
Phosphorus Modification of Iron: Mechanistic Insights into Ammonia Synthesis on Fe2P Catalyst


Abstract
1. Introduction
2. Results and Discussion
2.1. Optimized Adsorbates and Their Binding Energies
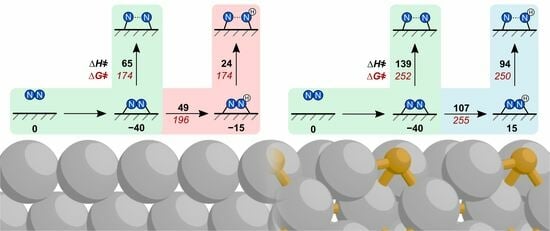
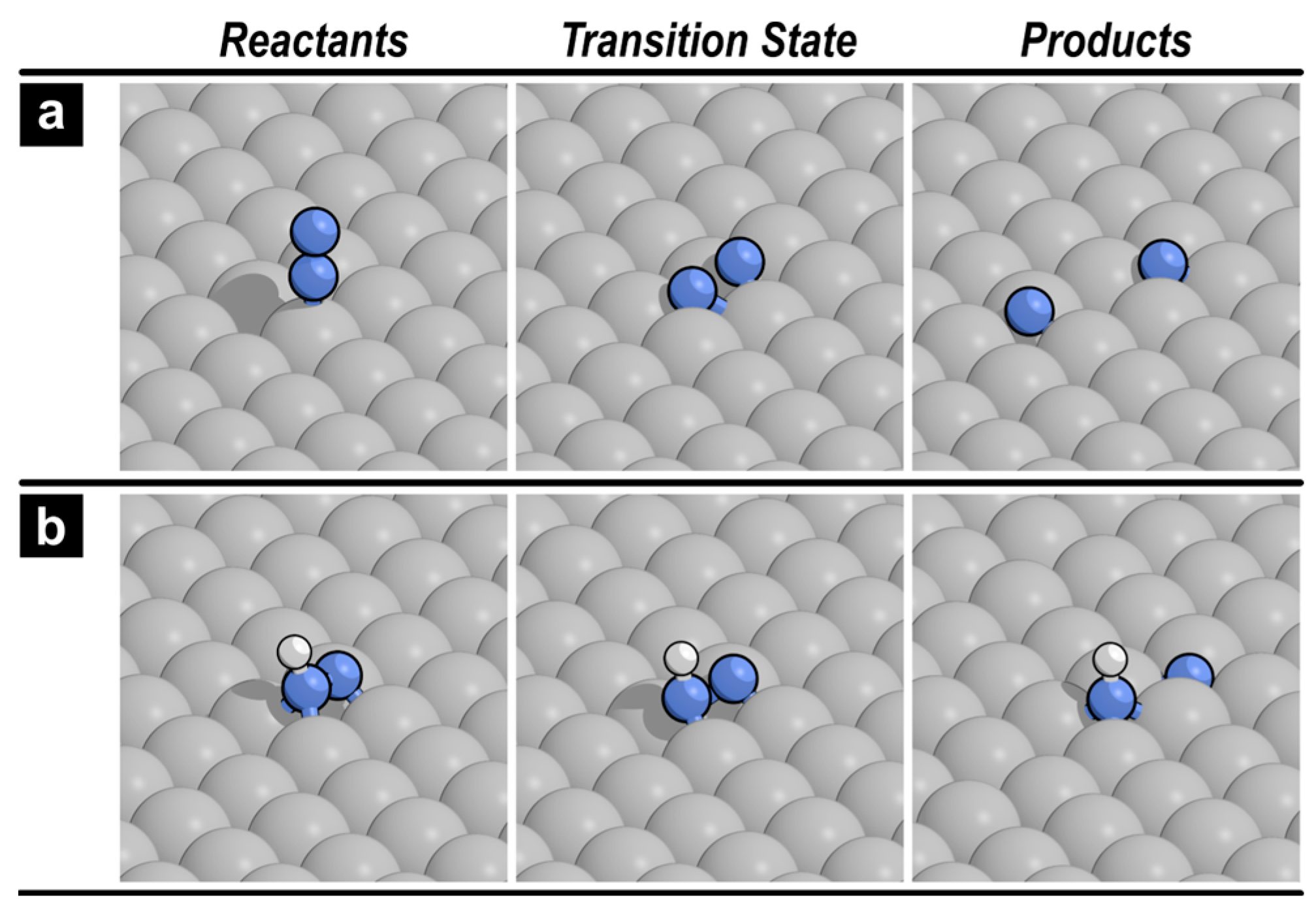
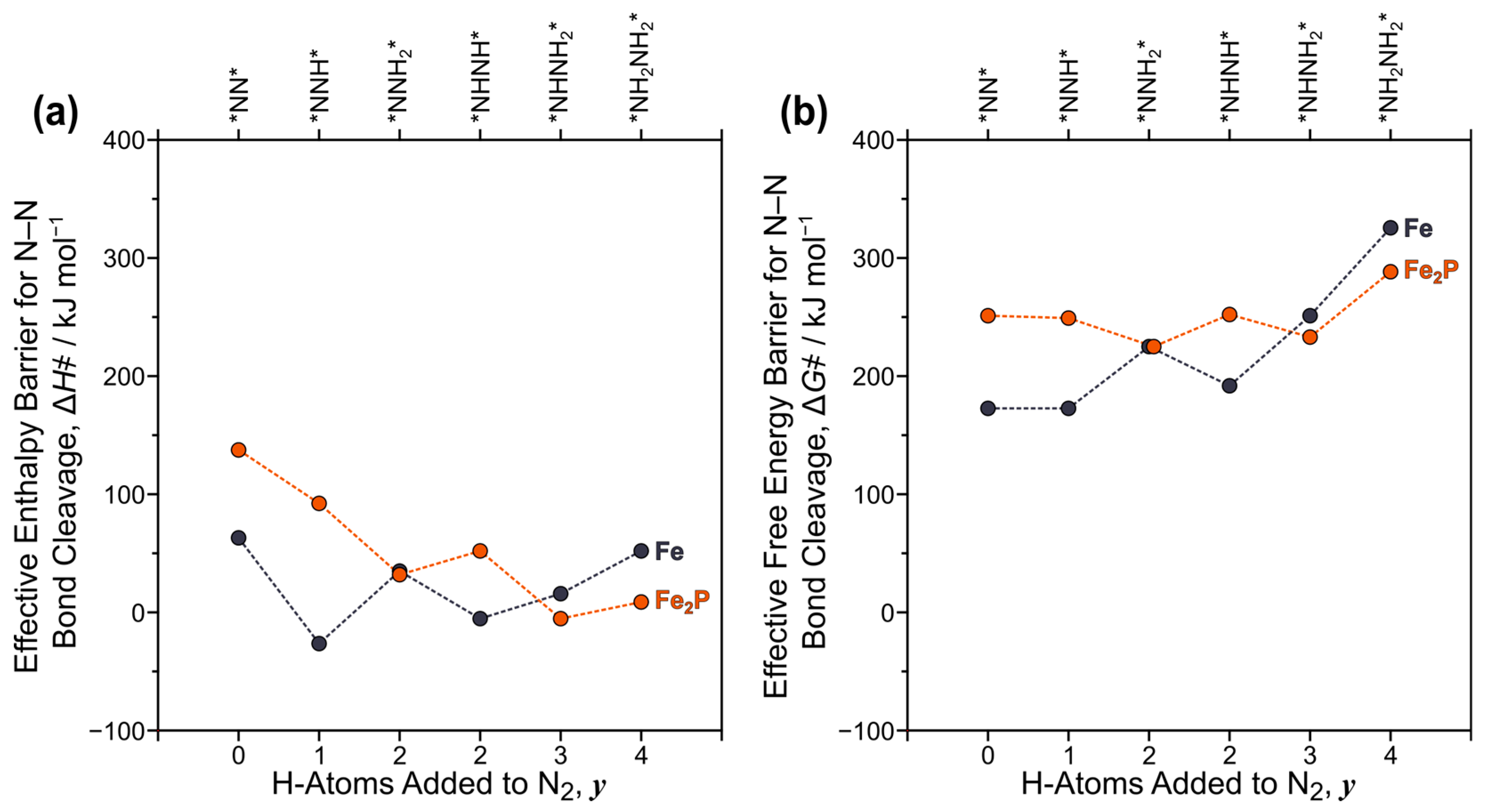
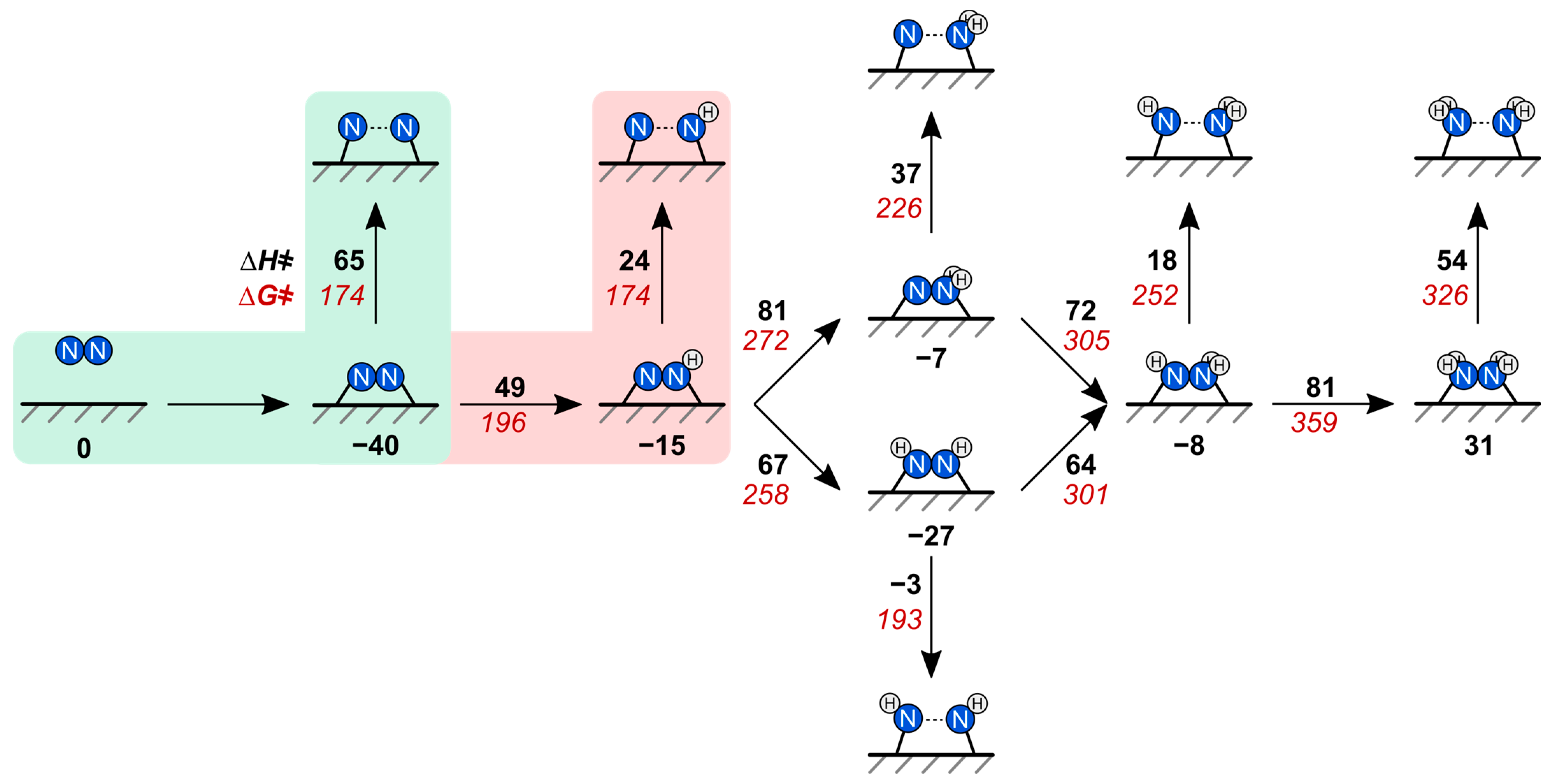
2.2. N–N Bond Activation Pathways on Fe(110)
2.3. N–N Bond Activation Pathways on Fe2P(001)
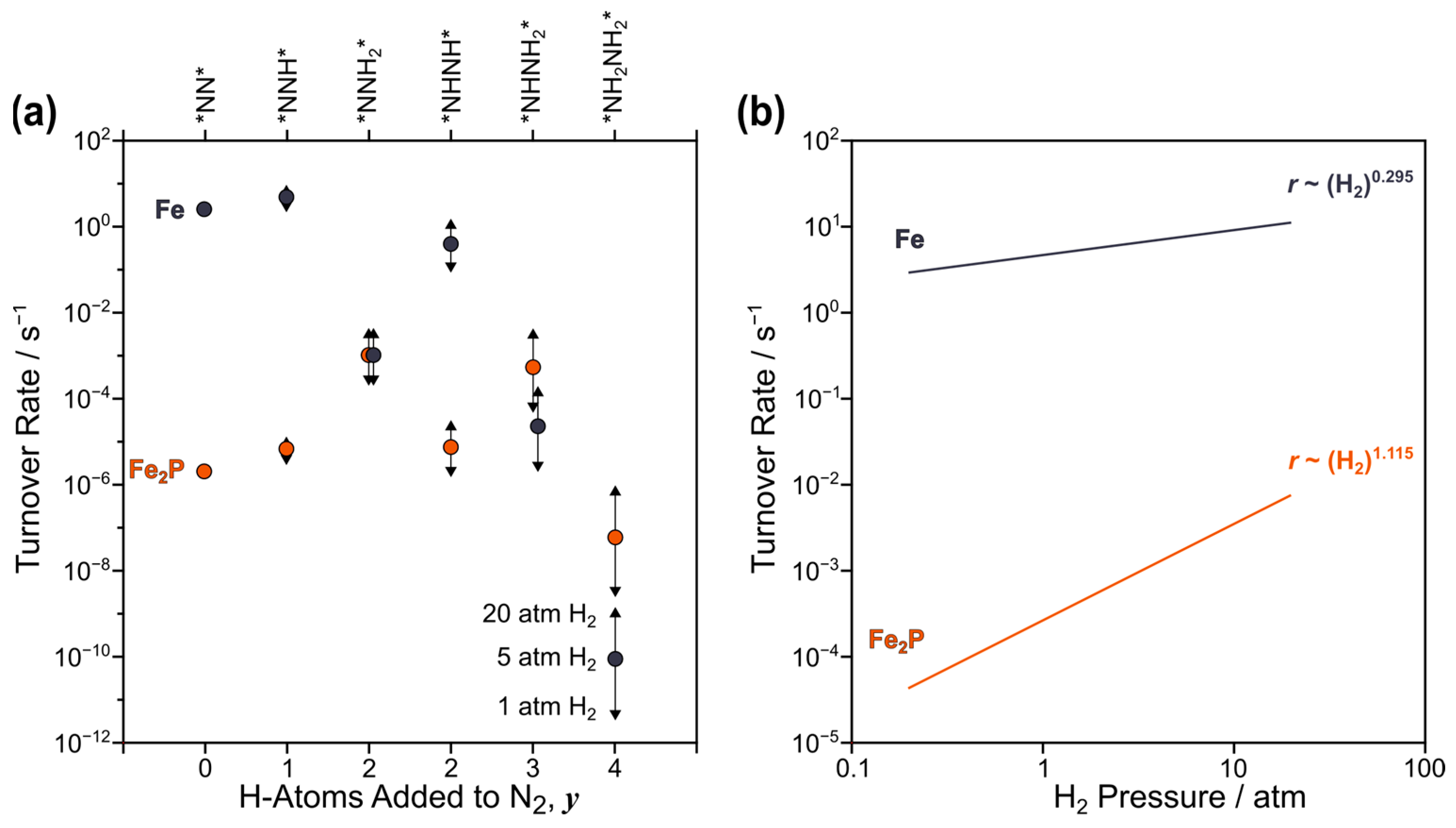
2.4. DFT-Predicted N–N Bond Cleavage Turnover Rate
3. Computational Methods
4. Conclusions
Supplementary Materials
Funding
Data Availability Statement
Conflicts of Interest
References
- MacFarlane, D.R.; Cherepanov, P.V.; Choi, J.; Suryanto, B.H.R.; Hodgetts, R.Y.; Bakker, J.M.; Ferrero Vallana, F.M.; Simonov, A.N. A Roadmap to the Ammonia Economy. Joule 2020, 4, 1186–1205. [Google Scholar] [CrossRef]
- Smil, V. Enriching the Earth: Fritz Haber, Carl Bosch, and the Transformation of World Food Production; MIT Press: Cambridge, MA, USA, 2004; ISBN 0262693135. [Google Scholar]
- Pfromm, P.H. Towards Sustainable Agriculture: Fossil-Free Ammonia. J. Renew. Sustain. Energy 2017, 9, 034702. [Google Scholar] [CrossRef]
- Boudart, M. Kinetics and Mechanism of Ammonia Synthesis. Catal. Rev. 1981, 23, 1–15. [Google Scholar] [CrossRef]
- Honkala, K.; Hellman, A.; Remediakis, I.N.; Logadottir, A.; Carlsson, A.; Dahl, S.; Christensen, C.H.; Nørskov, J.K. Ammonia Synthesis from First-Principles Calculations. Science 2005, 307, 555–558. [Google Scholar] [CrossRef] [PubMed]
- Schlögl, R. Catalytic Synthesis of Ammonia—A “Never-Ending Story”? Angew. Chem. Int. Ed. 2003, 42, 2004–2008. [Google Scholar] [CrossRef] [PubMed]
- Ertl, G. Surface Science and Catalysis—Studies on the Mechanism of Ammonia Synthesis: The P. H. Emmett Award Address. Catal. Rev. 1980, 21, 201–223. [Google Scholar] [CrossRef]
- Logadóttir, Á.; Nørskov, J.K. Ammonia Synthesis over a Ru(0001) Surface Studied by Density Functional Calculations. J. Catal. 2003, 220, 273–279. [Google Scholar] [CrossRef]
- Vojvodic, A.; Medford, A.J.; Studt, F.; Abild-Pedersen, F.; Khan, T.S.; Bligaard, T.; Nørskov, J.K. Exploring the Limits: A Low-Pressure, Low-Temperature Haber-Bosch Process. Chem. Phys. Lett. 2014, 598, 108–112. [Google Scholar] [CrossRef]
- Logadottir, A.; Rod, T.H.; Nørskov, J.K.; Hammer, B.; Dahl, S.; Jacobsen, C.J.H. The Brønsted-Evans-Polanyi Relation and the Volcano Plot for Ammonia Synthesis over Transition Metal Catalysts. J. Catal. 2001, 197, 229–231. [Google Scholar] [CrossRef]
- Munter, T.R.; Bligaard, T.; Christensen, C.H.; Nørskov, J.K. BEP Relations for N2 Dissociation over Stepped Transition Metal and Alloy Surfaces. Phys. Chem. Chem. Phys. 2008, 10, 5202–5206. [Google Scholar] [CrossRef]
- Nørskov, J.K.; Bligaard, T.; Hvolbæk, B.; Abild-Pedersen, F.; Chorkendorff, I.; Christensen, C.H. The Nature of the Active Site in Heterogeneous Metal Catalysis. Chem. Soc. Rev. 2008, 37, 2163–2171. [Google Scholar] [CrossRef] [PubMed]
- Medford, A.J.; Vojvodic, A.; Hummelshøj, J.S.; Voss, J.; Abild-Pedersen, F.; Studt, F.; Bligaard, T.; Nilsson, A.; Nørskov, J.K. From the Sabatier Principle to a Predictive Theory of Transition-Metal Heterogeneous Catalysis. J. Catal. 2015, 328, 36–42. [Google Scholar] [CrossRef]
- Nørskov, J.K.; Bligaard, T.; Logadottir, A.; Bahn, S.; Hansen, L.B.; Bollinger, M.; Bengaard, H.; Hammer, B.; Sljivancanin, Z.; Mavrikakis, M.; et al. Universality in Heterogeneous Catalysis. J. Catal. 2002, 209, 275–278. [Google Scholar] [CrossRef]
- Jacobsen, C.J.H.; Dahl, S.; Clausen, B.G.S.; Bahn, S.; Logadottir, A.; Nørskov, J.K. Catalyst Design by Interpolation in the Periodic Table: Bimetallic Ammonia Synthesis Catalysts. J. Am. Chem. Soc. 2001, 123, 8404–8405. [Google Scholar] [CrossRef] [PubMed]
- Zeinalipour-Yazdi, C.D.; Hargreaves, J.S.J.; Catlow, C.R.A. Nitrogen Activation in a Mars–van Krevelen Mechanism for Ammonia Synthesis on Co3Mo3N. J. Phys. Chem. C 2015, 119, 28368–28376. [Google Scholar] [CrossRef]
- Jacobsen, C.J.H. Novel Class of Ammonia Synthesis Catalysts. Chem. Commun. 2000, 1057–1058. [Google Scholar] [CrossRef]
- Wang, P.; Chang, F.; Gao, W.; Guo, J.; Wu, G.; He, T.; Chen, P. Breaking Scaling Relations to Achieve Low-Temperature Ammonia Synthesis through LiH-Mediated Nitrogen Transfer and Hydrogenation. Nat. Chem. 2017, 9, 64–70. [Google Scholar] [CrossRef]
- Vojvodic, A.; Calle-Vallejo, F.; Guo, W.; Wang, S.; Toftelund, A.; Studt, F.; Martínez, J.I.; Shen, J.; Man, I.C.; Rossmeisl, J.; et al. On the Behavior of Brønsted-Evans-Polanyi Relations for Transition Metal Oxides. J. Chem. Phys. 2011, 134, 244509. [Google Scholar] [CrossRef]
- Somorjai, G.A.; Materer, N. Surface Structures in Ammonia Synthesis. Top. Catal. 1994, 1, 215–231. [Google Scholar] [CrossRef]
- Qian, J.; An, Q.; Fortunelli, A.; Nielsen, R.J.; Goddard, W.A. Reaction Mechanism and Kinetics for Ammonia Synthesis on the Fe(111) Surface. J. Am. Chem. Soc. 2018, 140, 6288–6297. [Google Scholar] [CrossRef]
- Hu, J.; Zhao, T.; Zhang, H.; Li, X.; Shi, A.; Li, X.; Wang, Q.; Hu, G. Fe2P Nanoparticle-Decorated Porous Biochar for High-Efficiency Electrosynthesis of Ammonia from Toxic Nitrite. Surf. Interfaces 2023, 38, 102818. [Google Scholar] [CrossRef]
- Chouki, T.; Machreki, M.; Rutkowska, I.A.; Rytelewska, B.; Kulesza, P.J.; Tyuliev, G.; Harb, M.; Azofra, L.M.; Emin, S. Highly Active Iron Phosphide Catalysts for Selective Electrochemical Nitrate Reduction to Ammonia. J. Environ. Chem. Eng. 2023, 11, 109275. [Google Scholar] [CrossRef]
- Witzke, M.E.; Almithn, A.; Conrad, C.L.; Hibbitts, D.D.; Flaherty, D.W. Mechanisms and Active Sites for C–O Bond Rupture within 2-Methyltetrahydrofuran over Ni, Ni12P5, and Ni2P Catalysts. ACS Catal. 2018, 8, 7141–7157. [Google Scholar] [CrossRef]
- Almithn, A.; Alhulaybi, Z. A Mechanistic Study of Methanol Steam Reforming on Ni2P Catalyst. Catalysts 2022, 12, 1174. [Google Scholar] [CrossRef]
- Almithn, A.; Alghanim, S.N.; Mohammed, A.A.; Alghawinim, A.K.; Alomaireen, M.A.; Alhulaybi, Z.; Hossain, S.S. Methane Activation and Coupling Pathways on Ni2P Catalyst. Catalysts 2023, 13, 531. [Google Scholar] [CrossRef]
- Waldt, C.; Montalvo-Castro, H.; Almithn, A.; Loaiza-Orduz, Á.; Plaisance, C.; Hibbitts, D. Role of Phosphorous in Transition Metal Phosphides for Selective Hydrogenolysis of Hindered C–O Bonds. J. Catal. 2023, 421, 403–418. [Google Scholar] [CrossRef]
- Mortensen, J.J.; Hansen, L.B.; Hammer, B.; Nørskov, J.K. Nitrogen Adsorption and Dissociation on Fe(111). J. Catal. 1999, 182, 479–488. [Google Scholar] [CrossRef]
- Logadottir, A.; Nørskov, J.K. The Effect of Strain for N2 Dissociation on Fe Surfaces. Surf. Sci. 2001, 489, 135–143. [Google Scholar] [CrossRef]
- Wang, T.; Tian, X.; Yang, Y.; Li, Y.-W.; Wang, J.; Beller, M.; Jiao, H. Coverage-Dependent N2 Adsorption and Its Modification of Iron Surfaces Structures. J. Phys. Chem. C 2016, 120, 2846–2854. [Google Scholar] [CrossRef]
- Witzke, M.E.; Almithn, A.; Conrad, C.L.; Triezenberg, M.D.; Hibbitts, D.D.; Flaherty, D.W. In Situ Methods for Identifying Reactive Surface Intermediates during Hydrogenolysis Reactions: C–O Bond Cleavage on Nanoparticles of Nickel and Nickel Phosphides. J. Am. Chem. Soc. 2019, 141, 16671–16684. [Google Scholar] [CrossRef]
- Egeberg, R.C.; Dahl, S.; Logadottir, A.; Larsen, J.H.; Nùrskov, J.K.; Chorkendorff, I. N2 Dissociation on Fe(1 1 0) and Fe/Ru(0 0 0 1): What Is the Role of Steps? Surf. Sci. 2001, 491, 183–194. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Ab Initio Molecular Dynamics for Liquid Metals. Phys. Rev. B 1993, 47, 558–561. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Hafner, J. Ab Initio Molecular-Dynamics Simulation of the Liquid-Metal–Amorphous-Semiconductor Transition in Germanium. Phys. Rev. B 1994, 49, 14251–14269. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient Iterative Schemes for Ab Initio Total-Energy Calculations Using a Plane-Wave Basis Set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Furthmüller, J. Efficiency of Ab-Initio Total Energy Calculations for Metals and Semiconductors Using a Plane-Wave Basis Set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Kravchenko, P.; Plaisance, C.; Hibbitts, D. A New Computational Interface for Catalysis. ChemRxiv 2019. [Google Scholar] [CrossRef]
- Blöchl, P.E. Projector Augmented-Wave Method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Joubert, D. From Ultrasoft Pseudopotentials to the Projector Augmented-Wave Method. Phys. Rev. B 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Hammer, B.; Hansen, L.B.; Nørskov, J.K. Improved Adsorption Energetics within Density-Functional Theory Using Revised Perdew-Burke-Ernzerhof Functionals. Phys. Rev. B 1999, 59, 7413–7421. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef]
- Zhang, Y.; Yang, W. Comment on “Generalized Gradient Approximation Made Simple”. Phys. Rev. Lett. 1998, 80, 890. [Google Scholar] [CrossRef]
- Wilburn, D.R.; Bassett, W.A. Hydrostatic Compression of Iron and Related Compounds; an Overview. Am. Mineral. 1978, 63, 591–596. [Google Scholar]
- Koumina, A.; Bacmann, M.; Fruchart, D.; Soubeyroux, J.-L.; Wolfers, P.; Tobola, J.; Kaprzyk, S.; Niziol, S.; Mesnaoui, M.; Zach, R. Crystallographic and Magnetic Properties of Fe2P. Ann. Chim. Sci. Des Mater. 1998, 23, 177–180. [Google Scholar] [CrossRef]
- Monkhorst, H.J.; Pack, J.D. Special Points for Brillouin-Zone Integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Pack, J.D.; Monkhorst, H.J. “Special Points for Brillouin-Zone Integrations”—A Reply. Phys. Rev. B 1977, 16, 1748–1749. [Google Scholar] [CrossRef]
- Henkelman, G.; Jónsson, H. Improved Tangent Estimate in the Nudged Elastic Band Method for Finding Minimum Energy Paths and Saddle Points. J. Chem. Phys. 2000, 113, 9978–9985. [Google Scholar] [CrossRef]
- Jónsson, H.; Mills, G.; Jacobsen, K.W. Nudged Elastic Band Method for Finding Minimum Energy Paths of Transitions. In Proceedings of the Classical and Quantum Dynamics in Condensed Phase Simulations; World Scientific: Singapore, 1998; pp. 385–404. [Google Scholar]
- Henkelman, G.; Jónsson, H. A Dimer Method for Finding Saddle Points on High Dimensional Potential Surfaces Using Only First Derivatives. J. Chem. Phys. 1999, 111, 7010–7022. [Google Scholar] [CrossRef]
- McQuarrie, D.A. Statistical Mechanics; University Science Books: Sausalito, CA, USA, 2000. [Google Scholar]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Fe(110) | Fe2P(001) | |||
|---|---|---|---|---|
| Species | Adsorption Mode | ΔEads | Adsorption Mode | ΔEads |
| kJ mol−1 | kJ mol−1 | |||
| N2* | M1 | −33 | M1 | −47 |
| N* | M3 | −576 | M3 | −518 |
| NH* | M3 | −465 | M3 | −426 |
| NH2* | M2 | −263 | M2 | −258 |
| NH3* | M1 | −52 | M1 | −64 |
| H* | M3 | −272 | M3 | −256 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Almithn, A. Phosphorus Modification of Iron: Mechanistic Insights into Ammonia Synthesis on Fe2P Catalyst. Molecules 2024, 29, 1894. https://doi.org/10.3390/molecules29081894
Almithn A. Phosphorus Modification of Iron: Mechanistic Insights into Ammonia Synthesis on Fe2P Catalyst. Molecules. 2024; 29(8):1894. https://doi.org/10.3390/molecules29081894
Chicago/Turabian StyleAlmithn, Abdulrahman. 2024. "Phosphorus Modification of Iron: Mechanistic Insights into Ammonia Synthesis on Fe2P Catalyst" Molecules 29, no. 8: 1894. https://doi.org/10.3390/molecules29081894
APA StyleAlmithn, A. (2024). Phosphorus Modification of Iron: Mechanistic Insights into Ammonia Synthesis on Fe2P Catalyst. Molecules, 29(8), 1894. https://doi.org/10.3390/molecules29081894