Abstract
A study on the functionalisation of 2-mercapto-5-methyl-1,3,4-thiadiazole has been conducted, yielding two series of products: 2-(ω-haloalkylthio)thiadiazoles and symmetrical bis-thiadiazoles, with variable chain lengths. The experimental conditions were optimised for each class of compounds by altering the base used and the reagents’ proportions, leading to the development of separate protocols tailored to their specific reactivity and purification needs. The target halogenide reagents and bis-thiadiazole ligands were obtained either as single products or as mixtures easily separable by chromatography. Characterisation of the products was performed using 1D and 2D NMR spectra in solution, complemented by single crystal X-ray diffraction (XRD) for selected samples, to elucidate their structural properties.
1. Introduction
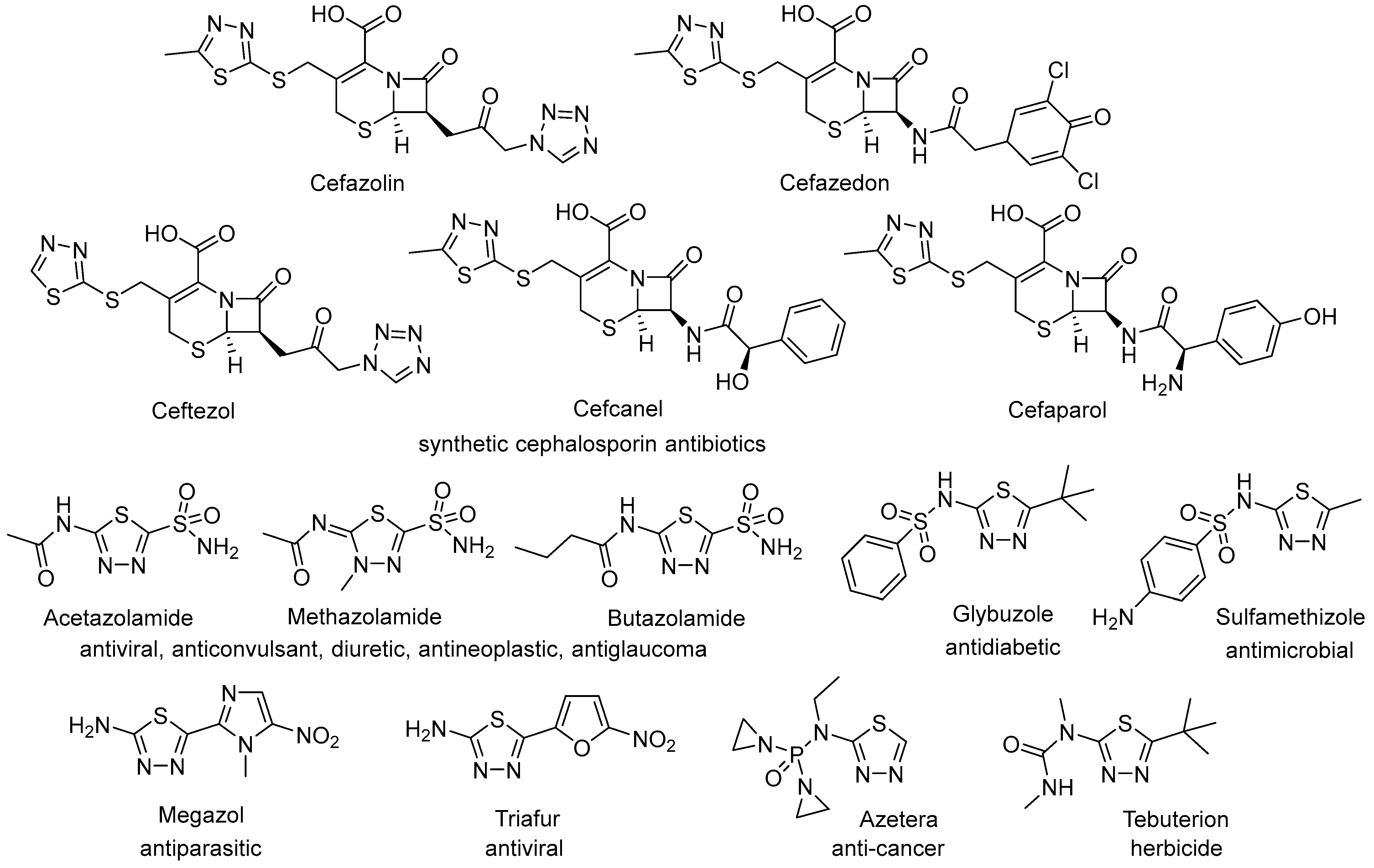
The 1,3,4-thiadiazole motif serves as a structural subunit in numerous products with remarkable biological activities [1,2,3,4,5,6,7,8,9], such as anticancer [10,11,12,13,14,15], antimicrobial [16,17,18], antiepileptic [19,20,21], among others. Many drugs featuring the 1,3,4-thiadiazole unit are commercially available and have become widely prescribed medications. Notable examples include a series of synthetic and semi-synthetic cephalosporin antibiotics, sulfonamide drugs with multifunctional activities, among a few others, illustrated in Figure 1. In addition to their diverse biological activities, derivatives of 1,3,4-thiadiazole have also been employed as ligands for metal complexes [22,23], in chelating resins [24], as components of azo dyes [25], and as lubricant additives [26], demonstrating their versatility across various applications.
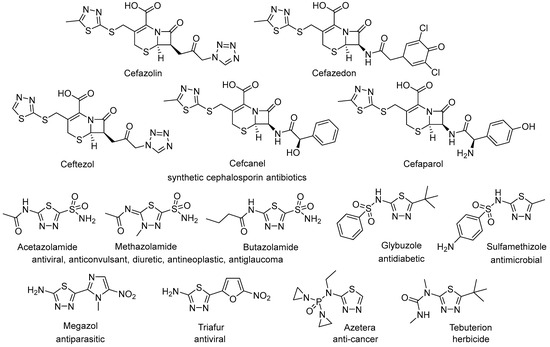
Figure 1.
Products available on the market containing a thiadiazole unit.
Several synthetic protocols for the preparation of 1,3,4-thiadiazole-containing bioactive compounds have been developed and summarised [2,8,17,27,28,29,30,31,32,33], the main part based on ring-closure reactions. Variable procedures are applied, like intramolecular cyclization of thiosemicarbazides [15,25] or potassium salt of hydrazinodithio formate [34] in concentrated sulphuric acid, condensation of thiosemicarbazide with carboxylic acids [35], benzoic acids [36,37], or N-arylsulfonylated amino acids [38], thiocarbohydrazides with aldehydes [39], hydrazinecarbodithioates or thiosemicarbazones with hydrazonoyl chlorides [40], sulfonamide diazonium chlorides with phenacyl thiocyanate [41], etc. At the same time, methods using an already built 1,3,4-thiadiazole unit are also exploited. The sulfonamides acetazolamide [42,43] and methazolamide [44] and similar bioactive products [45,46,47] are obtained by functionalisation of 5-amino-2-mercapto-1,3,4-thiadiazole, while the synthetic protocols for the preparation of cephalosporin antibiotics cefazolin [48,49], cefazedon [50,51], cefcanel [52], and cefaparol [53] involve 2-mercapto-5-methyl-1,3,4-thiadiazole. To the best of our knowledge, the synthesis of ω-haloalkylthio derivatives, which can be used as alkylating agents to obtain libraries of products, is not reported in the literature.
Sulphur-containing ligands, on the other hand, have shown remarkable coordination properties [54,55,56,57,58,59,60,61] and have displayed a wide range of applications [62,63,64,65,66,67,68]. Among them, heterocyclic thioethers have exhibited a broad spectrum of bioactivities [69,70,71,72] and efficiency as adsorbents for lead ion removal from water [73], ligands in N-heterocyclic carbene (NHC) complexes [74,75,76,77], metal–organic frame-works [78,79,80,81,82], etc. As a very particular example, it is shown that symmetrical bis-1,3,4-thiadiazole-containing bis-thioethers tune the framework formation of silver(I) coordination architectures [83]. Ligands with methylene and ethylene linkers are obtained and it is found that the prolongation of the hydrocarbon bridge results in increased flexibility and diversity in coordination modes.
Herein, we present a study on the functionalisation of 2-mercapto-5-methyl-1,3,4-thiadiazole with α,ω-dihalogenides with variable chain lengths, leading to 2-(ω-haloalkylthio) thiadiazoles and/or symmetrical bis-thiadiazoles, with an emphasis on the optimisation of conditions for the synthesis of both alkylation agents (bromides and chlorides) and bis-heterocyclic ligands.
2. Results and Discussion
The functionalisation of 2-mercapto-5-methyl-1,3,4-thiadiazole (1) with α,ω-dihalogenoalkanes was performed by applying the one-step procedure shown in Scheme 1. The efforts were initially directed towards optimisation of the protocols for the synthesis of halogenide reagents. To obtain the methylene-bridged compound 2, the starting thiadiazole (1) was first reacted with dibromomethane (DBM). Solvent, base, reagents’ proportions, and temperature were varied and it was observed that the bis-thiadiazole 6 was the only product in all cases, while not even traces of bromide 2a were detected. Selected examples are presented in Table 1, entries 1–3. As seen, notwithstanding the distinction in the reaction conditions, ligand 6 was isolated in commensurable good yields; 74–76%. In an attempt to override the bis-alkylation and to obtain the corresponding alkylating agent, chloride (2b), the transformation was performed in dichloromethane used both as reagent and solvent, i.e., in vast excess. Since it is well documented that dichloromethane can act as a double alkylating agent towards amines in the presence of sodium hydroxide as a base [83], weaker organic bases were applied in the experiments. Independently of the latter, compound 6 was again the predominant product, but together with chloride 2b, which was isolated in up to 25% yield (entries 4 and 5).
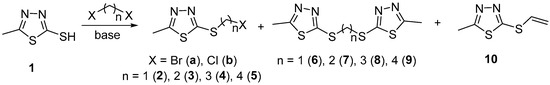
Scheme 1.
Synthesis of the alkylating agents 2–5 and bis-thiadiazole ligands 6–9.

Table 1.
Synthesis of the target reagents 2–5 and bis-thiadiazoles 6–9; selected experiments.
Further attempts were focused on the preparation of ethylene-bridged compounds. The reaction was carried out with two equiv. of dibromoethane (DBE) in order to facilitate the mono-substitution process. Variable results were obtained depending on the base used. When using potassium carbonate in acetonitrile at room temperature (entry 6) the bromide 3a was isolated in only 13% yield in parallel with 30% of bis-thiadiazole 7 and 12% of a side-product, olefin 10. The latter was obtained quantitatively by using potassium hydroxide in refluxing ethanol (98% yield), the protocol reported in the literature [23] for the preparation of 7. This fact is an indication that the heating has to be avoided at least when using an excess of the reagent, which hinders the bis-alkylation process. Contrary, when we performed the reaction with triethylamine in ether, both products were isolated in high common yield (entry 7); the bromide 3a being predominant. The reaction with dichloroethane (DCE), used both as reagent and solvent, led to a quantitative yield of the desired chloride 3b (entry 10), as already reported by us [84].
The reactions with two equiv. of dibromopropane (DBP) and dibromobutane (DBB) were carried out in ether at room temperature and with triethylamine as a base. In both cases, the desired products 4a (entry 11) and 5a (entry 15) were obtained in good yields along with the corresponding bis-thiadiazoles 8 and 9 as mixtures easily separable by chromatography. No olefin formation was detected under these particular conditions. Dichloropropane (DCP) and dichlorobutane (DCB), used both as reagents and solvents, in the presence of triethylamine as a base led, like in the case of 3b, to the corresponding chlorides 4b (entry 14) and 5b (entry 18) as sole products with 93% and 98% yield, respectively.
It should be noted that both bromides, 3a–5a, and chlorides, 3b–5b, are not stable enough and decompose after prolonged storage at room temperature; the process being faster in bromides.
Further attempts were focused on the optimisation of the protocols for the synthesis of bis-thiadiazole ligands 7–9. In order to tune the reaction output towards predominant formation of disubstituted products, the experiments were performed by alteration of the thiadiazole/reagent proportions, namely with close to a 2:1 ratio instead of 1:2. For all three ligands, the 2:1.1 ratio was found to be the most effective and the bis-thiadiazoles 7 (entry 9), 8 (entry 13), and 9 (entry 17) were obtained in 69%, 73%, and 78% yield, respectively. Thus, it can be summarised that only methylene-bridged thiadiazole is obtained as a sole product, while the corresponding bromides are also formed with the lengthening of the hydrocarbon bridge.
These results show that the efficiency of the synthetic protocols for both alkylating agents and symmetrical ligands is strongly dependent on the length of the hydrocarbon bridge and the type of the halogen atom. It is clear that the transformations with dihalogenomethanes lead to complete or predominant bis-substitution, while with the prolongation of the linker length the reaction output follows a common pattern. Bromides are always obtained as mixtures with the corresponding bis-thiadiazole, except for the methylene-linked compound 2a, where not even traces of the desired product are detected. Chlorides are formed, by using a vast excess of the reagent as a minor component, from dichloromethane (2b) or as sole products from the rest of dichlorides (3b–5b) in excellent yields. Bis-thiadiazole ligands are generated as the only products when using dibromomethane as a reagent (6) independently of the conditions, and as major components with the remaining bromides when using an excess of the starting mercaptothiadiazole.
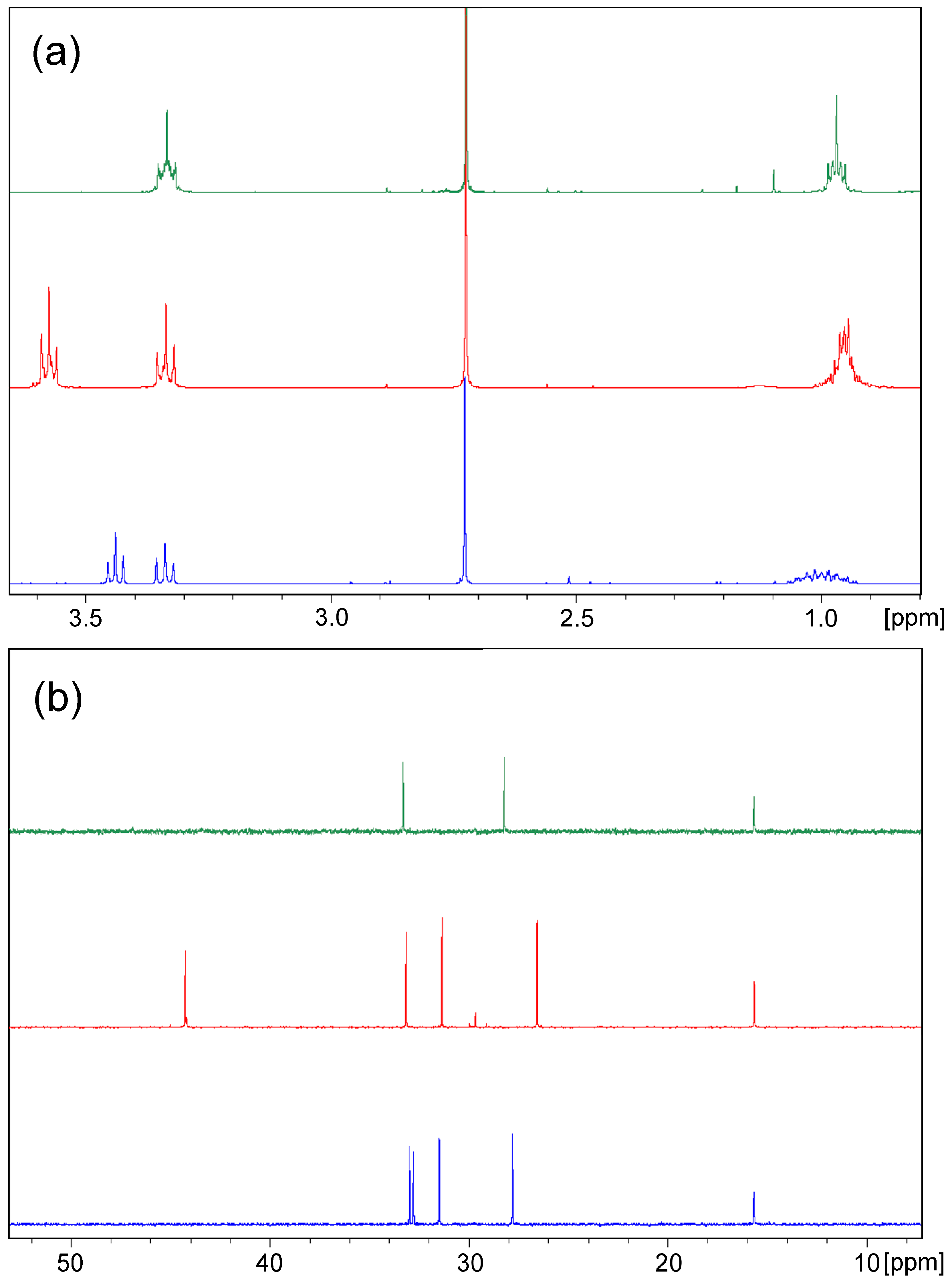
The structures of the products are assigned by 1D and 2D NMR spectra and confirmed by single crystal XRD of selected samples. The proton NMR spectra of the products with identical bridge length follow a characteristic pattern (Table S1); very similar to the chemical shifts of the signals for the methylene groups connected with sulphur but with double integral intensity in the spectrum of bis-thiadiazole compared to that of the corresponding halogenide. The main difference in carbon spectra is reasonably in the chemical shift of methylene group signals due to the influence of the halogen atom. The signals for the methylene groups directly connected to halogen are shifted up-field in bromides and down-field in chlorides, while those for methylene groups neighbouring to sulphur possess commensurable shifts, excluding methylene-bridged chloride 2b. The observed pattern is illustrated by the example of the butylene-linked compounds 5a, 5b, and 9 in Figure 2.
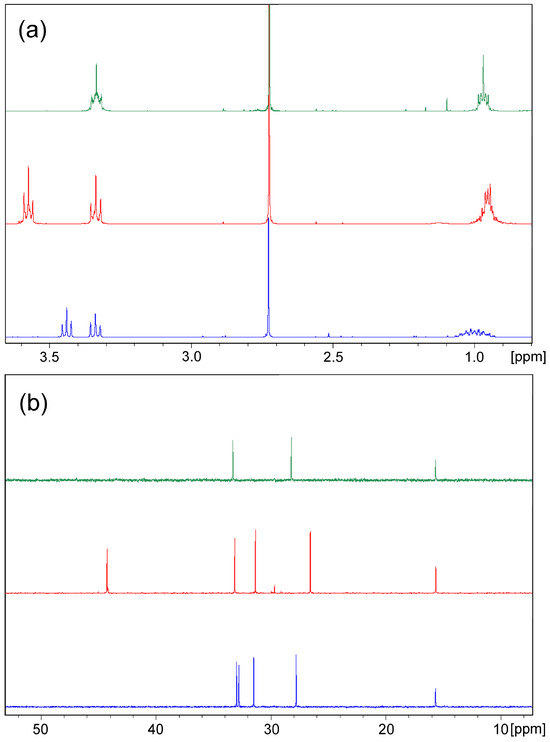
Figure 2.
Aliphatic area of 1H (a) and 13C (b) NMR spectra of compounds 5a (blue), 5b (red), and 9 (green).
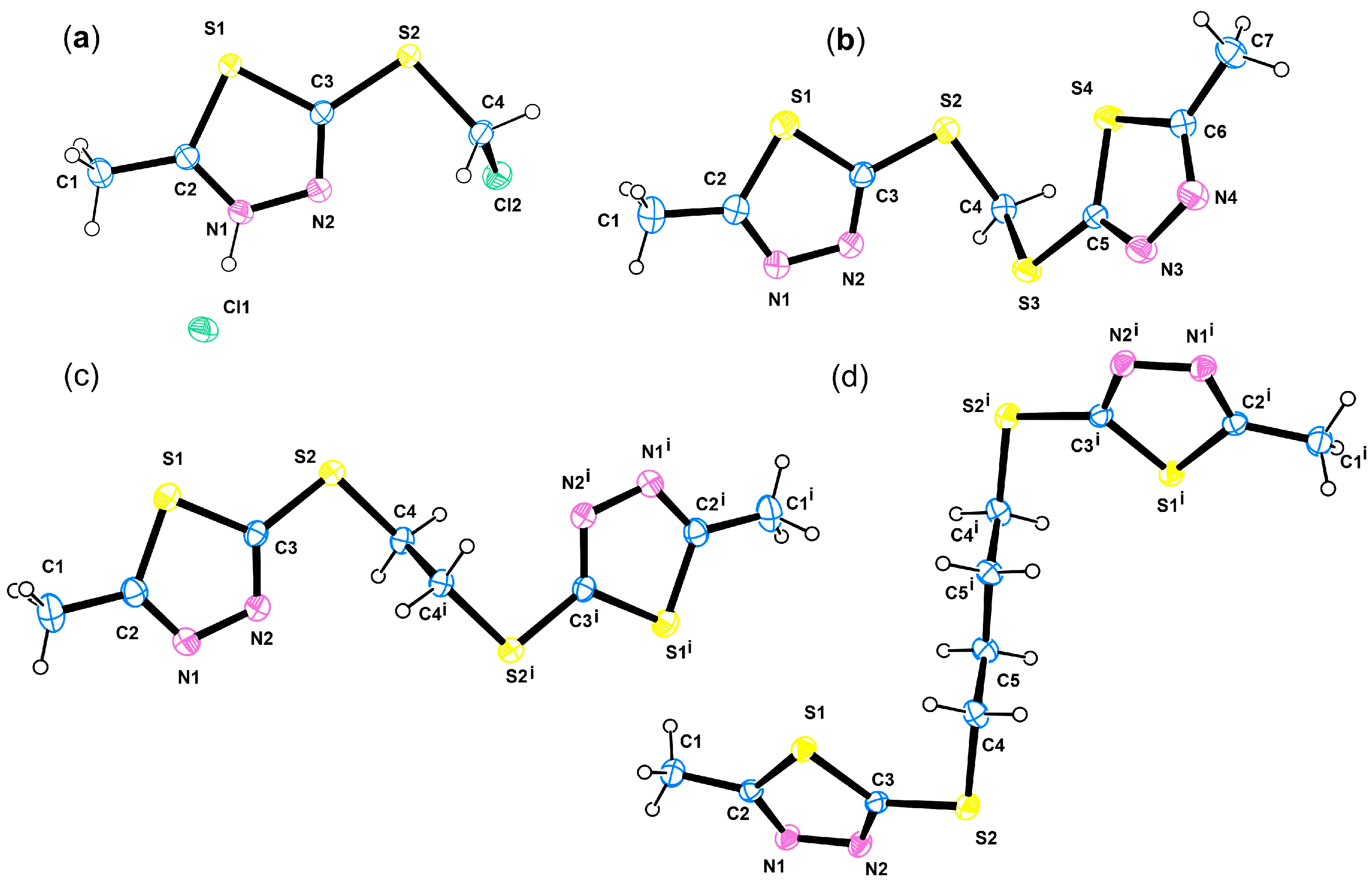
Single crystals for X-ray diffraction (XRD) analysis were grown through slow evaporation from unsaturated solutions of each compound; the colourless oil 2b being crystallised as hydrochloride. ORTEP views, depicting the molecules present in the asymmetric unit of the crystal structure, are provided in Figure 3, while essential data collection and crystallographic refinement parameters are presented in Table S2.
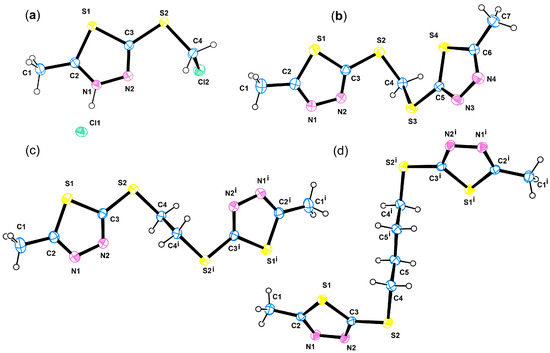
Figure 3.
ORTEP views of 2b hydrochloride (a), 6 (b), 7 (c), and 9 (d).
Compound 2b has one 2-mercapto-5-methyl-1,3,4-thiadiazole moiety while compounds 6, 7, and 9 comprise two such moieties bridged by methyl, ethyl, and butyl linkers. The crystal structures of 2b, 6, 7, and 9 disclosed a conserved geometry of the active 1,3,4-thiadiazole motif (Figure 4a). Indeed, the values of the bond distances and angles for the 1,3,4-thiadiazole are comparable to those of similar compounds [85,86,87].
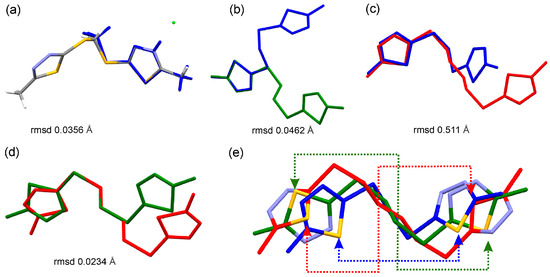
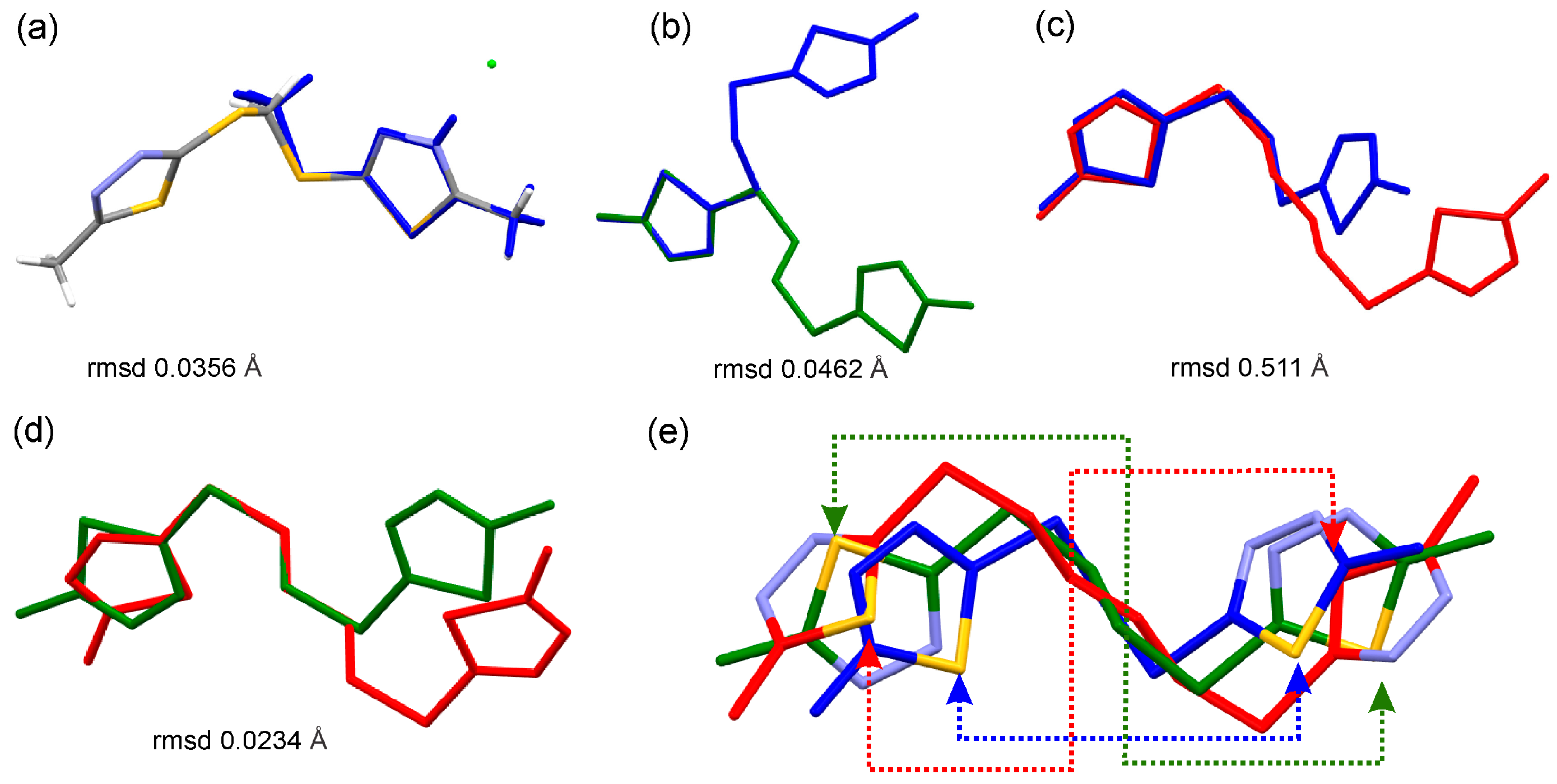
Figure 4.
Overlay of (a) the molecules of 2b hydrochloride (grey) and 6 (blue) based on the 1,3,4-thiadiazole moiety; (b) compounds 6 (blue) and 7 (green), (c) 6 (blue) and 9 (red), (d) 7 (green) and 9 (red) disclosing the conserved geometry of 1,3,4-thiadiazole moiety and a variable molecular structure due to the variable C–C chain lengths; and (e) overlay of 6, 7, and 9, the dashed lines highlight the different rotations of the thiadiazole around the C–S bond, the arrows point at S atoms.
In 2b, with the exception of the Cl2 all other atoms lie basically on the mean plane of the 1,3,4-thiadiazole moiety (Figure S1a) disclosing a distinct electronic conjugation. In 6, the angle between the mean planes of the 1,3,4-thiadiazole moieties is 65.9° (Figure S2). Further analysis of the molecular structure of 6 reveals that the rotation of the 1,3,4-thiadiazole ring along the S2–C3 and S3–C5 bond is permitted, resulting in two distinct arrangements of the thiadiazole rings around the S2/C4/S3 linker plane. The first arrangement of the 1,3,4-thiadiazole in 6 features a N2 atom pointing to the linker S2/C4/S3, thus it is closer to the linker plane. The second arrangement of the 1,3,4-thiadiazole in 6 the S4 is closer to the S2/C4/S3 plane (Figure S1b), e.g., it is pointing toward the linker. In compounds 7 and 9, the asymmetric unit features half of the molecule, the second half is generated through symmetry operation. The result is that the rotation of the 1,3,4-thiadiazole moiety along the S–C bond is not permitted for the specific crystal structure of 7 and for the specific crystal structure of 9. This man-made, introduced restriction has a limited individual crystallographic validity, as the comparison of the molecules of 7 and 9 demonstrates that 1,3,4-thiadiazole moieties have adopted different orientations. In compound 7, the N2 atom is closer to the 2(CH2) spacer, while in 9, the S atom is closer to the 4(CH2) spacer linking the two thiadiazole moieties (Figure 4e and Figure S1c,d).
It is noteworthy that the melting points of bis-thiadiazoles deviate from the anticipated trend, which suggests a decrease with the elongation of the (CH2)n bridge. Contrary to expectations, the melting points demonstrate an ascending sequence as follows: propylene (compound 8; oily solid) has the lowest melting point, followed by methylene (compound 6; 88.6–89.0 °C) and butylene (compound 9; 93.4–93.9 °C), and ethylene (compound 7; 132.9–133.6 °C) has the highest. This observation indicates that the melting points of these compounds are significantly influenced by their spatial orientation, allowing for their categorisation into two distinct groups based on the number of methylene units in the linker: compounds with an odd number (6 and 8) and those with an even number (7 and 9) of methylene groups. This differentiation suggests a clear relationship between spatial configuration and melting behaviour.
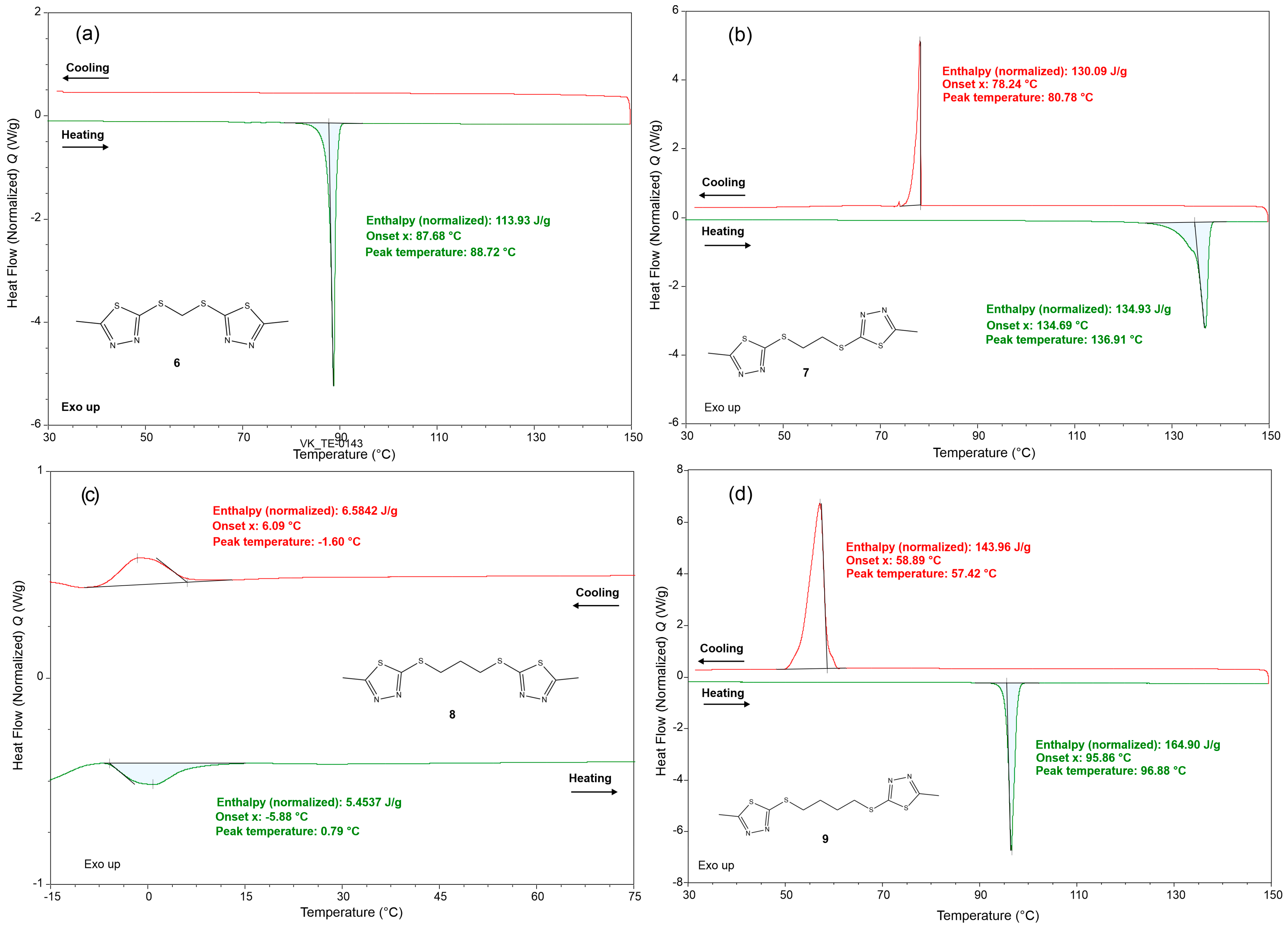
The DSC thermograms of compounds 6, 7, 8, and 9 are presented in Figure 5.
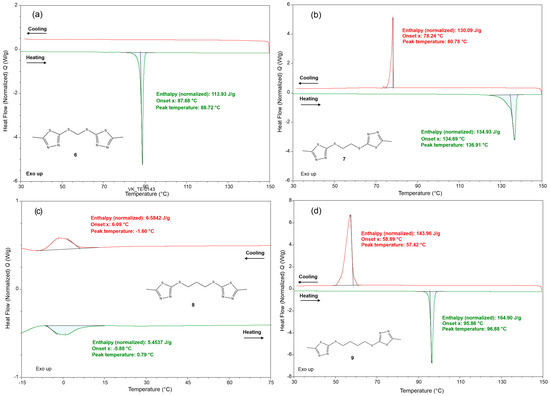
Figure 5.
DSC thermograms of (a) compound 6, (b) compound 7, (c) compound 8, and (d) compound 9; the heating and cooling curves are given in green and red, respectively.
The heating and cooling curves are given in green and red, respectively. The thermograms of compounds 6, 7, and 9 (Figure 5a,b,d) reveal that their crystal structures are conserved up to the melting points expressed with sharp and intensive endothermic peaks with maxima at 89 °C (H = 113.93 J/g), 137 °C (H = 134.93 J/g), and 97 °C (H = 164.90 J/g), respectively. No other thermal effects, such as phase transitions and decomposition, are observed for 6, 7, and 9 upon heating to 150 °C. The cooling of 6, 7, and 9 (from 150 to 30 °C) discloses exothermic effects only for 7 and 9 that are related to recrystallisation of the melt. This is confirmed by the similar values for the crystallisation and melting peak areas but also by the fact that the degree of supercooling (the difference between the onset temperatures of melting and crystallisation) does not exceed 50 K. For 6, the recrystallisation is not registered within the used temperature range. At ambient conditions, compound 8 appears as a viscous oil. It was observed that it solidifies when stored in a refrigerator (6–8 °C). Thus, the DSC heating and cooling range for compound 8 was adjusted to the range –15 to 75 °C. The thermogram for 8 reveals that the liquid-to-solid transition is represented by a broad and shallow exothermic effect with a maximum at −1.6 °C starting from 6 °C. The heating from −15 to 75 °C of 8 produces only one endothermic effect, onset at −5.88 °C and maximum at 0.79 °C.
3. Materials and Methods
3.1. General
All reagents were purchased from Aldrich (St. Louis, MO, USA), Merck (Rahway, NJ, USA), and Fluka (Buchs, Switzerland) and were used without any further purification. The deuterated chloroform was purchased from Deutero GmbH (Kastellaun, Germany). Fluka silica gel (TLC-cards 60778 with fluorescent indicator 254 nm) was used for TLC chromatography and Rf-value determination. Merck Silica gel 60 (0.040–0.063 mm) was used for flash chromatography purification of the products. The melting points were determined in capillary tubes on an SRS MPA100 OptiMelt (Sunnyvale, CA, USA) automated melting point system with heating rate of 1 °C per min. The NMR spectra were recorded on Bruker Avance II+ 600 or Bruker Avance NEO 400 spectrometers (Rheinstetten, Germany) in CDCl3; the chemical shifts are quoted in ppm in δ-values against tetramethylsilane (TMS) as an internal standard and the coupling constants were calculated in Hz. The assignment of the signals is confirmed by applying two-dimensional HSQC and HMBC techniques. The spectra were processed with the Topspin 3.6.3 program. The IR spectra were measured on a Shimadzu IR Spirit FT-IR spectrometer (Shimadzu Corporation, Columbia, SC, USA) using QATR-S as a single-reflection ATR measurement attachment. The mass spectra were recorded using a Q Exactive Plus Hybrid Quadrupole-Orbitrap Mass Spectrometer, Thermo Scientific (HESI HRMS) in positive mode. The spectra were processed by the Thermo Scientific FreeStyle program version 1.8 SP1 (Thermo Fisher Scientific Inc., Waltham, MA, USA). DSC experiments were performed on Discovery DSC 250 (TA Instruments, New Castle, DE, USA). Samples between 2 and 5 mg were heated in closed aluminum pans from 30 to 150 °C and then cooled back to 30 °C for compounds 6, 7, and 9, while for 8 the range was −15 to 75 °C. In all cases, the heating and cooling rate was 4 °C·min−1 under nitrogen flow of 30 mL·min−1.
3.2. Reaction with α,ω-Dibromoalkanes
To a solution of 2-mercapto-5-methyl-1,3,4-thiadiazole (4 mmol) and base (8 mmol) in variable solvent (20–40 mL), α,ω-dibromoalkane (2–8 mmol) was added and the mixture was stirred at rt or at reflux for different time (cf. Table 1). The solid residue (if formed) was filtered off and the solvent was removed in vacuo. The crude mixture was purified by flash chromatography on a silica gel with a gradient of polarity from DCM to 5% acetone in DCM.
Bromide 3a: Rf 0.46 (1% acetone in DCM); colourless solid; m. p. 34.1–34.4 °C; 1H NMR 2.738 (s, 3H, CH3), 3.724 (m, 2H, CH2-S), 3.743 (m, 2H, CH2-Br); 13C NMR 15.66 (CH3), 29.69 (CH2-Br), 35.18 (CH2-S), 163.69 (Cq-2), 165.50 (Cq-5); IR (ATR) 1433, 1413, 1381, 1200, 1178, 1078, 1057, 618, 601 cm−1; HR-MS (HESI+) m/z calcd. for C5H7BrN2S2+ [M + H]+ 238.9307, found 238.9308, ∆ = 0.1 mDa.
Bromide 4a: Rf 0.30 (1% acetone in DCM); colourless oil; 1H NMR 2.372 (m, 2H, C-CH2-C), 2.729 (s, 3H, CH3), 3.451 (t, 2H, J 6.8, CH2-S), 3.551 (t, 2H, J 6.3, CH2-Br); 13C NMR 15.65 (CH3), 31.66 (CH2-Br), 31.72 (C-CH2-C), 32.09 (CH2-S), 164.79 (Cq-2), 165.12 (Cq-5); IR (ATR) 1499, 1205, 1184, 1050, 986, 655, 505 cm−1; HR-MS (HESI+) m/z calcd. for C6H9BrN2S2+ [M + H]+ 252.9463, found 252.9460, ∆ = −0.3 mDa.
Bromide 5a: Rf 0.32 (1% acetone in DCM); colourless oil; 1H NMR 1.970 (m, 2H, CH2-C-S), 2.032 (m, 2H, CH2-C-Br), 2.726 (s, 3H, CH3), 3.337 (t, 2H, J 7.0, CH2-S), 3.438 (t, 2H, J 6.4, CH2-Br); 13C NMR 15.64 (CH3), 27.78 (CH2-C-S), 31.47 (CH2-C-Br), 32.76 (CH2-Br), 32.97 (CH2-S), 164.94 (Cq-5), 165.22 (Cq-2); IR (ATR) 1452, 1428, 1382, 1308, 1187, 1085, 732, 646, 606 cm−1; HR-MS (HESI+) m/z calcd. for C7H11BrN2S2+ [M + H]+ 266.9620, found 266.9618, ∆ = −0.2 mDa.
Bis-thiadiazole 6: Rf 0.51 (5% acetone in DCM); colourless solid; m. p. 88.6–89.0 °C (lit. [23] 78–80 °C); 1H NMR 2.758 (s, 6H, CH3), 5.197 (s, 2H, CH2); 13C NMR 15.76 (CH3), 36.88 (CH2), 163.60 (Cq-2), 165.91 (Cq-5); IR (ATR) 1379, 1187, 1074, 1035, 814, 717, 669, 612 cm−1.
Bis-thiadiazole 7: Rf 0.43 (5% acetone in DCM); colourless solid; m. p. 132.9–133.6 °C (lit. [23] 135–136 °C); 1H NMR 2.734 (s, 6H, CH3), 3.759 (s, 4H, CH2); 13C NMR 15.68 (CH3), 33.00 (CH2), 164.45 (Cq-2), 165.34 (Cq-5); IR (ATR) 1372, 1184, 1064, 1025, 729, 616 cm−1.
Bis-thiadiazole 8: Rf 0.14 (5% acetone in DCM); colourless oil; 1H NMR 2.328 (quint, 2H, J 7.0, C-CH2-C), 2.726 (s, 6H, CH3), 3.445 (t, 4H, J 7.0, 2 CH2-S); 13C NMR 15.63 (CH3), 28.63 (C-CH2-C), 32.39 (2 CH2-S), 164.93 (Cq-2), 165.10 (Cq-5); IR (ATR) 1379, 1185, 1065, 1038, 759, 615 cm−1; HR-MS (HESI+) m/z calcd. for C9H12N4S4+ [M + H]+ 305.0018, found 305.0016, ∆ = −0.2 mDa.
Bis-thiadiazole 9: Rf 0.14 (5% acetone in DCM); colourless solid; m. p. 93.4–93.9 °C; 1H NMR 1.968 (m, 4H, 2 C-CH2-C), 2.723 (s, 6H, CH3), 3.333 (m, 4H, 2 CH2-S); 13C NMR 15.64 (CH3), 28.20 (C-CH2-C), 33.29 (2 CH2-S), 164.92 (Cq-5), 165.31 (Cq-2); IR (ATR) 1389, 1194, 1068, 1048, 652, 608 cm−1; HR-MS (HESI+) m/z calcd. for C10H14N4S4+ [M + H]+ 319.0174, found 319.0175, ∆ = 0.1 mDa.
Olefin 10: Rf 0.63 (5% acetone in DCM); colourless oil; 1H NMR 2.754 (s, 3H, CH3), 5.633 (d, 1H, 3J 9.4, CH2cis=), 5.681 (d, 1H, 3J 16.7, CH2trans=), 6.882 (dd, 1H, 3Jcis 9.4, 3Jtrans 16.8, CH=); 13C NMR 15.72 (CH3), 120.30 (CH2=), 126.90 (CH=), 163.72 (Cq-2), 165.81 (Cq-5); IR (ATR) 1590, 1385, 1188, 1065, 1045, 953, 625, 603 cm−1; HR-MS (HESI+) m/z calcd. for C5H6N2S2+ [M + H]+ 159.0045, found 159.0045, ∆ = 0 mDa.
3.3. Reaction with α,ω-Dichloroalkanes
To a solution of 2-mercapto-5-methyl-1,3,4-thiadiazole (4 mmol) in α,ω-dichloroalkane (30 mL; 10 mL for 4b and 5b), a base (8 mmol) was added and the mixture was stirred at rt for 24 h (cf. Table 1). The solid residue (if formed) was filtered off and the solvent was removed in vacuo. The crude mixture was purified by flash chromatography on a silica gel with a gradient of polarity from DCM to 5% acetone in DCM.
Chloride 2b: Rf 0.46 (1% acetone in DCM); colourless oil; 1H NMR 2.793 (s, 3H, CH3), 5.252 (s, 2H, CH2); 13C NMR 15.82 (CH3), 47.29 (CH2), 161.33 (Cq-2), 166.91 (Cq-5); IR (ATR) 1449, 1378, 1267, 1198, 1050, 739, 623 cm−1; HR-MS (HESI+) m/z calcd. for C4H5ClN2S2+ [M + H]+ 180.9655, found 180.9655, ∆ = 0 mDa.
Chloride 3b: cf. ref. [84].
Chloride 4b: Rf 0.69 (5% acetone in DCM); colourless oil; 1H NMR 2.293 (m, 2H, C-CH2-C), 2.727 (s, 3H, CH3), 3.453 (t, 2H, J 6.9, CH2-S), 3.692 (t, 2H, J 6.3, CH2-Cl); 13C NMR 15.63 (CH3), 30.89 (CH2-S), 31.69 (C-CH2-C), 43.12 (CH2-Cl), 164.84 (Cq-2), 165.10 (Cq-5); IR (ATR) 1441, 1268, 736 cm−1; HR-MS (HESI+) m/z calcd. for C6H9ClN2S2+ [M + H]+ 208.9968, found 208.1547, ∆ = 0 mDa.
Chloride 5b: Rf 0.66 (5% acetone in DCM); colourless oil; 1H NMR 1.945 (m, 4H, 2 C-CH2-C), 2.724 (s, 3H, CH3), 3.337 (t, 2H, J 6.9, CH2-S), 3.573 (t, 2H, J 6.2, CH2-Cl); 13C NMR 15.62 (CH3), 26.54 (CH2-C-S), 31.33 (CH2-C-Cl), 33.13 (CH2-S), 44.24 (CH2-Cl), 164.92 (Cq-5), 165.24 (Cq-2); IR (ATR) 1432, 1382, 1187, 1070, 1035, 649 cm−1; HR-MS (HESI+) m/z calcd. for C7H11ClN2S2+ [M + H]+ 223.0125, found 223.0125, ∆ = 0 mDa.
3.4. Crystallography
Single crystals of compounds 2b, 6, 7, and 9 with suitable size and diffracting quality were mounted on glass capillaries or nylon Cryoloop (Hampton research, Aliso Viejo, CA, USA). Diffraction data for 2b were collected on a SupernovaDual diffractometer equipped with an Atlas CCD detector, while diffraction data for 6 were collected on a Bruker D8 Venture diffractometer equipped with a PHOTON II CPAD detector. Both diffractometers operate with a micro-focus sealed X-ray source, generating MoKα radiation (0.71073 Å). The collected data were processed with CrysAlisPro (41.117a-64bit) [88] (for 2b) or APEX4 (2022.1-1) [89] (for 6) programs. The structures were solved with intrinsic phasing methods and refined using the full-matrix least-squares method of F2 using ShelxT (2018.1) [90] and ShelxL [91] as implemented in the OLEX2-ver.1.5 graphical interface [92]. All non-hydrogen atoms were located successfully from a Fourier map and were refined anisotropically. Hydrogen atoms were placed on calculated positions riding on the parent carbon atoms (Ueq = 1.2 for C-Hmethyl = 0.93 Å and C-Hmethylenic = 0.97 Å) while those riding on heteroatoms (N1 in 2b) were placed from a difference Fourier map and refined freely. Ortep-3v2 software [93] was used to prepare the figures visualizing the molecules present in the in the asymmetric unit. Crystallography Open Database (COD) entries 3000465, 3000466, 3000493, and 3000494 contain the supplementary crystallographic data for this paper. These data, accessed on 30 January 2024, can be obtained free of charge via http://www.crystallography.net (accessed on 18 April 2024).
4. Conclusions
A series of 2-(ω-bromoalkylthio)-5-methyl-1,3,4-thiadiazoles, 2-(ω-chloroalkylthio)-5-methyl-1,3,4-thiadiazoles and symmetrical bis-thiadiazoles possessing variable chain lengths are obtained via optimised protocols and are characterised by 1D and 2D NMR spectra and by single crystal XRD of selected samples.
It is observed that the reaction output is strongly dependent both on the alkyl chain length and on the type of the halogen atoms. The symmetrical methylene-bridged bis-thiadiazole is isolated as a sole or major product from the reactions with dibromomethane and dichloromethane, respectively. Bromides and bis-thiadiazoles are formed as mixtures when using dibromoethane, dibromopropane, and dibromobutane; the ratios being dependent on the reagents’ proportions. Bromides are the predominant products when using a 2 molar excess of dibromides, while the opposite relation is achieved with a reversed ratio. Chlorides are isolated in quantitative yields from reactions with dichloroethane, dichloropropane, and dichlorobutane, used both as reagents and solvents.
Despite the fact that the chlorides 3b–5b can be obtained quantitatively, the bromides 3a–5a are also of interest because they are more reactive as alkylating agents. The obtained reagents offer unlimited opportunities for the preparation of targets by alkylation of diverse molecules. On the other hand, symmetrical bis-thiadiazoles, which are isolated in up to 80% yield, can find broad applications as polynuclear ligands in coordination chemistry, including in MOFs preparation.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/molecules29091938/s1, NMR data (Table S1), Crystallographic data collection and refinement parameters (Table S2), Crystallography (Figures S1 and S2), and original NMR, IR and HR-MS spectra (Section S1).
Author Contributions
The synthetic experiments and NMR analysis were accomplished by V.B.K. The single crystal XRD analyses were performed by R.I.R. and B.L.S. The HESI HRMS spectra were conducted by Z.S.P. All authors contributed in the discussion of the results and in the manuscript writing. All authors have read and agreed to the published version of the manuscript.
Funding
Financial support from The EU, COST Action CA22147 European metal-organic framework network: combining research and development to promote technological solutions (EU4MOFs), and by the Bulgarian Ministry of Education and Science, the Operational Program “Science and Education for Smart Growth” 2014–2020, co-financed by European Union through the European Structural and Investment Funds, under the Projects Centre of Excellence “National centre of mechatronics and clean technologies”, project BG05M2OP001-1.001-0008 (for Bruker D8venture XRD equipment), and Centre of Competence “Sustainable utilization of bio-resources and waste of medicinal and aromatic plants for innovative bioactive products”, project BG05M2OP001-1.002-0012 (for Q Exactive Plus Hybrid Quadrupole-Orbitrap MS equipment), is gratefully acknowledged.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data of the current study are available from the corresponding authors on reasonable request.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Jain, A.K.; Sharma, S.; Vaidya, A.; Ravichandran, V.; Agrawal, R.K. 1,3,4-Thiadiazole and its Derivatives: A Review on Recent Progress in Biological Activities. Chem. Biol. Drug Des. 2013, 81, 557–576. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Li, C.-Y.; Wang, X.-M.; Yang, Y.-H.; Zhu, H.-L. 1,3,4-Thiadiazole: Synthesis, reactions, and applications in medicinal, agricultural, and materials chemistry. Chem. Rev. 2014, 114, 5572–5610. [Google Scholar] [CrossRef] [PubMed]
- Khalilullah, H.; Khan, M.U.; Mahmood, D.; Akhtar, J.; Osman, G. 1,3,4-Thiadiazole: A biologically active scaffold. Int. J. Pharm. Pharm. Sci. 2014, 6, 8–15. [Google Scholar]
- Matysiak, J. Biological and pharmacological activities of 1,3,4-thiadiazole based compounds. Mini Rev. Med. Chem. 2015, 15, 762–775. [Google Scholar] [CrossRef] [PubMed]
- Joseph, L.; George, M.; Mathews, P. A review on various biological activities of 1,3,4- thiadiazole derivatives. J. Pharm. Chem. Biol. Sci. 2015, 3, 329–345. [Google Scholar]
- Han, X.; Yu, Y.L.; Hu, Y.S.; Liu, X.H. 1,3,4-thiadiazole: A privileged scaffold for drug design and development. Curr. Top. Med. Chem. 2021, 21, 2546–2573. [Google Scholar] [CrossRef] [PubMed]
- Irfan, A.; Ullah, S.; Anum, A.; Jabeen, N.; Zahoor, A.F.; Kanwal, H.; Kotwica-Mojzych, K.; Mojzych, M. Synthetic transformations and medicinal significance of 1,2,3-thiadiazoles derivatives: An update. Appl. Sci. 2021, 11, 5742. [Google Scholar] [CrossRef]
- Schatz, J.; Gogić, K.; Benkert, T. 1,3,4-Thiadiazoles. In Comprehensive Heterocyclic Chemistry IV, 4th ed.; Elsevier Science: Amsterdam, The Netherlands, 2022; Volume 5, Chapter 5.10; pp. 407–447. [Google Scholar]
- Anthwal, T.; Paliwal, S.; Nain, S. Diverse Biological Activities of 1,3,4-Thiadiazole Scaffold. Chemistry 2022, 4, 1654–1671. [Google Scholar] [CrossRef]
- Aliabadi, A. 1,3,4-Thiadiazole based anticancer agents. Anti-Cancer Agents Med. Chem. 2016, 16, 1301–1314. [Google Scholar] [CrossRef]
- Raj, V.; Rai, A.; Saha, S. Human Cancer cell line based approach of 1,3,4-thiadiazole and its fused ring: A comprehensive review. Anti-Cancer Agents Med. Chem. 2017, 17, 500–523. [Google Scholar] [CrossRef]
- Raj, V.; Rai, A.; Singh, A.K.; Keshari, A.K.; Trivedi, P.; Ghosh, B.; Kumar, U.; Kumar, D.; Saha, S. Discovery of Novel 2-Amino-5-(Substituted)-1,3,4-Thiadiazole Derivatives: New Utilities for Colon Cancer Treatment. Anti-Cancer Agents Med. Chem. 2018, 18, 719–738. [Google Scholar] [CrossRef] [PubMed]
- Janowska, S.; Paneth, A.; Wujec, M. Cytotoxic properties of 1,3,4-Thiadiazole derivatives—A review. Molecules 2020, 25, 4309. [Google Scholar] [CrossRef] [PubMed]
- Obakachi, V.A.; Kushwaha, B.; Kushwaha, N.D.; Mokoena, S.; Ganai, A.M.; Pathan, T.K.; van Zyl, W.E.; Karpoormath, R. Synthetic and anti-cancer activity aspects of 1,3,4-thiadiazole containing bioactive molecules: A concise review. J. Sulfur Chem. 2021, 42, 670–691. [Google Scholar] [CrossRef]
- Janowska, S.; Khylyuk, D.; Bielawska, A.; Szymanowska, A.; Gornowicz, A.; Bielawski, K.; Noworól, J.; Mandziuk, S.; Wujec, M. New 1,3,4-Thiadiazole Derivatives with Anticancer Activity. Molecules 2022, 27, 1814. [Google Scholar] [CrossRef] [PubMed]
- Serban, G.; Stanasel, O.; Serban, E.; Bota, S. 2-Amino-1,3,4-thiadiazole as a potential scaffold for promising antimicrobial agents. Drug Des. Devel. Ther. 2018, 12, 1545–1566. [Google Scholar] [CrossRef]
- Barbosa, G.A.D.; de Aguiar, A.P. Synthesis of 1,3,4-thiadiazole derivatives and microbiological activities: A review. Rev. Virtual Quim. 2019, 11, 806–848. [Google Scholar] [CrossRef]
- Kumar, D.; Kumar, H.; Kumar, V.; Deep, A.; Sharma, A.; Marwaha, M.G.; Marwaha, R.K. Mechanism-based approaches of 1,3,4 thiadiazole scaffolds as potent enzyme inhibitors for cytotoxicity and antiviral activity. Med. Drug Discov. 2023, 17, 100150. [Google Scholar] [CrossRef]
- Sahoo, B.M.; Dinda, S.C.; Ravi Kumar, B.V.V.; Panda, J.R.; Brahmkshatriya, P.S. Molecular Docking Study, Green Synthesis and Pharmacological Evaluation of 1,3,4-thiadiazole Derivatives as Potential Antiepileptic Agents. Mini-Rev. Med. Chem. 2013, 13, 2076–2081. [Google Scholar] [CrossRef]
- Raj, V.; Rai, A.; Singh, M.; Kumar, R.; Kumar, A.; Kumar, V.; Sharma, S.K. Recent Update on 1,3,4-Thiadiazole Derivatives: As Anticonvulsant Agents. Am. Res. J. Pharm. 2015, 1, 34–61. [Google Scholar]
- Anthwal, T.; Nain, S. 1,3,4-Thiadiazole Scaffold: As Anti-Epileptic Agents. Front. Chem. 2022, 9, 671212. [Google Scholar] [CrossRef]
- Barboiu, M.; Cimpoesu, M.; Guran, C.; Supuran, C. Synthesis and Biological Activity of Metal Complexes of 5-(2-Aminoethyl)-2-Amino-1,3,4-Thiadiazole. Met.-Based Drugs 1996, 3, 227–232. [Google Scholar] [CrossRef]
- Zheng, Y.; Du, M.; Li, J.; Zhang, R.; Bu, X. Tuning the framework formation of silver(i) coordination architectures with heterocyclic thioethers. Dalton Trans. 2003, 8, 1509–1514. [Google Scholar] [CrossRef]
- Zhou, H.; Zheng, J.; Wang, H.; Wang, J.; Song, X.; Cao, Y.; Xiong, C. Preparation of a novel chloromethylated polystyrene-2-mercapto-1,3,4-thiadiazole chelating resin and its adsorption properties and mechanism for separation and recovery of Hg(II) from aqueous solutions. Water Sci. Technol. 2017, 76, 1915–1924. [Google Scholar] [CrossRef]
- Kudelko, A.; Olesiejuk, M.; Luczynski, M.; Swiatkowski, M.; Sieranski, T.; Kruszynski, R. 1,3,4-Thiadiazole-Containing Azo Dyes: Synthesis, Spectroscopic Properties and Molecular Structure. Molecules 2020, 25, 2822. [Google Scholar] [CrossRef]
- Hipler, F.; Fischer, R.A.; Müller, J. Matrix-isolation pyrolysis investigation of mercapto-functionalized 1,3,4-thiadiazoles: Thermal stability of thiadiazole lubricant additives. Phys. Chem. Chem. Phys. 2005, 7, 731–737. [Google Scholar] [CrossRef]
- Kornis, G.I. Comprehensive Heterocyclic Chemistry II; Katritzky, A.R., Scriven, E.F.V., Rees, C.W., Eds.; Elsevier Science Ltd.: Amsterdam, The Netherlands, 1996; Volume 4, Chapter 4.10; pp. 379–408. [Google Scholar]
- Koutentis, P.A.; Constantinides, C.P. Comprehensive Heterocyclic Chemistry III; Katritzky, A.R., Scriven, E.F.V., Rees, C.W., Eds.; Elsevier Science Ltd.: Amsterdam, The Netherlands, 2008; Volume 5, Chapter 5.10; pp. 567–605. [Google Scholar]
- Manimaran, T.; Anand Raj, M.; Jishala, M.I.; Gopalasatheeskumar, K. Review on substituted 1, 3, 4 thiadiazole compounds. Int. J. Pharm. Anal. Res. 2017, 6, 222–231. [Google Scholar]
- Ram, V.J.; Sethi, A.; Nath, M.; Pratap, R. Five-Membered Heterocycles, The Chemistry of Heterocycles, Nomenclature and Chemistry of Three-to-Five Membered Heterocycles; Ram, V.J., Sethi, A., Nath, M., Pratap, R., Eds.; Elsevier Ltd.: Amsterdam, The Netherlands, 2019; Chapter 5; pp. 149–478. [Google Scholar]
- Shamroukh, A.H.; Hegab, M.I. A Review on synthesis, therapeutic, and computational studies of substituted 1, 3, 4 thiadiazole derivatives. Egypt. J. Chem. 2020, 63, 4387–4408. [Google Scholar]
- Kumar, D.; Aggarwal, N.; Kumar, V.; Chopra, H.; Marwaha, R.K.; Sharma, R. Emerging synthetic strategies and pharmacological insights of 1,3,4-thiadiazole derivatives: A comprehensive review. Future Med. Chem. 2024, 16, 563–581. [Google Scholar] [CrossRef]
- Kadu, N.S.; Masand, V.H. Synthesis of 1,3,4-thiadiazole derivative using appropriate reaction conditions. Int. J. Res. Appl. Sci. Engin. Technol. 2022, 10, 312–321. [Google Scholar] [CrossRef]
- Chen, Z.; Xu, W.; Liu, K.; Yang, S.; Fan, H.; Bhadury, P.S.; Huang, D.-Y.; Zhang, Y. Synthesis and antiviral activity of 5-(4-chlorophenyl)-1,3,4-thiadiazole sulfonamides. Molecules 2010, 15, 9046–9056. [Google Scholar] [CrossRef]
- Kumar, B.S.; Reddy, P.R.; Ravindranath, L.R.K.R.; Prasad, A.R.G.; Mallika, A. Synthesis, characterization and in vitro santimicrobial evaluation of sulphonyl urea derivatives as potential inhibitors of beta-ketoacyl-acyl carrier protein synthase III (FabH). Acta Univ. 2015, 25, 12–21. [Google Scholar]
- Alam, J.; Alam, O.; Ali, R.; Mohd; Naim, J.; Khan, S.A. Synthesis and anti-inflammatory activity of some new thiadiazole linked pyrazole benzene sulphonamides as cyclooxygenase inhibitors. Orient. J. Chem. 2015, 31, 1873–1885. [Google Scholar] [CrossRef]
- Cristina, A.; Leonte, D.; Vlase, L.; Csaba Bencze, L.; Imre, S.; Marc, G.; Apan, B.; Mogoșan, C.; Zaharia, V. Heterocycles 48. Synthesis, characterization and biological evaluation of imidazo[2,1-b][1,3,4]thiadiazole derivatives as anti-inflammatory agents. Molecules 2018, 23, 2425. [Google Scholar] [CrossRef]
- Shafique, M.; Hameed, S.; Naseer, M.M.; Al-Masoudi, N.A. Synthesis of new chiral 1,3,4-thiadiazole-based di- and tri-arylsulfonamide residues and evaluation of in vitro anti-HIV activity and cytotoxicity. Mol. Divers. 2018, 22, 957–968. [Google Scholar] [CrossRef]
- Almandil, N.B.; Taha, M.; Gollapalli, M.; Rahim, F.; Ibrahim, M.; Mosaddik, A.; Anouar, E.H. Indole bearing thiadiazole analogs: Synthesis, β-glucuronidase inhibition and molecular docking study. BMC Chem. 2019, 13, 14. [Google Scholar] [CrossRef]
- Gomha, S.M.; Edrees, M.M.; Muhammad, Z.A.; El-Reedy, A.A.M. 5-(Thiophen-2-yl)-1,3,4-thiadiazole derivatives: Synthesis, molecular docking and in vitro cytotoxicity evaluation as potential anticancer agents. Drug Des. Devel. Ther. 2018, 12, 1511–1523. [Google Scholar] [CrossRef]
- Qurban, J.; Gouda, M.A. Synthesis and cytotoxic activity of some new sulfa drugs containing thiadiazole. Indian J. Heterocycl. Chem. 2021, 31, 559–564. [Google Scholar]
- Vardanyan, R.S.; Hruby, V.J. Antiepileptic Drugs. In Synthesis of Essential Drugs; Elsevier B.V.: Amsterdam, The Netherlands, 2006; Chapter 9; pp. 125–133. [Google Scholar]
- Vardanyan, R.S.; Hruby, V.J. Diuretics. In Synthesis of Essential Drugs; Elsevier B.V.: Amsterdam, The Netherlands, 2006; Chapter 21; pp. 277–293. [Google Scholar]
- Vardanyan, R.S.; Hruby, V.J. Antimicrobial Drugs. In Synthesis of Essential Drugs; Elsevier B.V.: Amsterdam, The Netherlands, 2006; Chapter 33; pp. 499–523. [Google Scholar]
- Chhajed, M.; Shrivastava, A.K.; Taile, V. Synthesis of 5-arylidine amino-1,3,4-thiadiazol-2-[(N-substituted benzyol)]sulphonamides endowed with potent antioxidants and anticancer activity induces growth inhibition in HEK293, BT474 and NCI-H226 cells. Med. Chem. Res. 2014, 23, 3049–3064. [Google Scholar] [CrossRef]
- El-Gazzar, M.G.; Zaher, N.H.; El-Tablawy, S.Y. Morphological changes of some pathogenic microbial strains induced by novel thiadiazole derivatives. Med. Chem. Res. 2014, 23, 1844–1854. [Google Scholar] [CrossRef]
- Çevik, U.A.; Celik, I.; İnce, U.; Maryam, Z.; Ahmad, I.; Patel, H.; Özkay, Y.; Kaplancıklı, Z.A. Synthesis, biological evaluation, and molecular modeling studies of new 1,3,4-thiadiazole derivatives as potent antimicrobial agents. Chem. Biodivers. 2023, 20, e202201146. [Google Scholar]
- Kariyone, K.; Harada, H.; Kurita, M.; Takano, T. Cefazolin, a new semisynthetic cephalosporin antibiotic. I. synthesis and chemical properties of cefazolin. J. Antibiot. 1970, 23, 131–136. [Google Scholar] [CrossRef]
- Hussain, S.A.; Mahmood, T.; Chawala, N. Improved and economical synthesis of cefazolin sodium. Pak. J. Pharm. 2012, 25, 23–26. [Google Scholar]
- Zhao, M. Method for Preparing Cefazedone. CN104230958, 24 December 2014. [Google Scholar]
- Minghua, L.; Zhangwei, G.; Yanxiao, F.; Qingli, L.; Yuqing, Z. Synthesis Technology of Cefazedone. CN110437256A, 12 November 2019. [Google Scholar]
- Ludescher, J.; Wieser, J. Cephalosporin Derivative. EP0463553A1, 2 January 1992. [Google Scholar]
- Dunn, G.L.; Hoover, J.R.E.; Berges, D.A.; Taggart, J.J.; Davis, L.D.; Dietz, E.M.; Jakas, D.R.; Yim, N.; Actor, P.; Uri, J.V.; et al. Orally active 7-phenylglycyl cephalosporins—Structure-activity studies related to cefatrizine (SK AND F 60771). J. Antibiot. 1976, 29, 65–80. [Google Scholar] [CrossRef]
- Ali, M.A.; Livingstone, S.E. Metal-complexes of sulphur-nitrogen chelating-agents. Coord. Chem. Rev. 1974, 13, 101–132. [Google Scholar]
- Mews, R.; Watson, P.G.; Lork, E. Three-coordinate sulphur(VI)-nitrogen species: An attempt to breathe some new life into an old topic. Coord. Chem. Rev. 1997, 158, 233–273. [Google Scholar] [CrossRef]
- Mensforth, E.J.; Hill, M.R.; Batten, S.R. Coordination polymers of sulphur-donor ligands. Inorg. Chim. Acta 2013, 403, 9–24. [Google Scholar] [CrossRef]
- Zhang, W.H.; Ren, Z.G.; Lang, J.P. Rational construction of functional molybdenum (tungsten)-copper-sulfur coordination oligomers and polymers from preformed cluster precursors. Chem. Soc. Rev. 2016, 45, 4995–5019. [Google Scholar] [CrossRef]
- Aarabi, F.; Naake, T.; Fernie, A.R.; Hoefgen, R. Coordinating sulfur pools under sulfate deprivation. Trends Plant Sci. 2020, 25, 1227–1239. [Google Scholar] [CrossRef]
- Haiduc, I. Inverse coordination metal complexes with oxalate and sulfur, selenium and nitrogen analogues as coordination centers. Topology and systematization. J. Coord. Chem. 2020, 73, 1619–1700. [Google Scholar] [CrossRef]
- Kamakura, Y.; Tanaka, D. Metal-organic frameworks and coordination polymers composed of sulfur-based nodes. Chem. Lett. 2021, 50, 523–533. [Google Scholar] [CrossRef]
- Seregin, I.V.; Kozhevnikova, A.D. Phytochelatins: Sulfur-containing metal(loid)-chelating ligands in plants. Int. J. Mol. Sci. 2023, 24, 2430. [Google Scholar] [CrossRef] [PubMed]
- Maiti, B.K.; Almeida, R.M.; Moura, I.; Moura, J.J.G. Rubredoxins derivatives: Simple sulphur-rich coordination metal sites and its relevance for biology and chemistry. Coord. Chem. Rev. 2017, 352, 379–397. [Google Scholar] [CrossRef]
- Colovic, M.B.; Vasic, V.M.; Djuric, D.M.; Krstic, D.Z. Sulphur-containing amino acids: Protective role against free radicals and heavy metals. Curr. Med. Chem. 2018, 25, 324–335. [Google Scholar] [CrossRef]
- Tanifuji, K.; Ohki, Y. Metal-Sulfur Compounds in N2Reduction and Nitrogenase-Related Chemistry. Chem. Rev. 2020, 120, 5194–5251. [Google Scholar] [CrossRef] [PubMed]
- Paradiso, V.; Capaccio, V.; Lamparelli, D.H.; Capacchione, C. Metal complexes bearing sulfur-containing ligands as catalysts in the reaction of CO2 with epoxides. Catalysts 2020, 10, 825. [Google Scholar] [CrossRef]
- Hu, X.; Huang, T.; Zhang, G.; Lin, S.; Chen, R.; Chung, L.-H.; He, J. Metal-organic framework-based catalysts for lithium-sulfur batteries. Coord. Chem. Rev. 2023, 475, 214879. [Google Scholar] [CrossRef]
- Song, Y.; Zou, L.; Wei, C.; Zhou, Y.; Hu, Y. Single-atom electrocatalysts for lithium–sulfur chemistry: Design principle, mechanism, and outlook. Carbon Energy 2023, 5, e286. [Google Scholar] [CrossRef]
- Tanifuji, K.; Ohta, S.; Ohki, Y.; Seino, H. Activation of unsaturated small molecules by bio-relevant multinuclear metal-sulfur clusters. Coord. Chem. Rev. 2023, 475, 214838. [Google Scholar] [CrossRef]
- Egly, J.; Bouché, M.; Chen, W.; Maisse-François, A.; Achard, T.; Bellemin-Laponnaz, S. Synthesis, structural characterization and anti-proliferative activity of (κ1-C)- and (κ2-C,S)-PtII complexes bearing thioether-functionalized N-heterocyclic carbenes. Eur. J. Inorg. Chem. 2018, 2018, 159–166. [Google Scholar] [CrossRef]
- Li, P.; Yang, Y.; Wang, X.; Wu, X. Recent achievements on the agricultural applications of thioether derivatives: A 2010–2020 decade in review. J. Heterocycl. Chem. 2021, 58, 1225–1251. [Google Scholar] [CrossRef]
- Bisht, S.; Kumar, L.; Kaul, G.; Akhir, A.; Saxena, D.; Chopra, S.; Karthik, R.; Goyal, N.; Batra, S. Synthesis and biological evaluation of substituted 3-isoxazolethioethers as antileishmanial and antibacterial agents. ChemistrySelect 2022, 7, e202201664. [Google Scholar] [CrossRef]
- Ozcan, I.; Akkoc, S.; Alici, H.; Capanlar, S.; Sahin, O.; Tahtaci, H. Novel thioether-bridged 2,6-disubstituted and 2,5,6-trisubstituted imidazothiadiazole analogues: Synthesis, antiproliferative activity, ADME, and molecular docking studies. Chem. Biodivers. 2023, 20, e202200884. [Google Scholar] [CrossRef] [PubMed]
- Rezania, H.; Vatanpour, V.; Salehi, E.; Gavari, N.; Shockravi, A.; Ehsani, M. Wholly heterocycles-based polyamide–sulfide containing pyridine and thiazole rings: A super-adsorbent polymer for lead removal. J. Polym. Environ. 2019, 27, 1790–1800. [Google Scholar] [CrossRef]
- Fliedel, C.; Braunstein, P. Thioether-functionalized N-heterocyclic carbenes: Mono- and bis-(S,CNHC) palladium complexes, catalytic C-C coupling, and characterization of a unique Ag4I4(S,CNHC)2 planar cluster. Organometallics 2010, 29, 5614–5626. [Google Scholar] [CrossRef]
- Liu, Y.; Kean, Z.S.; d’Aquino, A.I.; Manrajd, Y.D.; Mendez-Arroyo, J.; Mirkin, C.A. Palladium(II) weak-link approach complexes bearing hemilabile N-heterocyclic carbene-thioether ligands. Inorg. Chem. 2017, 56, 5902–5910. [Google Scholar] [CrossRef]
- Mechrouk, V.; Maisse-François, A.; Bellemin-Laponnaz, S.; Achard, T. β-Alkylation through dehydrogenative coupling of primary alcohols and secondary alcohols catalyzed by thioether-functionalized n-heterocyclic carbene ruthenium complexes. Eur. J. Inorg. Chem. 2023, 26, e202300188. [Google Scholar] [CrossRef]
- Shi, X.; Yi, A.; Liu, Q.; Zhang, Y.; Lin, S.; Lu, X. Nonplanar π-conjugated sulfur heterocyclic quinone polymer cathode for air-rechargeable zinc/organic battery with simultaneously boosted output voltage, rate capability, and cycling life. ACS Nano 2023, 17, 25005–25013. [Google Scholar] [CrossRef] [PubMed]
- Li, J.-J.; Wei, L.-L.; Han, Z.; Xu, L.; Li, L.-K. Syntheses, crystal structures, thermal and photoluminescent properties of four coordination complexes with a heterocyclic thioether carboxylate ligand. Z. Anorg. Allg. Chem. 2015, 641, 2490–2497. [Google Scholar] [CrossRef]
- Yoon, M.; Moon, D. New Zr (IV) based metal-organic framework comprising a sulfur-containing ligand: Enhancement of CO2 and H2 storage capacity. Microporous Mesoporous Mat. 2015, 215, 116–122. [Google Scholar] [CrossRef]
- Li, M.-T.; Kong, N.; Lan, Y.Q.; Su, Z.-M. Sulfur-containing bimetallic metal organic frameworks with multi-fold helix as anode of lithium ion batteries. Dalton Trans. 2018, 47, 4827–4832. [Google Scholar] [CrossRef]
- Li, X.; Ma, W.; Li, H.; Zhang, Q.; Liu, H. Sulfur-functionalized metal-organic frameworks: Synthesis and applications as advanced adsorbents. Coord. Chem. Rev. 2020, 408, 213191. [Google Scholar] [CrossRef]
- Dubskikh, V.A.; Kovalenko, K.A.; Nizovtsev, A.S.; Lysova, A.A.; Samsonenko, D.G.; Dybtsev, D.N.; Fedin, V.P. Enhanced adsorption selectivity of carbon dioxide and ethane on porous metal–organic framework functionalized by a sulfur-rich heterocycle. Nanomaterials 2022, 12, 4281. [Google Scholar] [CrossRef] [PubMed]
- Mills, J.E.; Maryanoff, C.A.; McComsey, D.F.; Stanzione, R.C.; Scott, L. The reaction of amines with methylene chloride. Evidence for rapid aminal formation from N-methylenepyrrolidinium chloride and pyrrolidine. J. Org. Chem. 1987, 52, 1857–1859. [Google Scholar] [CrossRef]
- Kurteva, V.; Rusew, R.; Shivachev, B. 4-Methyl-7-((2-((5-methyl-1,3,4-thiadiazol-2-yl)thio)ethyl)thio)-coumarin. Molbank 2022, 2022, M1491. [Google Scholar] [CrossRef]
- Singh, V.N.; Sharma, S. Stereoselective synthesis and characterization of monocyclic cis-β-lactams containing 5-methyl-1,3,4-thiadiazole-2-thiol moiety. J. Heterocycl. Chem. 2021, 58, 2163–2173. [Google Scholar] [CrossRef]
- Al-Mouqdady, O.D.; Al-Janabi, A.S.; Hatshan, M.R.; Al-Jibori, S.A.; Fiahan, A.S.; Wagner, C. Synthesis, characterization, anti-bacterial and anticancer activities of Palladium(II) mixed ligand complexes of 2-mercapto-5-methyl-1,3,4-thiadiazole (HmtzS) and phosphines. Crystal structure of [Pd(mtzS)2(dppf)].H2O.EtOH. J. Mol. Struct. 2022, 1264, 133219. [Google Scholar] [CrossRef]
- Zheng, Q.; Borsley, S.; Nichol, G.S.; Duarte, F.; Cockroft, S.L. The energetic significance of metallophilic interactions. Angew. Chem., Int. Ed. 2019, 131, 12747–12753. [Google Scholar] [CrossRef]
- CrysAlis PRO; Rigaku Oxford Diffraction Ltd.: Yarnton, UK, 2021.
- Bruker, S. APEX3 V2016. 9-0, SAINT V8. 37A; Bruker AXS Inc.: Madison, WI, USA, 2016; Volume 2013, p. 2014. [Google Scholar]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Section C 2015, 71, 3–8. [Google Scholar]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. A 2008, 64, 112–122. [Google Scholar] [CrossRef]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Farrugia, L.J. WinGX and ORTEP for Windows: An update. J. Appl. Crystallogr. 2012, 45, 849–854. [Google Scholar] [CrossRef]
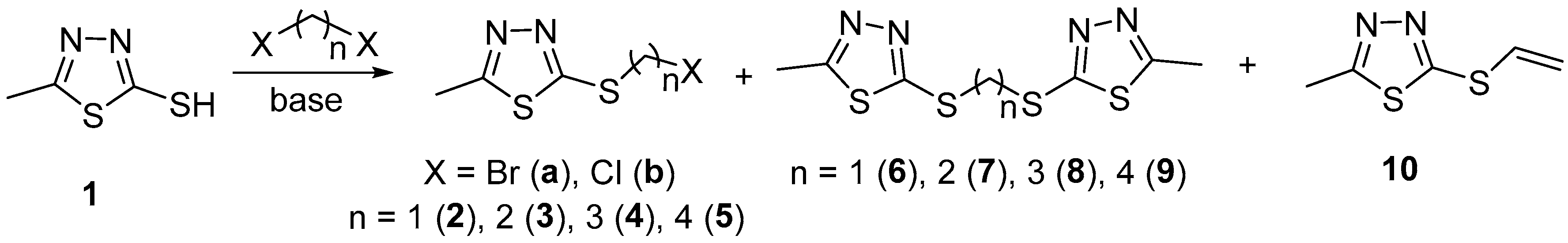
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).