
Synthesis and Structural Studies of peri-Substituted Acenaphthenes with Tertiary Phosphine and Stibine Groups †

 ,
,  , , and
, , and
Abstract
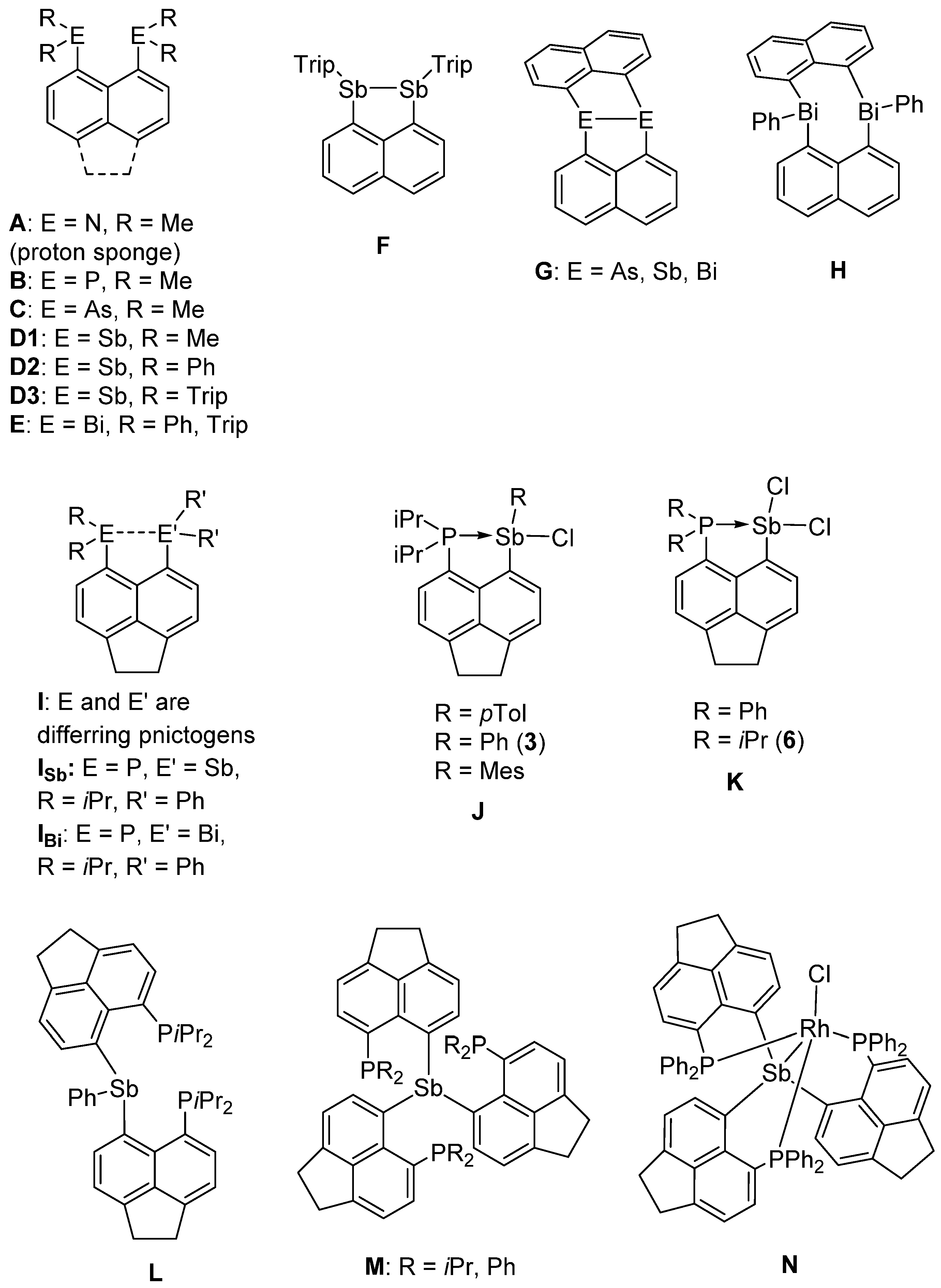
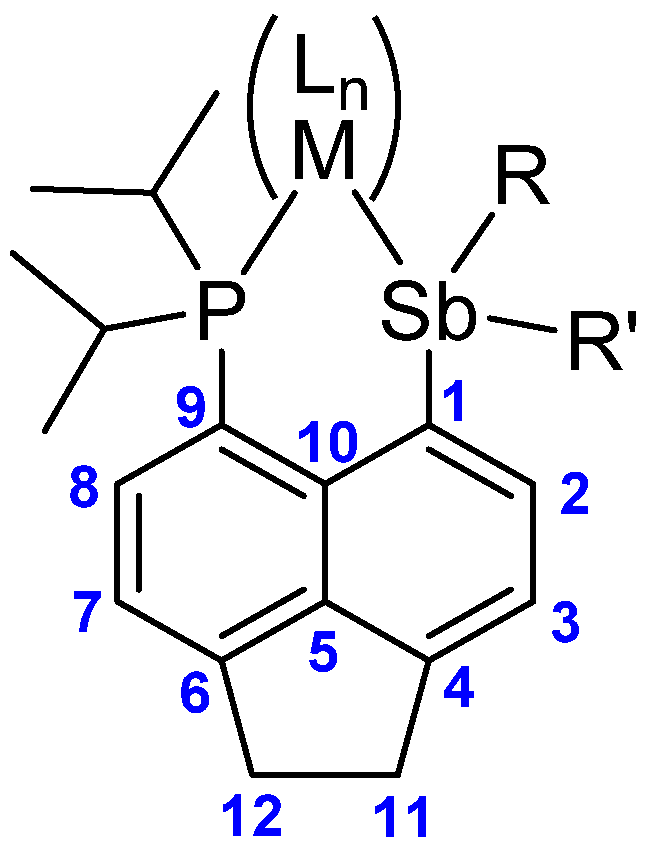
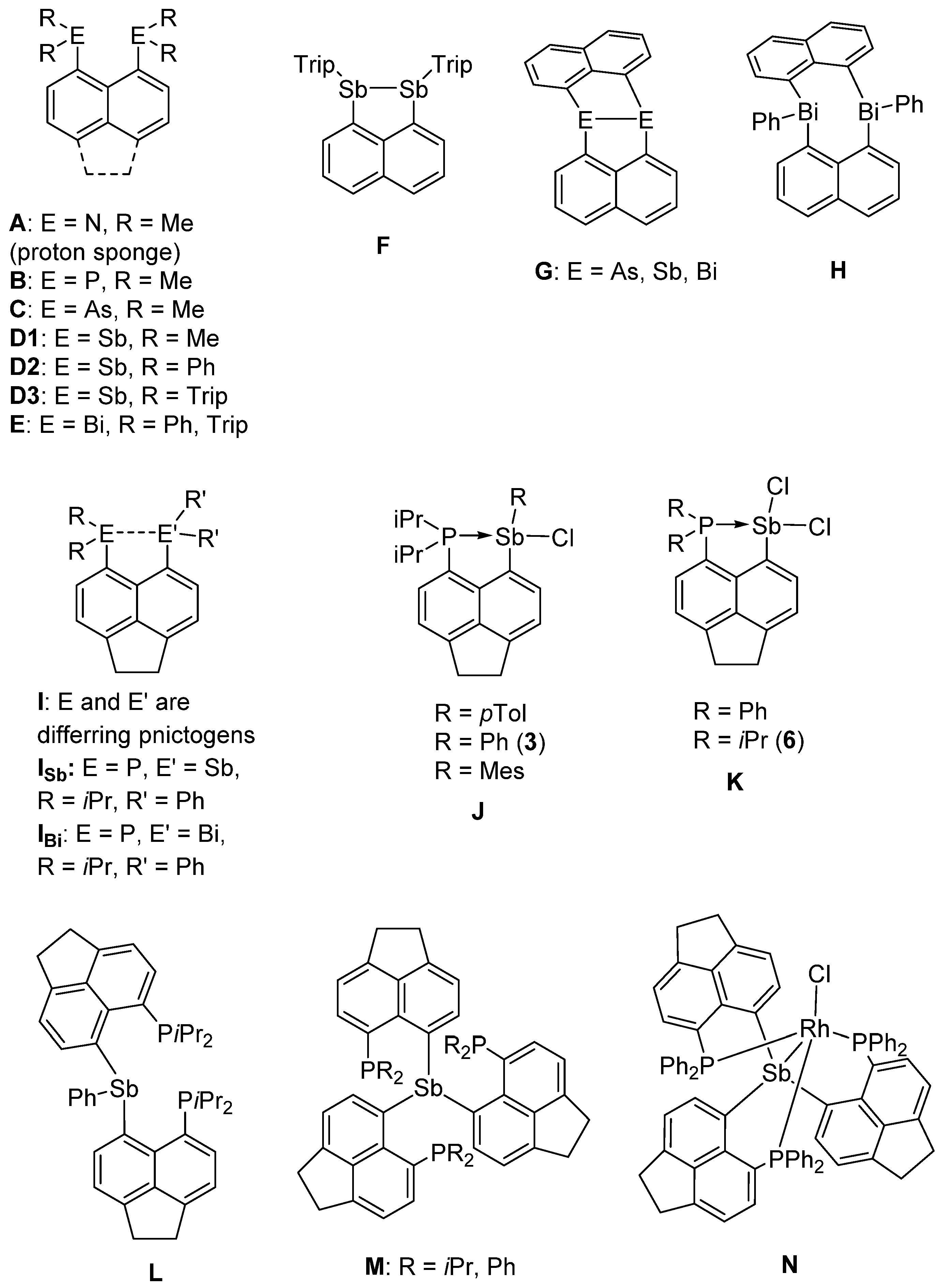
:1. Introduction
2. Results and Discussion
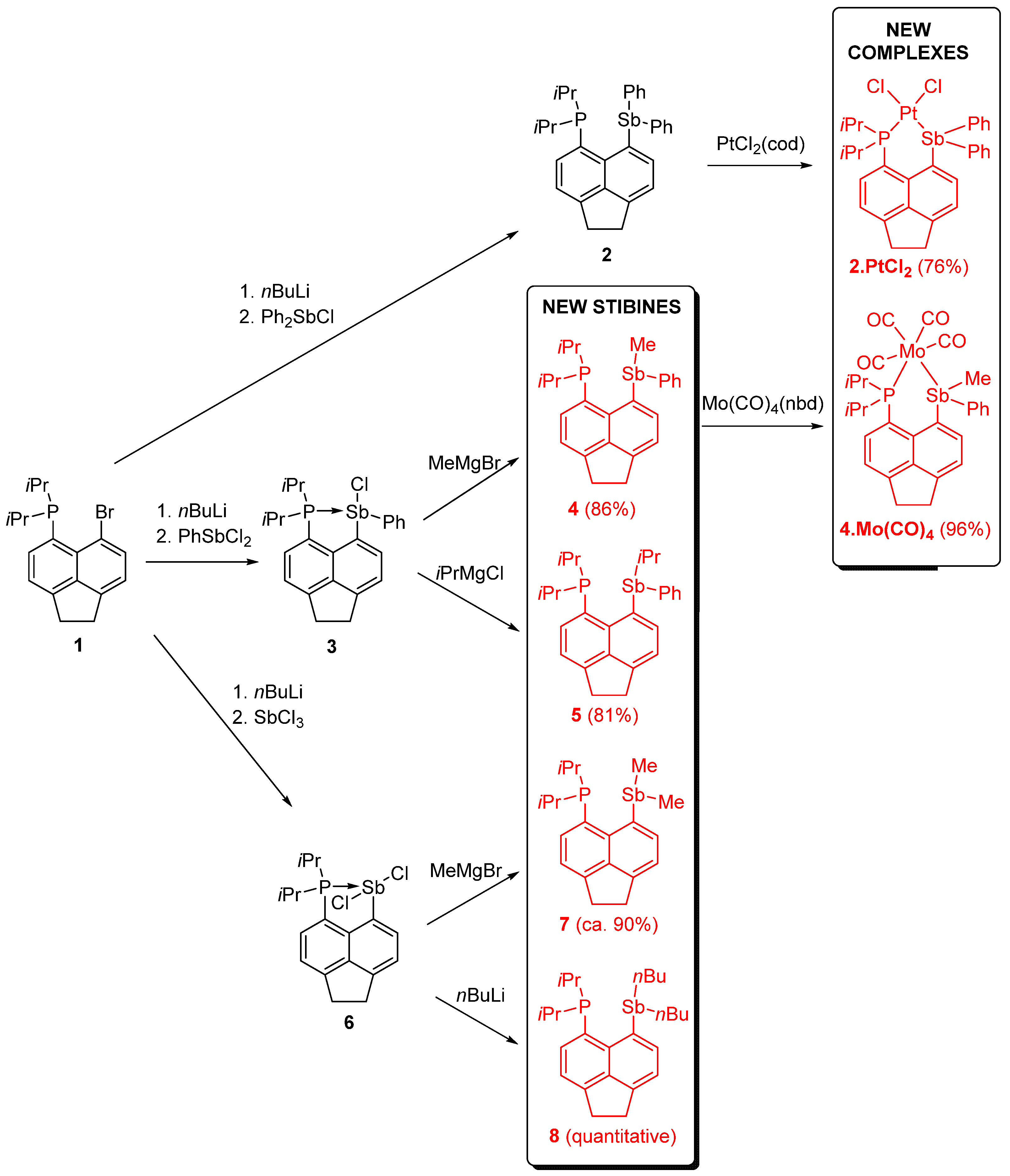
2.1. Synthesis and Spectroscopic Properties of the Tertiary Stibines 4, 5, 7, and 8
2.2. Synthesis and Spectroscopic Properties of Tertiary Stibine Metal Complexes 2.PtCl2 and 4.Mo(CO)4
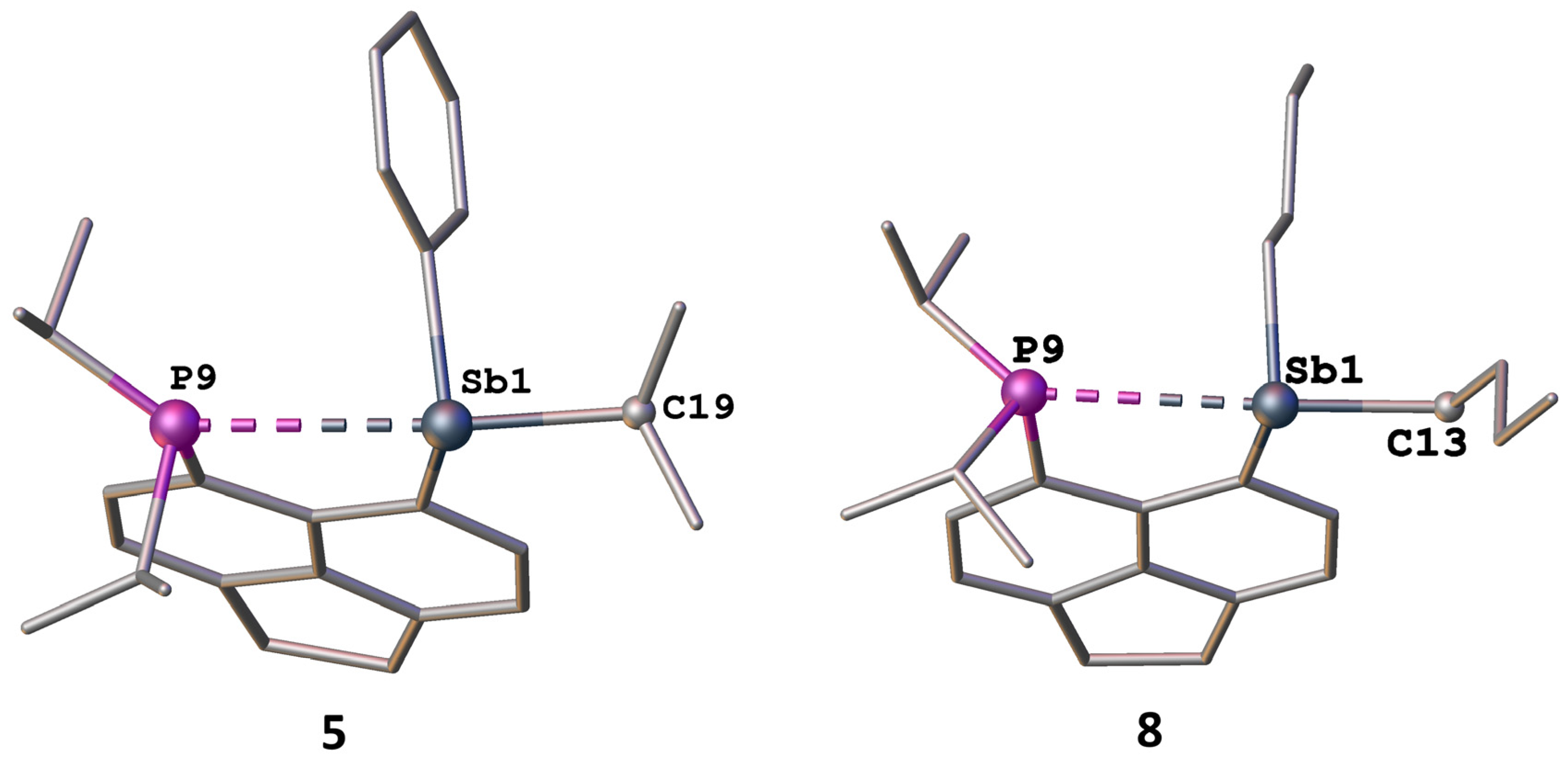
2.3. Structural Discussion
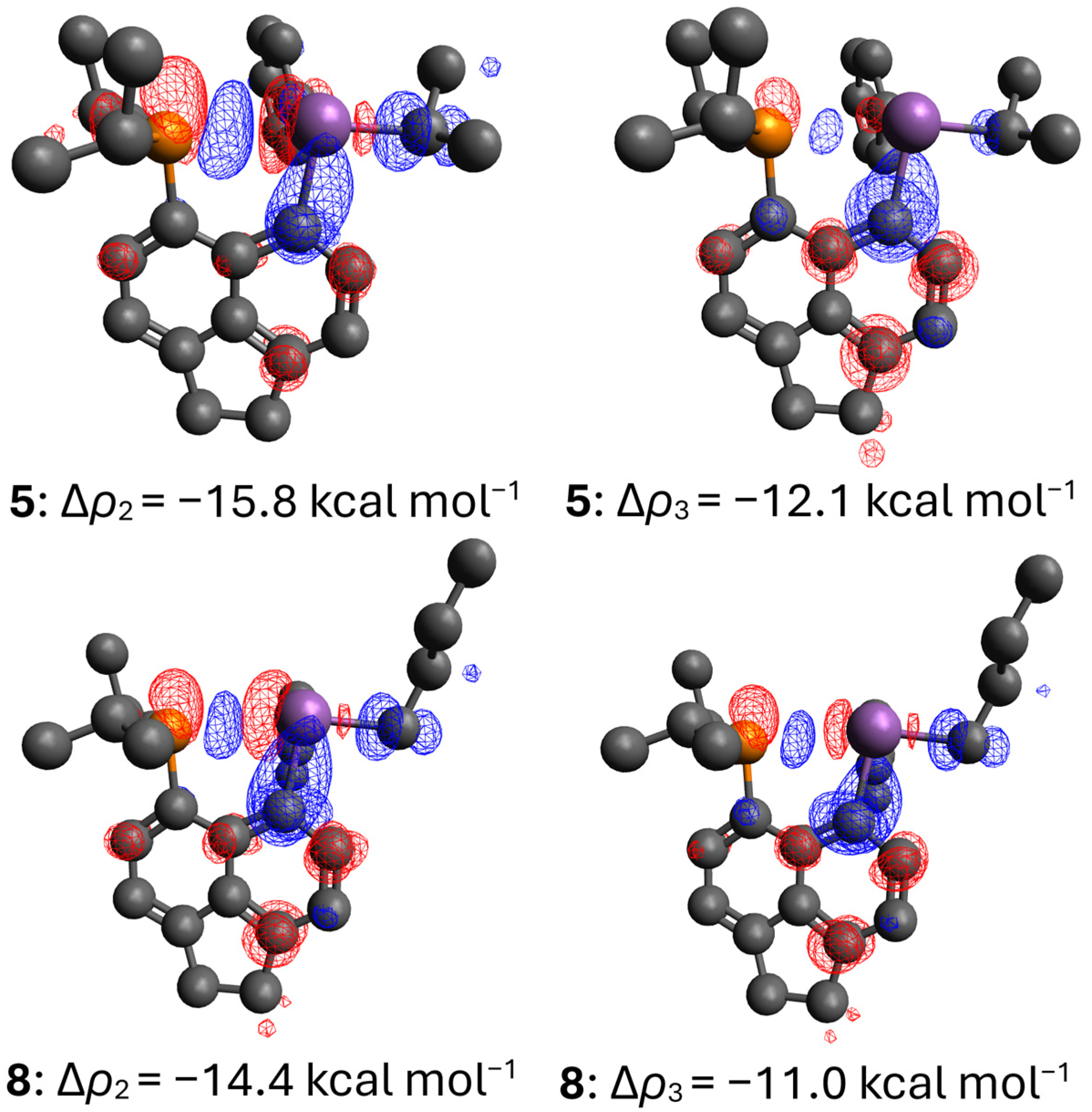
2.4. Computational Analysis
3. Experimental Section
3.1. General Considerations
3.2. NMR Spectroscopy
3.3. Other Analyses
3.4. [iPr2P-Ace-SbPh2]PtCl2, 2.PtCl2
3.5. iPr2P-Ace-Sb(Ph)Me, 4
3.6. [iPr2P-Ace-Sb(Ph)Me]Mo(CO)4, 4.Mo(CO)4
3.7. iPr2P-Ace-Sb(Ph)iPr, 5
3.8. iPr2P-Ace-SbMe2, 7
3.9. iPr2P-Ace-Sb(nBu)2, 8
3.10. X-ray Diffraction
3.11. Computational Methodology
3.11.1. Geometry Optimisations and QTAIM Analysis
3.11.2. EDA-NOCV Analysis
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References and Note
- Champness, N.R.; Levason, W. Coordination chemistry of stibine and bismuthine ligands. Coord. Chem. Rev. 1994, 133, 115–217. [Google Scholar] [CrossRef]
- Levason, W.; Reid, G. Developments in the coordination chemistry of stibine ligands. Coord. Chem. Rev. 2006, 250, 2565–2594. [Google Scholar] [CrossRef]
- Alder, R.W.; Bowman, P.S.; Steele, W.R.S.; Winterman, D.R. The remarkable basicity of 1,8-bis(dimethylamino)naphthalene. Chem. Commun. 1968, 723–724. [Google Scholar] [CrossRef]
- Costa, T.; Schmidbaur, H. 1,8-Naphthalindiylbis(dimethylphosphan): Konsequenzen sterischer Hinderung für Methylierung und Borylierung. Chem. Berichte 1982, 115, 1374–1378. [Google Scholar] [CrossRef]
- Karaçar, A.; Thönnessen, H.; Jones, P.G.; Bartsch, R.; Schmutzler, R. 1,8-Bis(phosphino)naphthalenes: Synthesis and molecular structures. Heteroat. Chem. 1997, 8, 539–550. [Google Scholar] [CrossRef]
- Yam, V.W.W.; Chan, C.L.; Choi, S.W.K.; Wong, K.M.C.; Cheng, E.C.C.; Yu, S.C.; Ng, P.-K.; Chan, W.-K.; Cheung, K.-K. Synthesis, photoluminescent and electroluminescent behaviour of four-coordinate tetrahedral gold(i) complexes. X-ray crystal structure of [Au(dppn)2]Cl. Chem. Commun. 2000, 53–54. [Google Scholar] [CrossRef]
- Karaçar, A.; Freytag, M.; Jones, P.G.; Bartsch, R.; Schmutzler, R. Platinum(II) Complexes of P-Chiral 1, 8-Bis(diorganophosphino)naphthalenes; Crystal Structures of dmfppn, rac-[PtCl2(dmfppn)], and rac-[PtCl2(dtbppn)] (dmfppn = 1, 8-di(methyl-pentafluorophenylphosphino)naphthalene and dtbppn = 1, 8-di(tert-butylphenylphosphino)naphthalene). Z. Anorg. Allg. Chem. 2002, 628, 533–544. [Google Scholar] [CrossRef]
- Kilian, P.; Knight, F.R.; Woollins, J.D. Synthesis of ligands based on naphthalene peri-substituted by Group 15 and 16 elements and their coordination chemistry. Coord. Chem. Rev. 2011, 255, 1387–1413. [Google Scholar] [CrossRef]
- Di Sipio, L.; Sindellari, L.; Tondello, E.; De Michelis, G.; Oleari, L. NiII complexes with 1,8-naphthalene bis(dimethylarsine). Coord. Chem. Rev. 1967, 2, 117–128. [Google Scholar] [CrossRef]
- Jura, M.; Levason, W.; Reid, G.; Webster, M. Preparation and properties of sterically demanding and chiral distibine ligands. Dalton Trans. 2008, 5774–5782. [Google Scholar] [CrossRef]
- Gehlhaar, A.; Wölper, C.; van der Vight, F.; Jansen, G.; Schulz, S. Noncovalent Intra- and Intermolecular Interactions in Peri-Substituted Pnicta Naphthalene and Acenaphthalene Complexes. Eur. J. Inorg. Chem. 2022, 2022, e202100883. [Google Scholar] [CrossRef]
- Gehlhaar, A.; Weinert, H.M.; Wölper, C.; Semleit, N.; Haberhauer, G.; Schulz, S. Bisstibane–distibane conversion via consecutive single-electron oxidation and reduction reaction. Chem. Commun. 2022, 58, 6682–6685. [Google Scholar] [CrossRef]
- Dzialkowski, K.; Gehlhaar, A.; Wolper, C.; Auer, A.A.; Schulz, S. Structure and Reactivity of 1,8-Bis(naphthalenediyl)dipnictanes. Organometallics 2019, 38, 2927–2942. [Google Scholar] [CrossRef]
- Ganesamoorthy, C.; Heimann, S.; Hölscher, S.; Haack, R.; Wölper, C.; Jansen, G.; Schulz, S. Synthesis, structure and dispersion interactions in bis(1,8-naphthalendiyl)distibine. Dalton Trans. 2017, 46, 9227–9234. [Google Scholar] [CrossRef] [PubMed]
- Gehlhaar, A.; Schiavo, E.; Wölper, C.; Schulte, Y.; Auer, A.A.; Schulz, S. Comparing London dispersion pnictogen–π interactions in naphthyl-substituted dipnictanes. Dalton Trans. 2022, 51, 5016–5023. [Google Scholar] [CrossRef] [PubMed]
- Chuit, C.; Corriu, R.J.P.; Monforte, P.; Reyé, C.; Declercq, J.-P.; Dubourg, A. Evidences for intramolecular N → P coordination in (8-dimethylamino-1-naphthyl) diphenylphosphane and derivatives. J. Organomet. Chem. 1996, 511, 171–175. [Google Scholar] [CrossRef]
- Biskup, D.; Bergmann, T.; Schnakenburg, G.; Gomila, R.M.; Frontera, A.; Streubel, R. Synthesis of a 1-aza-2-phospha-acenaphthene complex profiting from coordination enabled chloromethane elimination. RSC Adv. 2023, 13, 21313–21317. [Google Scholar] [CrossRef] [PubMed]
- Chalmers, B.A.; Bühl, M.; Athukorala Arachchige, K.S.; Slawin, A.M.Z.; Kilian, P. Structural, Spectroscopic and Computational Examination of the Dative Interaction in Constrained Phosphine–Stibines and Phosphine–Stiboranes. Chem.-A Eur. J. 2015, 21, 7520–7531. [Google Scholar] [CrossRef]
- Nejman, P.S.; Curzon, T.E.; Bühl, M.; McKay, D.; Woollins, J.D.; Ashbrook, S.E.; Cordes, D.B.; Slawin, A.M.Z.; Kilian, P. Phosphorus–Bismuth Peri-Substituted Acenaphthenes: A Synthetic, Structural, and Computational Study. Inorg. Chem. 2020, 59, 5616–5625. [Google Scholar] [CrossRef]
- Bergsch, J.U.; Slawin, A.M.Z.; Kilian, P.; Chalmers, B.A. Phosphine–Stibine and Phosphine–Stiborane peri-Substituted Donor–Acceptor Complexes. Molbank 2023, 2023, M1653. [Google Scholar] [CrossRef]
- Hupf, E.; Lork, E.; Mebs, S.; Chęcińska, L.; Beckmann, J. Probing Donor–Acceptor Interactions in peri-Substituted Diphenylphosphinoacenaphthyl–Element Dichlorides of Group 13 and 15 Elements. Organometallics 2014, 33, 7247–7259. [Google Scholar] [CrossRef]
- Chalmers, B.A.; Meigh, C.B.E.; Nejman, P.S.; Bühl, M.; Lébl, T.; Woollins, J.D.; Slawin, A.M.Z.; Kilian, P. Geminally Substituted Tris(acenaphthyl) and Bis(acenaphthyl) Arsines, Stibines, and Bismuthine: A Structural and Nuclear Magnetic Resonance Investigation. Inorg. Chem. 2016, 55, 7117–7125. [Google Scholar] [CrossRef] [PubMed]
- Furan, S.; Hupf, E.; Boidol, J.; Brunig, J.; Lork, E.; Mebs, S.; Beckmann, J. Transition metal complexes of antimony centered ligands based upon acenaphthyl scaffolds. Coordination non-innocent or not? Dalton Trans. 2019, 48, 4504–4513. [Google Scholar] [CrossRef] [PubMed]
- Batsanov, S.S. Van der Waals Radii of Elements. Inorg. Mater. 2001, 37, 871–885. [Google Scholar] [CrossRef]
- Cordero, B.; Gomez, V.; Platero-Prats, A.E.; Reves, M.; Echeverria, J.; Cremades, E.; Barragan, F.; Alvarez, S. Covalent radii revisited. Dalton Trans. 2008, 2832–2838. [Google Scholar] [CrossRef] [PubMed]
- You, D.; Smith, J.E.; Sen, S.; Gabbaï, F.P. A Stiboranyl Platinum Triflate Complex as an Electrophilic Catalyst. Organometallics 2020, 39, 4169–4173. [Google Scholar] [CrossRef]
- You, D.; Gabbaï, F.P. Unmasking the Catalytic Activity of a Platinum Complex with a Lewis Acidic, Non-innocent Antimony Ligand. J. Am. Chem. Soc. 2017, 139, 6843–6846. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Gabbaï, F.P. Solution and Solid-State Photoreductive Elimination of Chlorine by Irradiation of a [PtSb]VII Complex. J. Am. Chem. Soc. 2014, 136, 10866–10869. [Google Scholar] [CrossRef] [PubMed]
- Piesch, M.; Gabbaï, F.P.; Scheer, M. Phosphino-Stibine Ligands for the Synthesis of Heterometallic Complexes. Z. Anorg. Allg. Chem. 2021, 647, 266–278. [Google Scholar] [CrossRef]
- Bader, R.F.W. Atoms in Molecules: A Quantum Theory; Clarendon Press: Oxford, UK, 1994. [Google Scholar]
- Bader, R.F. A quantum theory of molecular structure and its applications. Chem. Rev. 1991, 91, 893–928. [Google Scholar] [CrossRef]
- Lepetit, C.; Fau, P.; Fajerwerg, K.; Kahn, M.L.; Silvi, B. Topological analysis of the metal-metal bond: A tutorial review. Coord. Chem. Rev. 2017, 345, 150–181. [Google Scholar] [CrossRef]
- Gervasio, G.; Bianchi, R.; Marabello, D. About the topological classification of the metal–metal bond. Chem. Phys. Lett. 2004, 387, 481–484. [Google Scholar] [CrossRef]
- Espinosa, E.; Alkorta, I.; Elguero, J.; Molins, E. From weak to strong interactions: A comprehensive analysis of the topological and energetic properties of the electron density distribution involving X–H⋯F–Y systems. J. Chem. Phys. 2002, 117, 5529–5542. [Google Scholar] [CrossRef]
- Espinosa, E.; Molins, E.; Lecomte, C. Hydrogen bond strengths revealed by topological analyses of experimentally observed electron densities. Chem. Phys. Lett. 1998, 285, 170–173. [Google Scholar] [CrossRef]
- Mitoraj, M.; Michalak, A. Natural orbitals for chemical valence as descriptors of chemical bonding in transition metal complexes. J. Mol. Model. 2007, 13, 347–355. [Google Scholar] [CrossRef] [PubMed]
- Michalak, A.; Mitoraj, M.; Ziegler, T. Bond Orbitals from Chemical Valence Theory. J. Phys. Chem. A 2008, 112, 1933–1939. [Google Scholar] [CrossRef] [PubMed]
- Mitoraj, M.P.; Michalak, A.; Ziegler, T. A Combined Charge and Energy Decomposition Scheme for Bond Analysis. J. Chem. Theory Comput. 2009, 5, 962–975. [Google Scholar] [CrossRef] [PubMed]
- Mitoraj, M.P.; Michalak, A. σ-Donor and π-Acceptor Properties of Phosphorus Ligands: An Insight from the Natural Orbitals for Chemical Valence. Inorg. Chem. 2010, 49, 578–582. [Google Scholar] [CrossRef] [PubMed]
- In the Orca 5.0.4 implementation of EDA-NOCV, the electrostatic and Pauli terms cannot be separated.
- Armarego, W.L.F.; Chai, C.L.L. Purification of Laboratory Chemicals, 6th ed.; Elsevier: Burlington, MA, USA, 2009. [Google Scholar]
- Chalmers, B.A.; Arachchige, K.S.A.; Prentis, J.K.D.; Knight, F.R.; Kilian, P.; Slawin, A.M.Z.; Woollins, J.D. Sterically Encumbered Tin and Phosphorus peri-Substituted Acenaphthenes. Inorg. Chem. 2014, 53, 8795–8808. [Google Scholar] [CrossRef]
- Fuller, A.L.; Scott-Hayward, L.A.S.; Li, Y.; Bühl, M.; Slawin, A.M.Z.; Woollins, J.D. Automated Chemical Crystallography. J. Am. Chem. Soc. 2010, 132, 5799–5802. [Google Scholar] [CrossRef]
- CrystalClear-SM Expert; v2.0 and 2.1; Rigaku Americas: The Woodlands, TX, USA, 2010; Rigaku Corporation: Tokyo, Japan, 2015.
- CrysAlisPro; v1.171.42.49; Rigaku Oxford Diffraction, Rigaku Corporation: Oxford, UK, 2022.
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. Sect. A Found. Crystallogr. 2008, 64, 112–122. [Google Scholar] [CrossRef]
- Beurskens, P.T.; Beurskens, G.; de Gelder, R.; Garcia-Granda, S.; Gould, R.O.; Israel, R.; Smits, J.M.M. DIRDIF-99; Crystallography Laboratory, University of Nijmegen: Nijmegen, The Netherlands, 1999. [Google Scholar]
- Palatinus, L.; Chapuis, G. SUPERFLIP—A computer program for the solution of crystal structures by charge flipping in arbitrary dimensions. J. Appl. Crystallogr. 2007, 40, 786–790. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- CrystalStructure; v4.3.0; Rigaku Americas: The Woodlands, TX, USA; Rigaku Corporation: Tokyo, Japan, 2018.
- Neese, F. Software update: The ORCA program system—Version 5.0. WIREs Comput. Mol. Sci. 2022, 12, e1606. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- van Lenthe, E.; Snijders, J.G.; Baerends, E.J. The zero-order regular approximation for relativistic effects: The effect of spin–orbit coupling in closed shell molecules. J. Chem. Phys. 1996, 105, 6505–6516. [Google Scholar] [CrossRef]
- Weigend, F.; Furche, F.; Ahlrichs, R. Gaussian basis sets of quadruple zeta valence quality for atoms H–Kr. J. Chem. Phys. 2003, 119, 12753–12762. [Google Scholar] [CrossRef]
- Schäfer, A.; Horn, H.; Ahlrichs, R. Fully optimized contracted Gaussian basis sets for atoms Li to Kr. J. Chem. Phys. 1992, 97, 2571–2577. [Google Scholar] [CrossRef]
- Schäfer, A.; Huber, C.; Ahlrichs, R. Fully optimized contracted Gaussian basis sets of triple zeta valence quality for atoms Li to Kr. J. Chem. Phys. 1994, 100, 5829–5835. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- Pantazis, D.A.; Chen, X.-Y.; Landis, C.R.; Neese, F. All-Electron Scalar Relativistic Basis Sets for Third-Row Transition Metal Atoms. J. Chem. Theory Comput. 2008, 4, 908–919. [Google Scholar] [CrossRef] [PubMed]
- Weigend, F. Accurate Coulomb-fitting basis sets for H to Rn. Phys. Chem. Chem. Phys. 2006, 8, 1057–1065. [Google Scholar] [CrossRef] [PubMed]
- Pantazis, D.A.; Neese, F. All-Electron Scalar Relativistic Basis Sets for the Lanthanides. J. Chem. Theory Comput. 2009, 5, 2229–2238. [Google Scholar] [CrossRef] [PubMed]
- Pantazis, D.A.; Neese, F. All-Electron Scalar Relativistic Basis Sets for the Actinides. J. Chem. Theory Comput. 2011, 7, 677–684. [Google Scholar] [CrossRef]
- Pantazis, D.A.; Neese, F. All-electron scalar relativistic basis sets for the 6p elements. Theor. Chem. Acc. 2012, 131, 1292. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef]
- Goerigk, L.; Grimme, S. A thorough benchmark of density functional methods for general main group thermochemistry, kinetics, and noncovalent interactions. Phys. Chem. Chem. Phys. 2011, 13, 6670–6688. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef] [PubMed]
- Adamo, C.; Barone, V. Toward reliable density functional methods without adjustable parameters: The PBE0 model. J. Chem. Phys. 1999, 110, 6158–6170. [Google Scholar] [CrossRef]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeersch, T.; Zurek, E.; Hutchison, G.R. Avogadro: An advanced semantic chemical editor, visualization, and analysis platform. J. Cheminform. 2012, 4, 17. [Google Scholar] [CrossRef]
- Chalmers, B.A.; Nejman, P.S.; Llewellyn, A.V.; Felaar, A.M.; Griffiths, B.L.; Portman, E.I.; Gordon, E.L.; Fan, K.J.H.; Woollins, J.D.; Buhl, M.; et al. A Study of Through-Space and Through-Bond J(PP) Coupling in a Rigid Nonsymmetrical Bis(phosphine) and Its Metal Complexes. Inorg. Chem. 2018, 57, 3387–3398. [Google Scholar] [CrossRef] [PubMed]
- Malkina, O.L.; Hierso, J.-C.; Malkin, V.G. Distinguishing “Through-Space” from “Through-Bonds” Contribution in Indirect Nuclear Spin–Spin Coupling: General Approaches Applied to Complex JPP and JPSe Scalar Couplings. J. Am. Chem. Soc. 2022, 144, 10768–10784. [Google Scholar] [CrossRef] [PubMed]
- Taylor, L.J.; Lawson, E.E.; Cordes, D.B.; Athukorala Arachchige, K.S.; Slawin, A.M.Z.; Chalmers, B.A.; Kilian, P. Synthesis and Structural Studies of Peri-Substituted Acenaphthenes with Tertiary Phosphine and Stibine Groups Dataset; University of St Andrews Research Portal: St Andrews, UK, 2024. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | 5 | 8 | 2.PtCl2∙CH2Cl2 | 4.Mo(CO)4 |
|---|---|---|---|---|
| peri-region distances (Å) | ||||
| P9···Sb1 | 3.172(3) | 3.218(2) | 3.357(4) | 3.3762(16) |
| P9–M1 | - | - | 2.248(4) | 2.5432(16) |
| Sb1–M1 | - | - | 2.4570(10) | 2.7007(6) |
| peri-region bond angles (°) | ||||
| P9···Sb1–C (quasi-linear) | 168.48(19) [a] | 167.14(16) [b] | - | - |
| P9–M1–Sb1 | - | - | 90.93(9) | 80.10(4) |
| Splay [c] | 15.1(12) | 16.3(9) | 16(2) | 17.2(8) |
| Out-of-plane displacements (Å) | ||||
| P9 | 0.256(6) | 0.202(5) | 0.509(13) | 0.571(6) |
| Sb1 | 0.064(6) | 0.213(5) | 0.788(13) | 0.406(6) |
| M1 | - | - | 0.428(17) | 1.007(7) |
| peri-region torsion angle (°) | ||||
| P9–C9···C1–Sb1 | 6.7(3) | 11.2(3) | 30.7(7) | 24.0(3) |
| Compound | Bond | ρ(r) | ∇2ρ(r) | Ei (kcal mol−1) | BD | |V(r)|/G(r) | ε |
|---|---|---|---|---|---|---|---|
| 5 | P9∙∙∙Sb1 | 0.0272 | 0.0349 | −4.6 | −0.108 | 1.25 | 0.0446 |
| C1–Sb1 | 0.1053 | 0.0620 | −34.9 | −0.455 | 1.76 | 0.0901 | |
| C9–P9 | 0.1600 | −0.0631 | −87.1 | −0.916 | 2.12 | 0.1394 | |
| Sb1–C13 | 0.1110 | 0.0652 | −38.0 | −0.472 | 1.76 | 0.0275 | |
| Sb1–C19 | 0.1020 | 0.0278 | −30.9 | −0.450 | 1.87 | 0.0330 | |
| 8 | P9∙∙∙Sb1 | 0.0246 | 0.0332 | −4.0 | −0.088 | 1.21 | 0.0536 |
| C1–Sb1 | 0.1038 | 0.0623 | −34.2 | −0.450 | 1.75 | 0.0933 | |
| C9–P9 | 0.1603 | −0.0637 | −87.3 | −0.917 | 2.12 | 0.1128 | |
| Sb1–C13 | 0.1018 | 0.0401 | −31.7 | −0.447 | 1.82 | 0.0412 | |
| Sb1–C17 | 0.1074 | 0.0347 | −34.1 | −0.466 | 1.85 | 0.0212 |
| Compound | ΔEint | ΔEsteric | ΔEorb | ΔEdisp |
|---|---|---|---|---|
| 5 | −226.67 | −40.55 | −173.78 | −12.34 |
| 8 | −221.92 | −39.61 | −171.04 | −11.27 |
| 2.PtCl2 | 4.Mo(CO)4 | 5 | 8 | |
|---|---|---|---|---|
| formula | C31H34Cl4PPtSb | C29H30MoO4PSb | C27H34PSb | C6H40PSb |
| fw | 896.24 | 691.22 | 511.29 | 505.33 |
| crystal description | Colourless block | Colourless prism | Yellow chip | Colourless chip |
| crystal size [mm3] | 0.09 × 0.06 × 0.03 | 0.17 × 0.15 × 0.04 | 0.10 × 0.08 × 0.06 | 0.12 × 0.10 × 0.03 |
| temperature [K] | 125 | 173 | 173 | 173 |
| space group | Pna21 | P | P21/n | C2/c |
| a [Å] | 14.797(2) | 10.1706(3) | 9.751(3) | 27.3960(13) |
| b [Å] | 18.166(3) | 12.6048(5) | 13.591(2) | 8.6563(4) |
| c [Å] | 11.8407(19) | 12.8276(7) | 19.091(5) | 22.5385(14) |
| α [°] | 76.520(14) | |||
| β [°] | 67.547(11) | 102.261(8) | 109.617(6) | |
| γ [°] | 67.048(12) | |||
| vol [Å]3 | 3182.8(9) | 1392.63(19) | 2472.3(11) | 5034.7(5) |
| Z | 4 | 2 | 4 | 8 |
| ρ (calc) [g/cm3] | 1.870 | 1.648 | 1.374 | 1.333 |
| μ [mm−1] | 5.626 | 1.507 | 1.189 | 1.167 |
| F(000) | 1728 | 688 | 1048 | 2096 |
| reflections collected | 24,780 | 24,150 | 29,608 | 28,282 |
| independent reflections (Rint) | 6286 (0.0886) | 4970 (0.1133) | 4528 (0.0604) | 22,368 (0.0755) |
| parameters, restraints | 347, 1 | 330, 0 | 297, 45 | 260, 0 |
| GoF on F2 | 1.106 | 1.081 | 1.109 | 0.720 |
| R1 [I > 2σ(I)] | 0.0505 | 0.0570 | 0.0620 | 0.0523 |
| wR2 (all data) | 0.0978 | 0.1494 | 0.1308 | 0.1151 |
| largest diff. peak/hole [e/Å3] | 0.88, −1.30 | 2.41, −0.72 | 0.67, −0.58 | 2.03, −1.39 |
| Flack parameter | 0.012(6) | - | - | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Taylor, L.J.; Lawson, E.E.; Cordes, D.B.; Athukorala Arachchige, K.S.; Slawin, A.M.Z.; Chalmers, B.A.; Kilian, P. Synthesis and Structural Studies of peri-Substituted Acenaphthenes with Tertiary Phosphine and Stibine Groups. Molecules 2024, 29, 1841. https://doi.org/10.3390/molecules29081841
Taylor LJ, Lawson EE, Cordes DB, Athukorala Arachchige KS, Slawin AMZ, Chalmers BA, Kilian P. Synthesis and Structural Studies of peri-Substituted Acenaphthenes with Tertiary Phosphine and Stibine Groups. Molecules. 2024; 29(8):1841. https://doi.org/10.3390/molecules29081841
Chicago/Turabian StyleTaylor, Laurence J., Emma E. Lawson, David B. Cordes, Kasun S. Athukorala Arachchige, Alexandra M. Z. Slawin, Brian A. Chalmers, and Petr Kilian. 2024. "Synthesis and Structural Studies of peri-Substituted Acenaphthenes with Tertiary Phosphine and Stibine Groups" Molecules 29, no. 8: 1841. https://doi.org/10.3390/molecules29081841
APA StyleTaylor, L. J., Lawson, E. E., Cordes, D. B., Athukorala Arachchige, K. S., Slawin, A. M. Z., Chalmers, B. A., & Kilian, P. (2024). Synthesis and Structural Studies of peri-Substituted Acenaphthenes with Tertiary Phosphine and Stibine Groups. Molecules, 29(8), 1841. https://doi.org/10.3390/molecules29081841