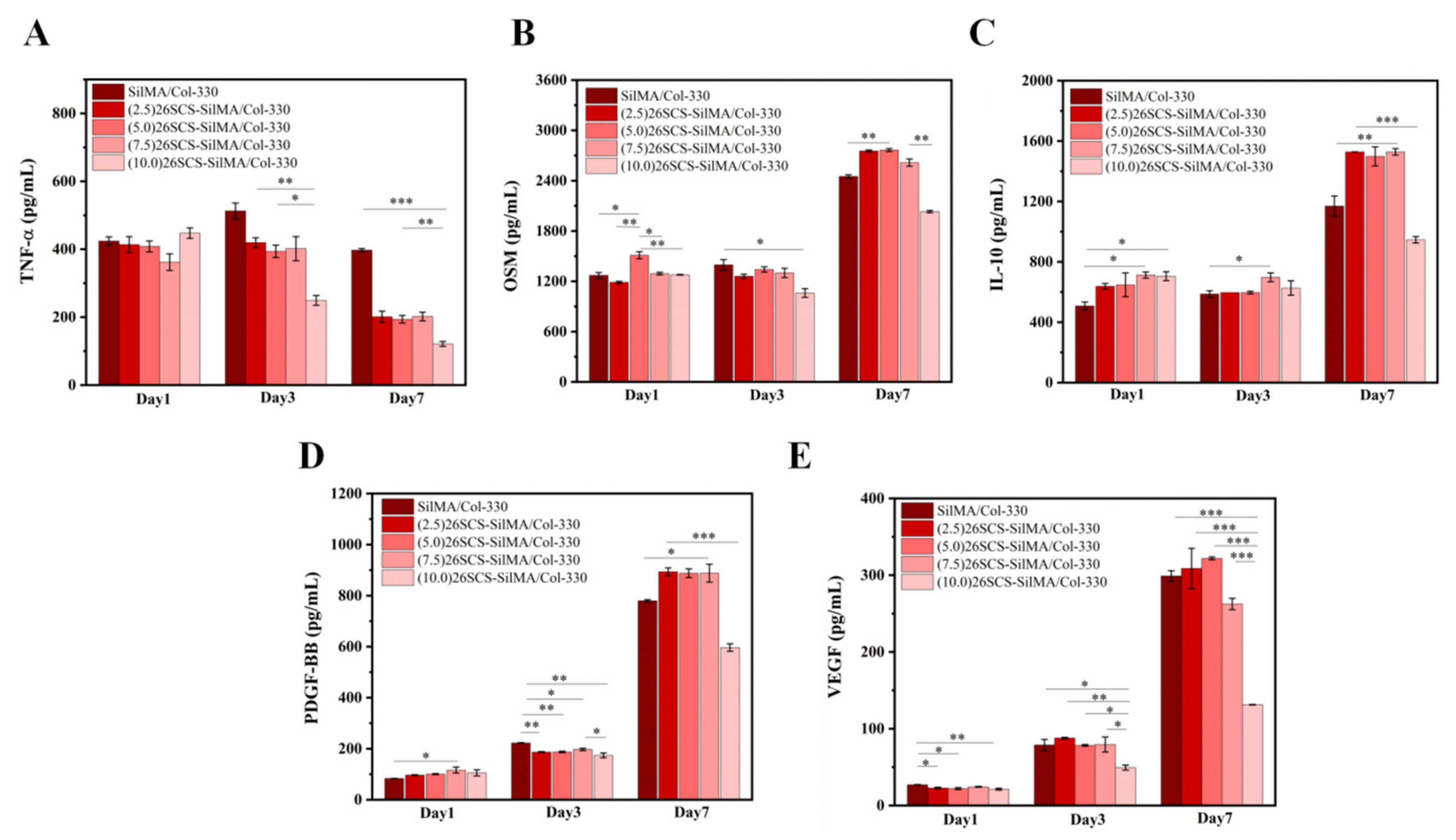
The macrophages (RAW264.7) were seeded at a density of 2 × 104 cells per well on 48-well plates covered with composite sponges, and SilMA/Col-330 was used as the control group. After 1, 3 and 7 days of incubation, supernatants were collected, and Mφ polarization related factors TNF-α, IL-1β, TGF-β1 and OSM, as well as angiogenesis-related factors VEGF and PDGF-BB, were determined by enzyme-linked immunosorbent assay (ELISA).
2.6.6. Cell Scratch Assay
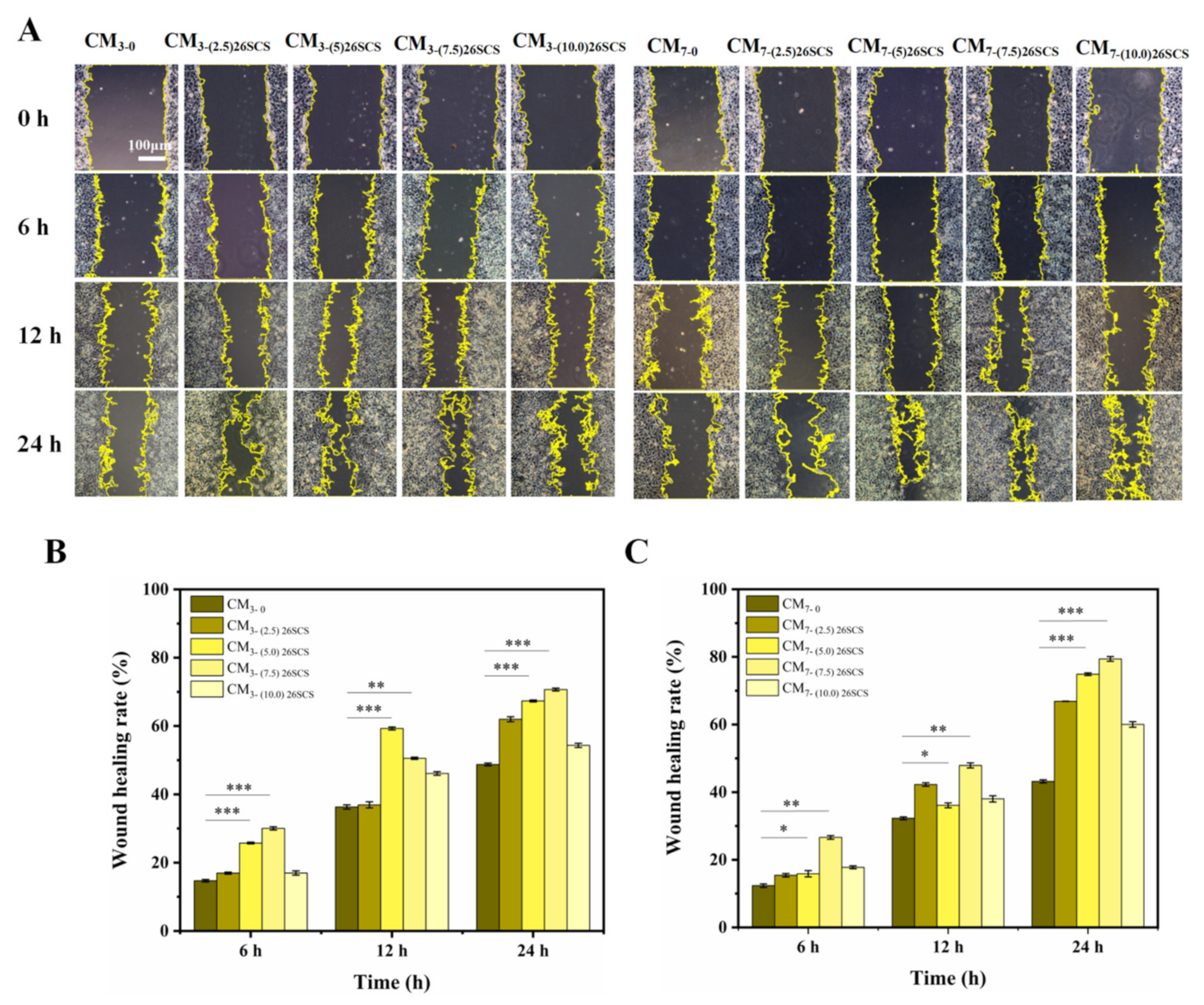
Macrophages (RAW264.7) were seeded at a density of 5 × 104 cells/well in 6-well plates coated with composite sponges. After 3 and 7 days of cultivation, the supernatant was taken and centrifuged at 12,000 r/min for 10 min to obtain conditioned medium CM (CM3/CM7-(0/2.5/5.0/7.5) 26SCS). Human umbilical vein endothelial cells (HUVECs) were seeded in 6-well plates at a density of 5 × 105 cells/well and incubated for 24 h. After forming a confluent monolayer, the cell monolayer was longitudinally scratched with a 200 µL sterile plastic tip. CMs were added to incubate for 0, 6, 12 and 24 h, and cell migration was measured using an FRD-6C microscope (FRD-6C, Cossim, Beijing, China). Five random fields were collected in each sample for analysis, and ImageJ (x64) 1.8.0 (National Institute of Health, Bethesda, MD, USA) was used to evaluate the wound area.
2.6.7. Tube Formation Assay
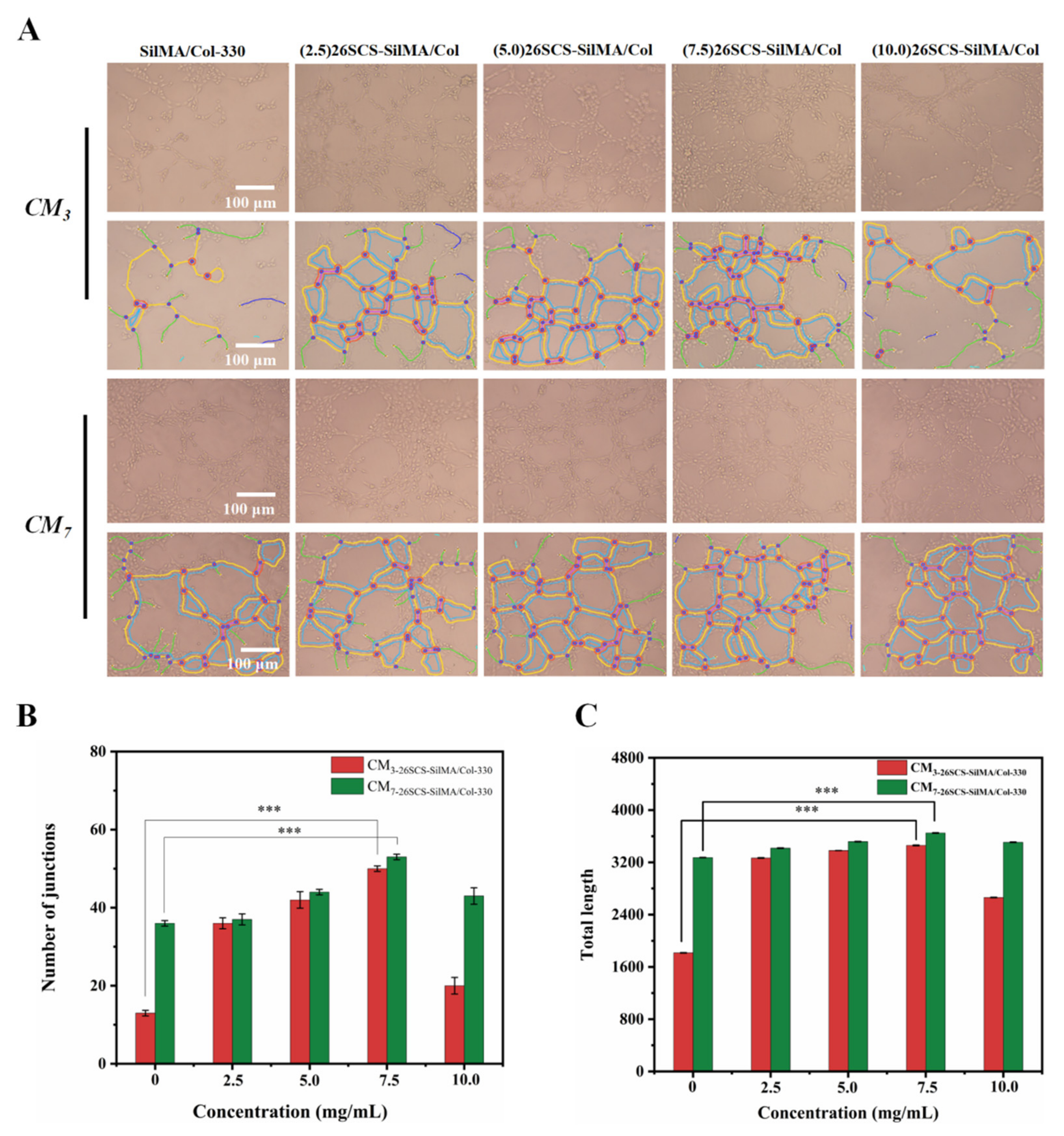
HUVECs were seeded on Matrigel at a density of 5 × 105 cells/well and incubated with CMs for 4 h. Network branches and lengths were calculated using images collected by the Angiogenesis Analyzer of ImageJ software (National Institute of Health, Bethesda, MD, USA).
2.7. In Vivo Wound Healing Analyses
2.7.1. Construction of Diabetic Rat Model
Male Sprague–Dawley rats (12 weeks old and 200–300 g weight) were purchased from Southern Medical University. All animal experiments involved in this study were authorized by the Laboratory Animal Ethics Committee of Jinan University, and all surgical operations were performed in compliance with national legal requirements for animal husbandry. Male Sprague–Dawley rats were fasted for 12 h, then intraperitoneally injected with 100 μL streptozotocin; the blank group was injected with the same dose of sodium citrate buffer. After 7 days, the rats’ fasting blood glucose levels were measured using a glucose meter. The diabetic rat model was considered to be successfully established when the blood glucose level exceeded 13.8 mmol/L.
2.7.2. Establishment of Back Wound Model in Diabetic Rats
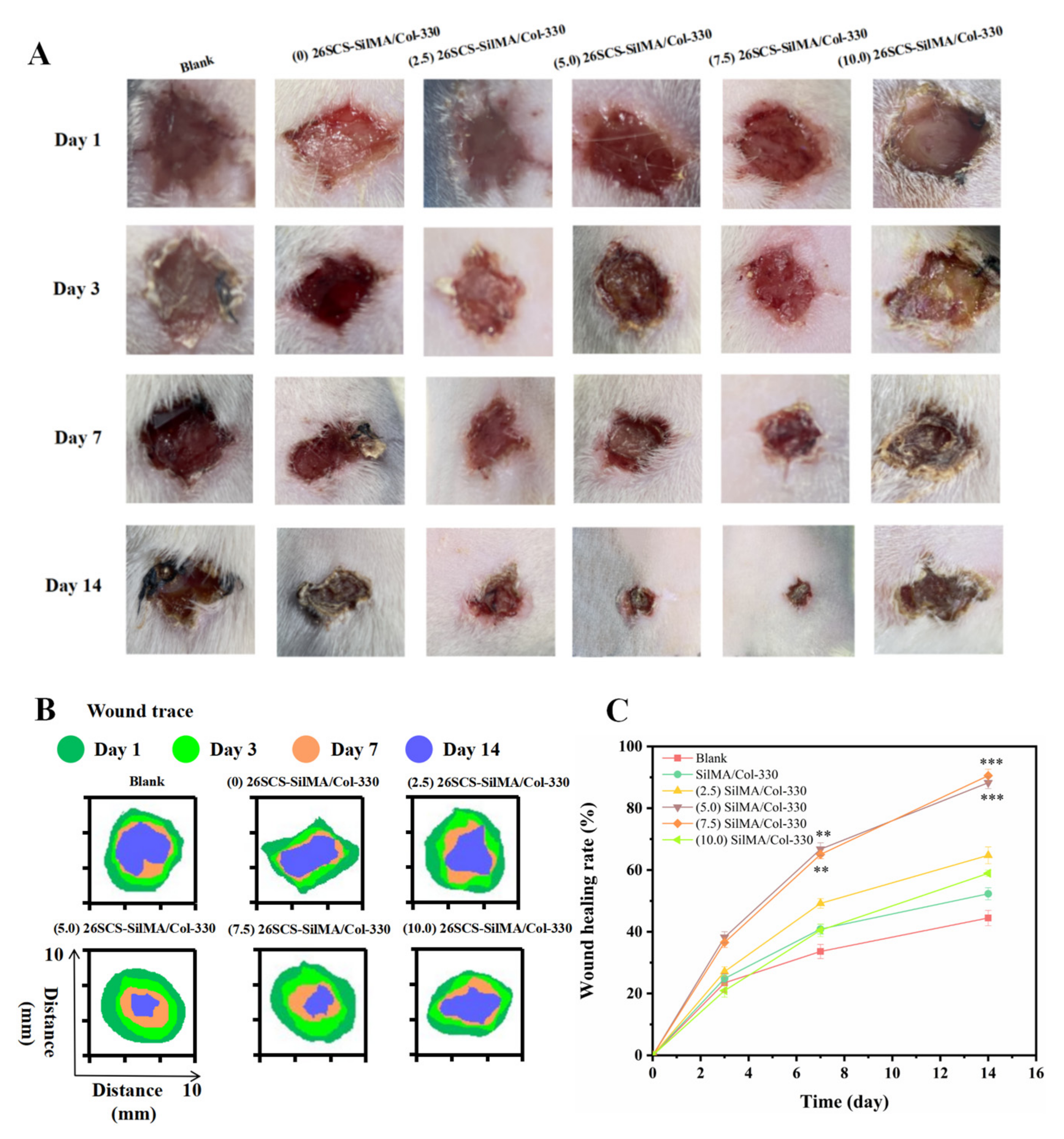
The streptozotocin-induced diabetic rats were operated on to establish a wound model, and the rats were randomly divided into six groups (five in each group). First, the rats were anesthetized by intraperitoneal injection of 0.15 mL 3% pentobarbital sodium. Then, the backs of all rats were shaved and sterilized, and a full-thickness oval skin wound model with a diameter of about 9 mm was built on the back of each rat. To prevent the rats from scratching the wound, the wound edges of the rats were treated with sterile erythromycin ointment. Finally, five groups of composite sponges ((0/2.5/5.0/7.5/10.0) 26SCS-SilMA/Col-330) were implanted into the wounds and wrapped with gauze. The blank group did not receive any treatment, and the wound was naturally repaired. Photos of the wounds were taken on days 1, 3, 7 and 14, and Image J was used to measure the wound area. The calculation formula for the wound healing rate (
W%) was defined as follows:
where
St is the area of the wound at the corresponding time point, and
S0 is the area of the initial wound.
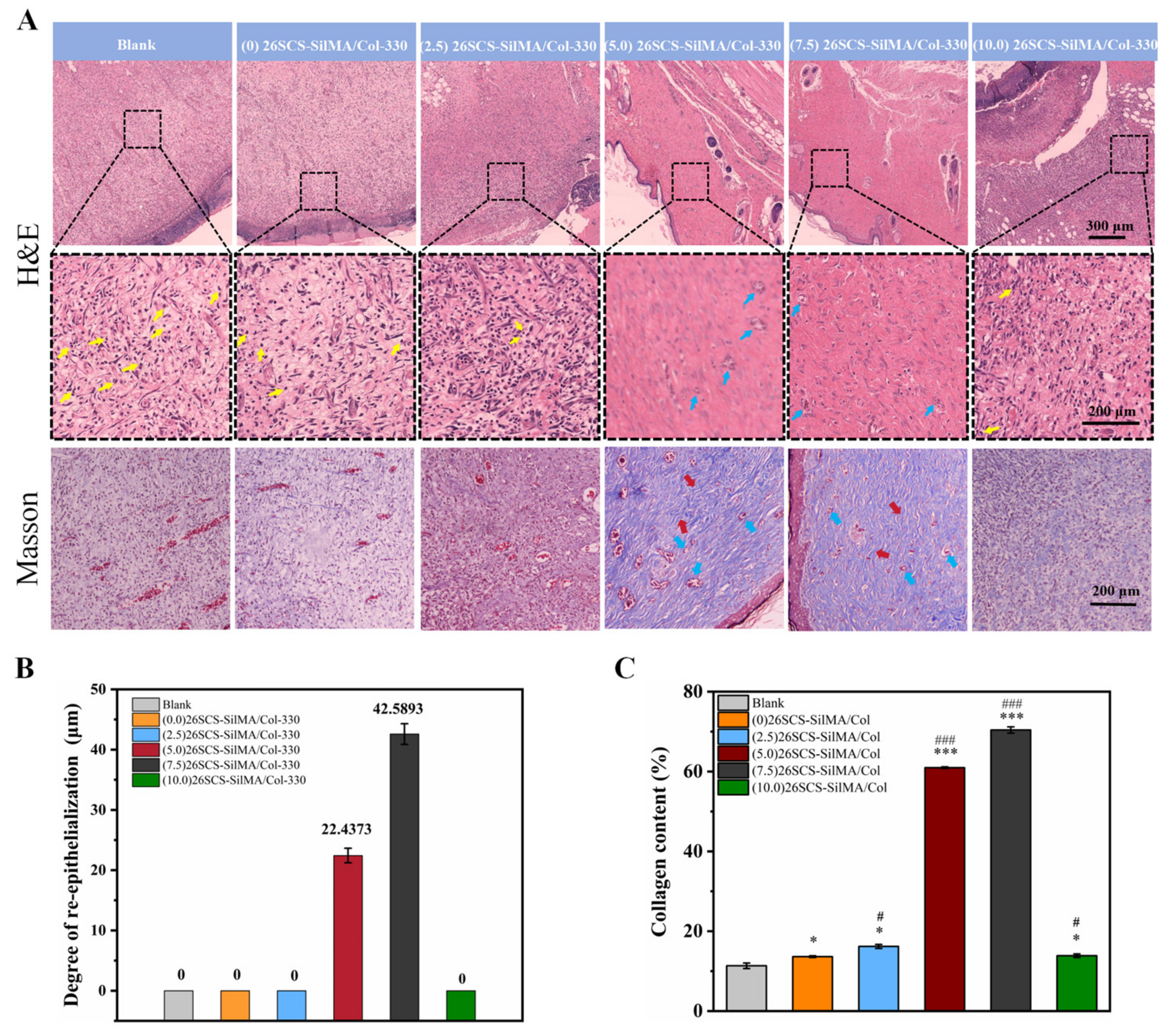
2.7.3. Histological Analysis
Wound tissue was collected on day 14, fixed with 4% paraformaldehyde and embedded in paraffin. The slides were stained with hematoxylin and eosin (H&E) or Masson’s trichrome; the stained sections were photographed under a microscope, and wound healing was observed.
2.7.4. Immunofluorescent Staining and Immunohistochemical Staining
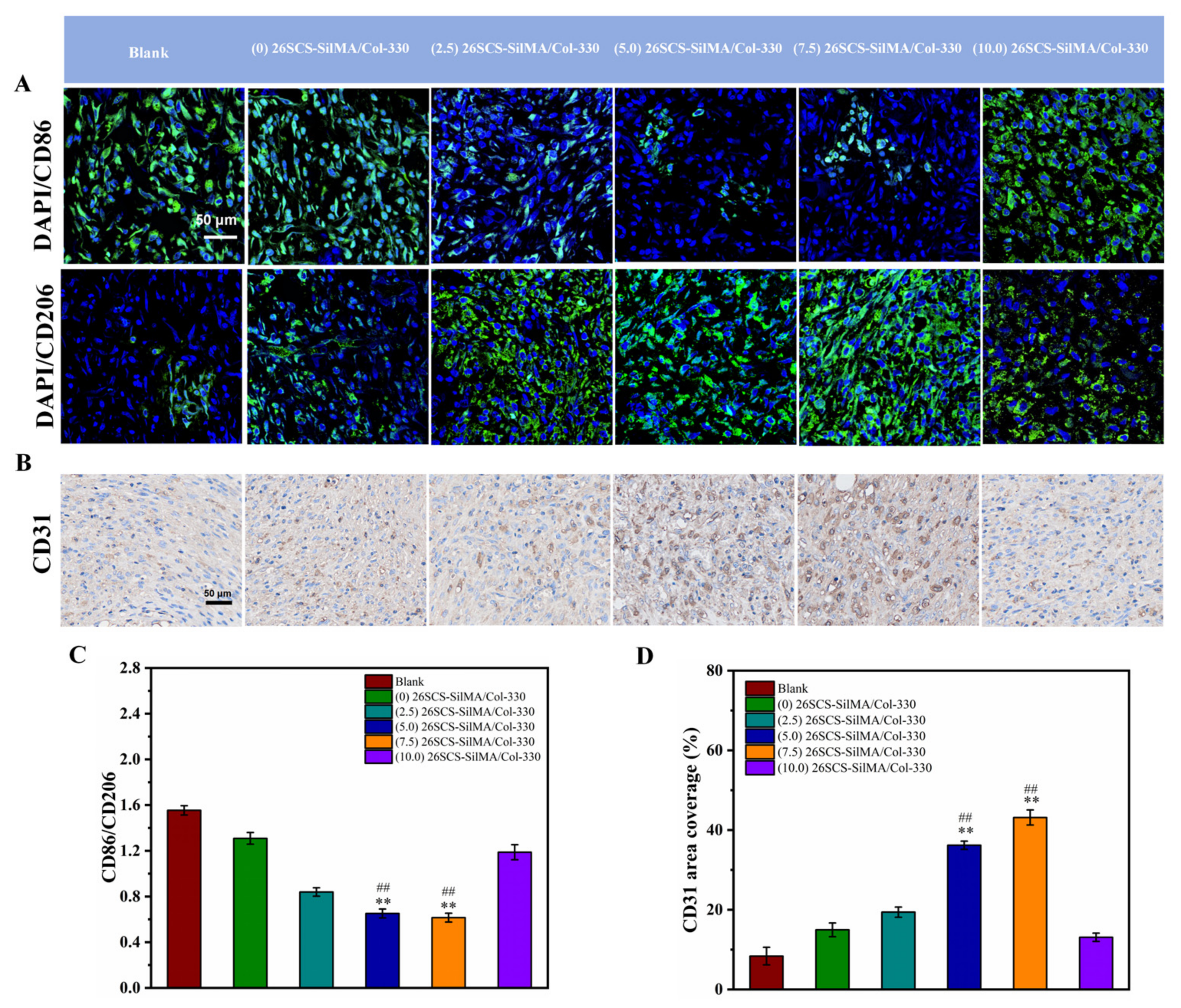
First, the paraffin sections of wound tissue were dewaxed, rehydrated and boiled in sodium citrate antigen repair solution for 15 min. Then, the sections were treated with rabbit anti-CD86 (1:300, HUABIO, Hangzhou, China) and rabbit anti-CD206 (1:500, HUABIO, China), incubated at 4 °C overnight, washed with PBS and incubated at room temperature for 2 h with DyLight fluorescently labeled secondary antibodies (1:400, Abbkine, Wuhan, China). Finally, the nucleus was stained with an anti-quenching agent containing DAPI. The immunofluorescence tissue was observed and photographed using a laser confocal microscope (Zeiss, Oberkochen, Germany, LSM 800). For immunohistochemistry, the sections were treated with rabbit anti-CD31 (abcam, Cambridge, UK), and the stained slices were scanned using a scanner (VS200, OlyVIA, Tokyo, Japan). Five slices were used in each experimental group, and the total defect area of each slice was quantitatively analyzed.
2.8. Statistical Analysis
All the data are expressed as means ± standard deviation (SD), and all statistical analysis was carried out using SPSS version 10.1 software (SPSS Inc., Chicago, IL, USA). Statistically significant differences (p < 0.05) between the various groups were adjusted using the Tukey–Kramer post hoc test.
4. Discussion
As is well known, macrophages play an essential role in driving wound inflammatory response [
40]. The abnormality of immune cells in diabetic wounds makes it difficult to suppress inflammation and keep the normal vascular system in a static state. To address this concern, we developed a bio-mimetic composite sponge dressing containing collagen and silk fibroin loaded with heparin-like polysaccharide (2-N, 6-O-sulfated chitosan), aiming to regulating the immune response of Mφs and prevent further M1 Mφ polarization, thus inducing effective angiogenesis in diabetic wounds.
The 26SCS-SilMA/Col-330 composite sponges prepared in this work exhibited a well-aligned lamellar structure with high porosity (more than 90%) and suitable layer spacing (295–325 μm), which not only provided active sites for cell adhesion and a cell-friendly micro-environment to promote cell infiltration but also provided mechanical support for the repair of damaged tissue [
41]. The water uptake of all 26SCS-loaded composite sponges exceeded 2000%, indicating an excellent absorptive property conducive to maintaining the humidity of the wound [
40].
Adequate vascular growth is critical in the healing process of diabetic wounds. In this work, the loading of 26SCS in the SilMA/Col sponges was designed to effectively induce angiogenesis by regulating the immune response of Mφs to up-regulate the expression of angiogenesis-related genes. It was found that the concentration of 26SCS had a significant impact on cell activity and Mφ phenotypic polarization. The concentration of 26SCS (Cscs) was only within the range of 1 ≤ Cscs≤ 3 mg/mL ((1)26SCS, (2)26SCS and (3)26SCS), L929 cells showed good cell compatibility and Mφs could guide appropriate inflammatory responses. Accordingly, we prepared four groups of 26SCS-loaded composite sponges by setting four different loading concentrations of 26SCS in SilMA/Col-330 sponges ((2.5/5.0/7.5/10.0) 26SCS-SilMA/Col-330) and studied the release behavior of 26SCS and the correlation between 26SCS release concentration, the Mφ polarization phenotype and angiogenesis.
The release profile shown in
Figure 3E indicates that 26SCS presented rapid release from the composite sponges in the initial stage (1–3 days) and basically maintained stability on day 4. The maximum release concentration of 26SCS in the (2.5/5.0/7.5) 26SCS-SilMA/Col-330 was within the rage of 1–3 mg/mL (
Figure 3E), while it was as high as 3.94 mg/mL in the (10.0) 26SCS-SilMA/Col-330, exceeding the appropriate range. The results of co-culture showed that both individual 26SCS medium and 26SCS-loaded composite sponges could induce M1/M2 phenotypic polarization and regulate the phenotypic transformation of Mφs. (1/2/3) 26SCS could effectively transform the Mφ phenotype from M1 to M2 by up-regulating the expression of pro-inflammatory gene TNF-α within 1–3 days and dramatically down-regulating the expression of
TNF-α while up-regulating the expressions of anti-inflammatory genes
OSM and
IL-10 on day 7. The trend of the expression level of pro-inflammatory gene
TNF-α in the SilMA/Col-330 and the 26SCS-SilMA/Col-330 was similar to that in 26SCS. However, the expression of
TNF-α in the 26SCS-SilMA/Col-330 was much lower than that in the SilMA/Col-330 during culture time. In particular,
TNF-α expression was significantly down-regulated in the 26SCS-SilMA/Col-330 on day 7, while a higher expression level was maintained in the SilMA/Col-330. Moreover, the expressions of anti-inflammatory genes
OSM and
IL-10 were dramatically up-regulated in the 26SCS-SilMA/Col-330 on day 7—much higher than that in the SilMA/Col-330. These findings indicate that the topological structure (porous and aligned lamellar structure) of composite sponges mainly triggered the pro-inflammatory response of Mφs in the early stage, leading to high expression of pro-inflammatory cytokine
TNF-α. With the release of 26SCS from the sponges, the pro-inflammatory phenotypes of Mφs were gradually suppressed and tended towards an anti-inflammatory phenotype. We suggest that loading 26SCS onto the SilMA/Col sponges not only enhanced the biological activity of the sponges, providing a friendly micro-environment for cells through sustainable release of 26SCS, but also enabled a synergistic effect between sponges and 26SCS to induce a favorable immune response. The results of co-culture reveal that 26SCS-SilMA/Col-330 sponges exhibited a prominent advantage over SilMA/Col-330 in regulating Mφ polarization to M2 and stimulating secretion of two typical angiogenic growth factors, namely
VEGF and
PDGF-BB. As shown in
Figure 6, the pro-inflammatory reaction of Mφs was initiated within 3 days, M1 Mφs were shifted to the M2 phenotype within 3 –7 days and the expressions of
VEGF and
PDGF-BB were significantly up-regulated on day 7, indicating that 26SCS-SilMA/Col-330 sponges could shift Mφ polarization to M2 in a timely manner to avoid excessive inflammation and elevated secretion of two typical angiogenic growth factors. Notably, the expressions levels of pro-inflammatory and anti-inflammatory factors, as well as
VEGF and
PDGF-BB, appeared higher in the (5.0/7.5) 26SCS-SilMA/Col-330 at different inflammatory stages, suggesting that 5 mg/mL and 7.5 mg/mL were the optimal loading concentrations of 26SCS to induce a positive inflammatory response. Correspondingly, CM7-(5.0/7.5) 26SCS exhibited a stronger vascularization ability than other groups. As shown in
Figure 7 and
Figure 8, the CM7-(5/7.5) 26SCS obviously promoted the migration speed of HUVECs and wound recovery and formed more capillary-like networks, more branches and longer tubes. In vivo data also revealed that 14 days post implantation, the (5.0/7.5) 26SCS SilMA/Col-330 groups exhibited fewer inflammatory cell aggregations, inflammation alleviation and faster re-epithelialization and formed more abundant collagen deposition, new hair follicles and sebaceous glands in the skin tissue. In addition, the granulation tissue implanted with the (5.0/7.5) 26SCS-SilMA/Col-330 showed a greater number of blood vessels than other groups.
We suggest that 26SCS efficiently induced angiogenesis by directing the immune response of Mφs with VEGF secretion, which is mainly attributed to the combined effect of sulfated amino groups (C2-NHSO
3−) and the polysaccharide chain structure. It has been reported that the polysaccharide chain structure in heparin can provide biological effects. Heparin can interact with
VEGF through its heparin-binding domain in the molecular structure and protect the biological activity of
VEGF. The 26SCS synthesized in this work was a heparin-like anionic polysaccharide with sulfated amino groups and a polysaccharide chain structure. Incorporation of 26SCS into the SilMA/Col-330 endowed the composite sponges with special bio-functions. As shown in
Figure 3E and
Figure 6, a large amount of 26SCS was released from the sponges around the 4th day, promoting the secretion of anti-inflammatory cytokines
IL-10 and
OSM, achieving timely transformation of Mφs from the M1 to M2 phenotype and preventing further polarization of M1-Mφ (the 7th day). Meanwhile, M2-Mφs secreted a large amount of angiogenic growth factors, and the 26SCS maintained the
VEGF activity via the heparin-binding domain, promoting angiogenesis of HUVECs.
Mφ polarization is known to be mediated by canonical signal transducers and activators of transcription (Stat) signaling pathways [
42]. The typical Stat signaling pathway is activated by
IL-4 or
IL-10 and tilts towards the M2 phenotype through the Stat6 or Stat3 pathway [
42]. OSM is a multifunctional cytokine [
43] that regulates many physiological processes in healthy individuals, such as by directly stimulating endothelial cells to influence the evolution of inflammatory responses [
44] and contributing to tissue remodeling processes (Tanaka and Miyajima, 2003) [
43]. The binding of active OSM to its receptor induces the phosphorylation of STAT3 (Fossey et al., 2011) [
45], inducing signaling through the JAK–STAT pathway (West, et al.) [
46]. In line with this knowledge, we proposed a possible mechanism by which 26SCS-SilMA/Col-330 sponges regulated Mφ polarization and promoted angiogenesis. In the first stage (1–3 days), macrophages, stimulated by physical sponge cues, were polarized into the M1 phenotype by secreting pro-inflammatory gene
TNF-α, which initiated the inflammatory response [
38,
40]. With the release of 26SCS from sponges and the increase in its concentration,
TNF-α expression was down-regulated, while anti-inflammatory cytokines
IL-10 and
OSM were highly expressed, which might activate the phosphorylation of Stat3 in Mφs [
34], inducing signaling through the Stat3 pathway [
47]. Accordingly, Mφs promptly converted the M1 to the M2 phenotype in the second stage (3–7 days), secreting anti-inflammatory cytokines and inhibiting the further development of inflammation. Highly expressed
IL-10 and
OSM stimulated Mφs to secrete a large amount of
VEGF and
PDGF-BB, promoting endothelial cell migration and inducing angiogenesis.
Here, we suggested that the regulation of inflammatory response and timely transition from the M1 to M2 phenotype could be achieved by regulating the release concentration and the release profile of 2626SC. The 26SCS-SilMA/Col-330 sponges without the addition of exogenous growth factors that we prepared in this work could become a bio-active material for the treatment of some inflammatory diseases.














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}