
Conformation Analysis and Stereodynamics of Symmetrically ortho-Disubstituted Carvacrol Derivatives

 and
and
Abstract
:1. Introduction
2. Results and Discussion
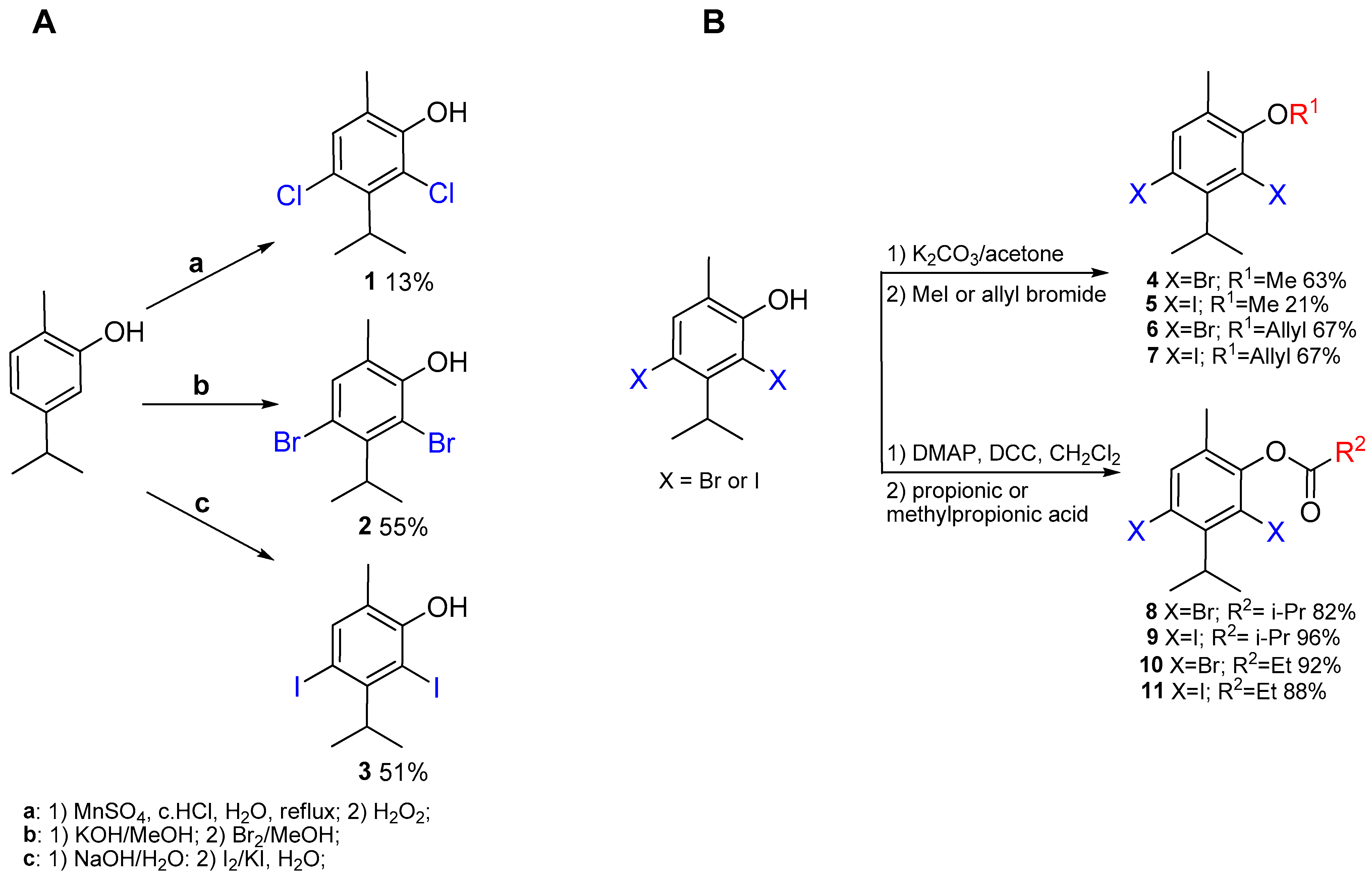
2.1. Synthesis of the Compounds
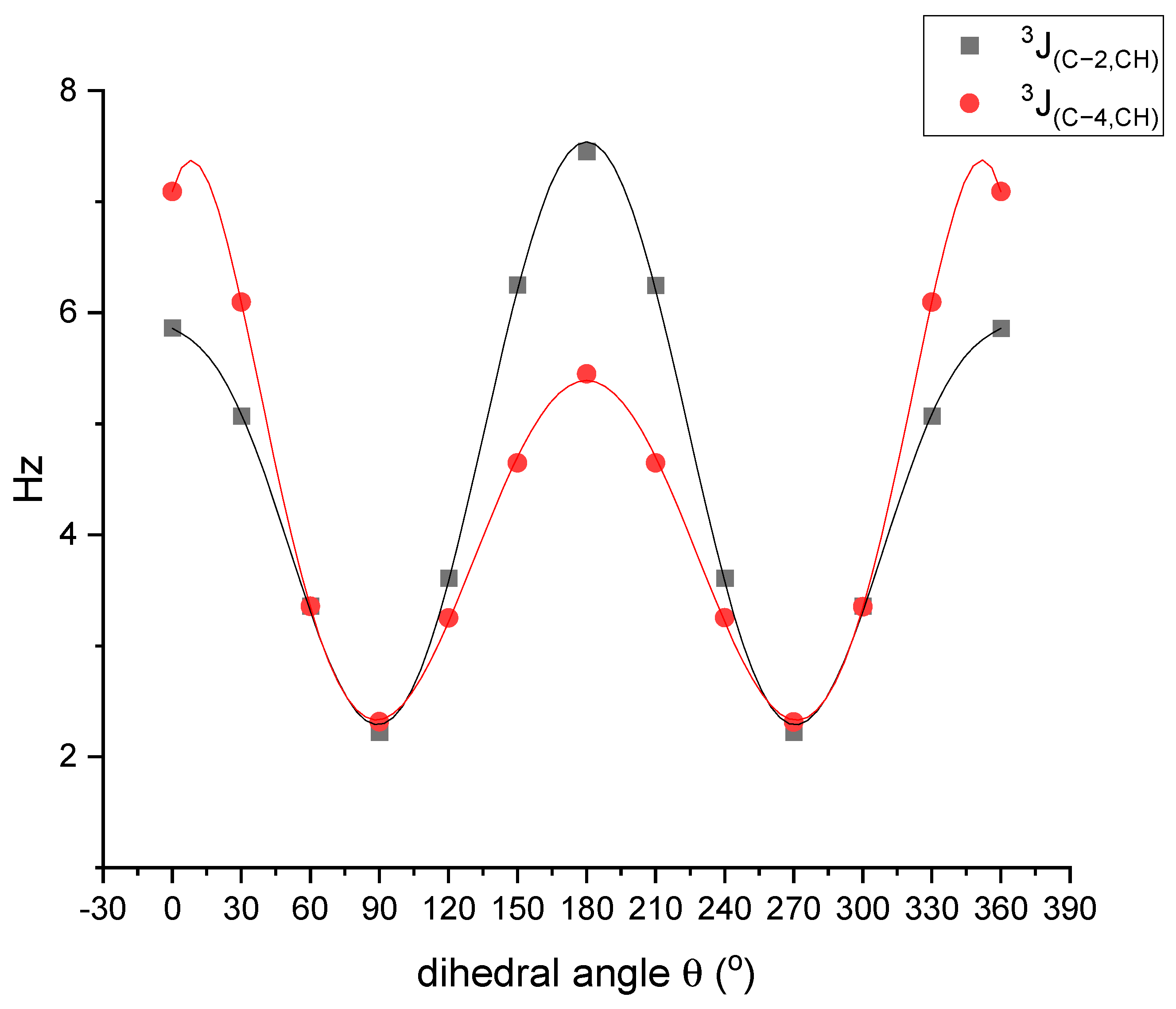
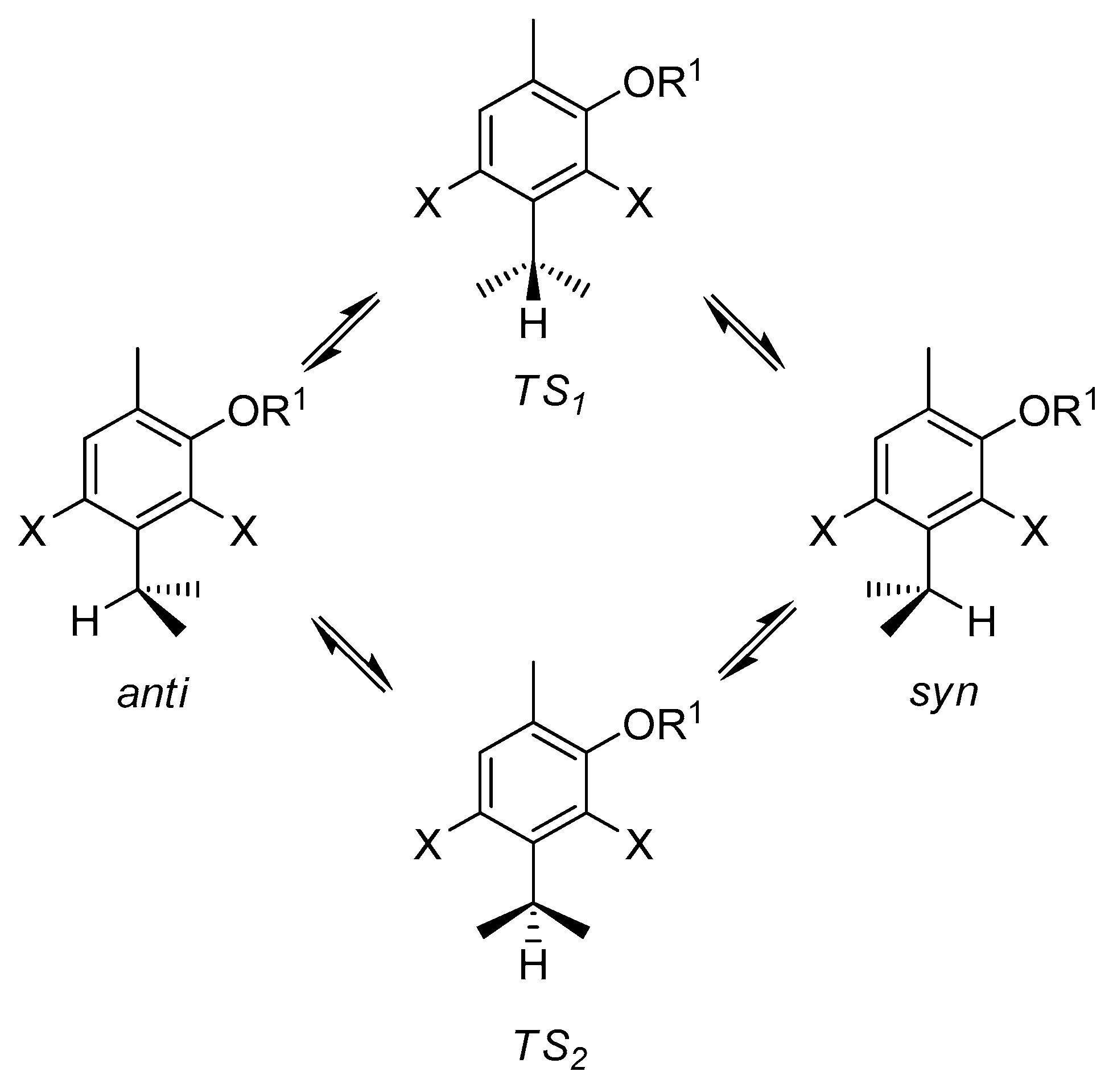
2.2. Conformational Analysis and Molecular Geometry
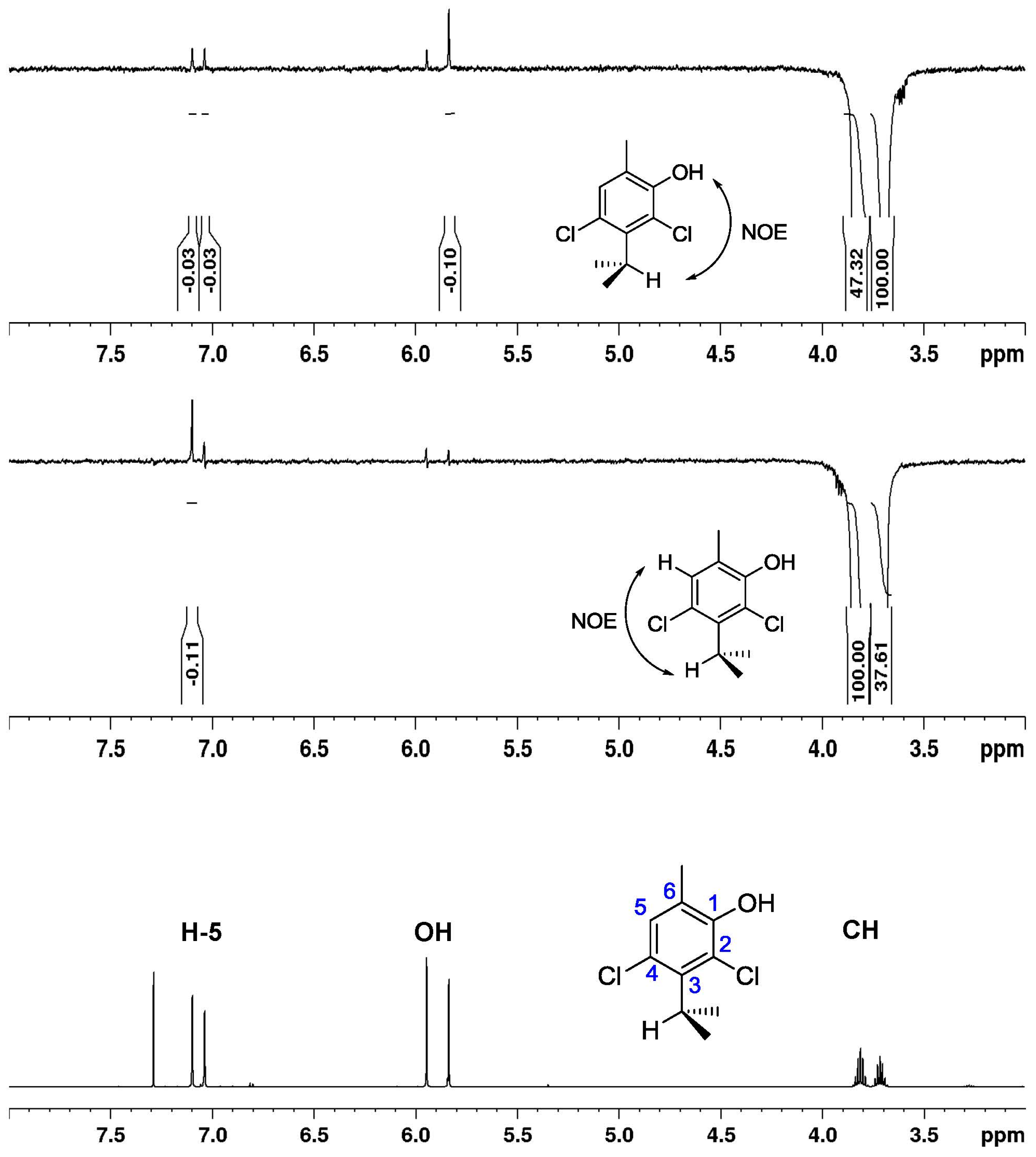
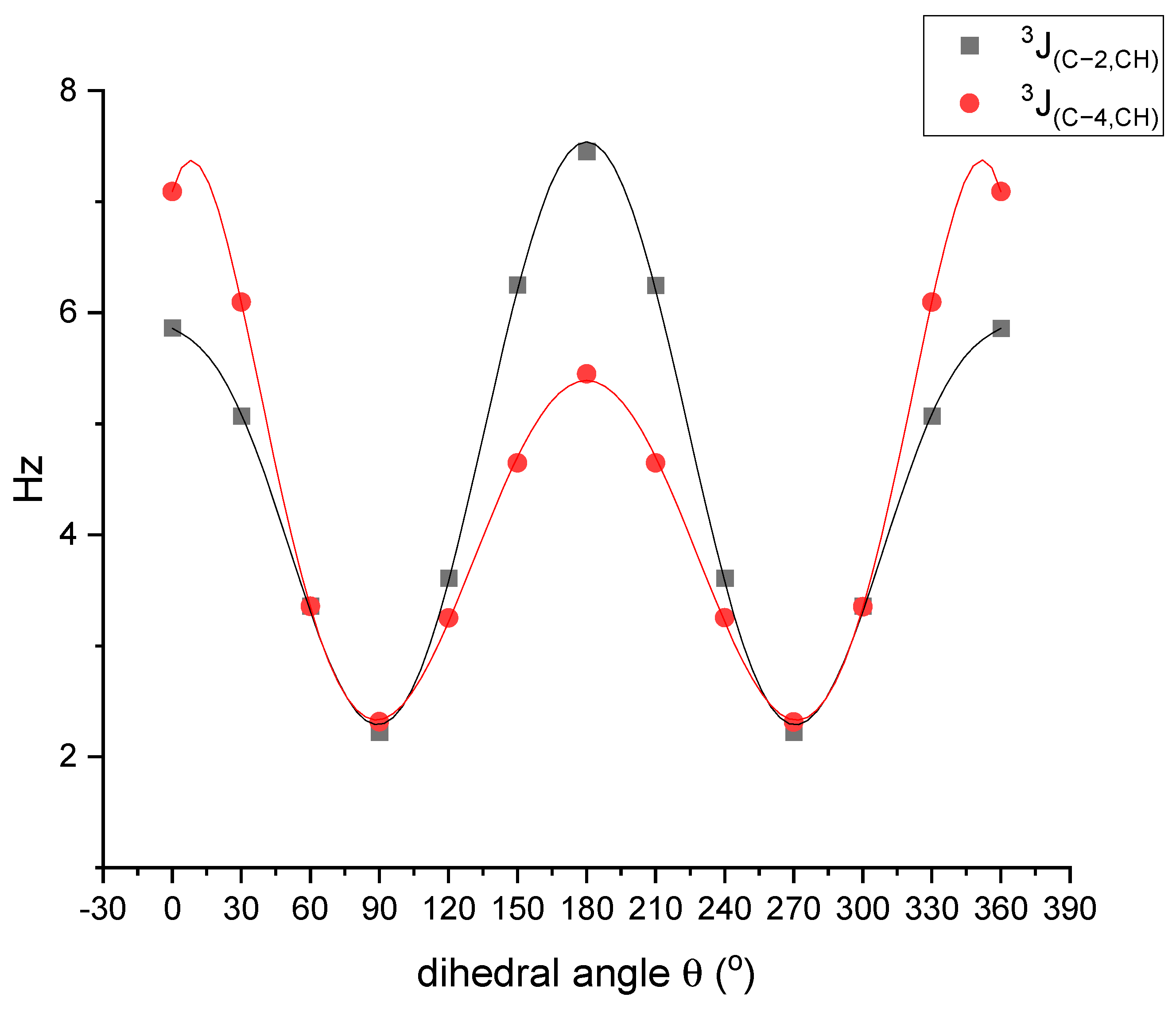
2.3. Dynamic NMR Spectroscopy
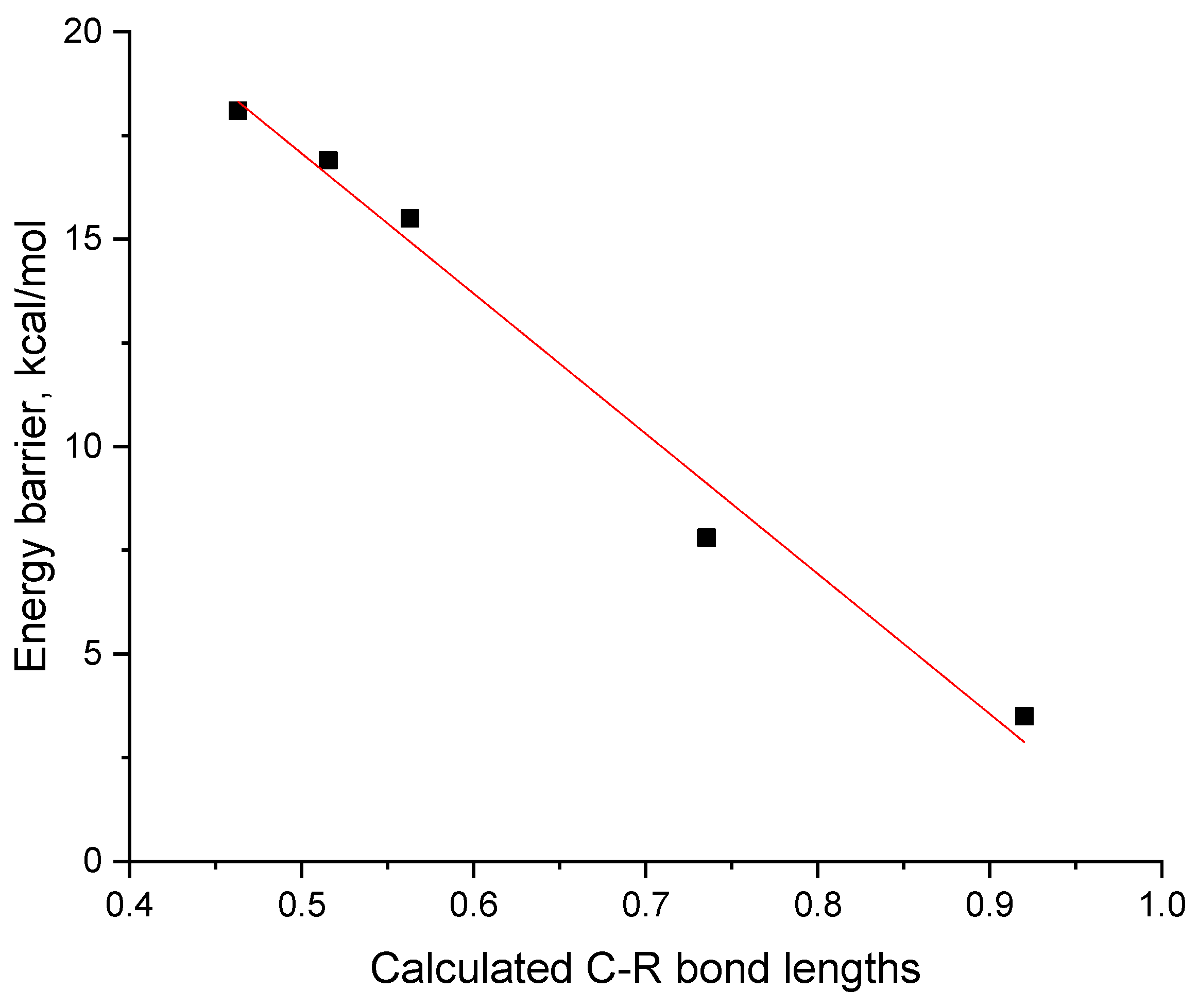
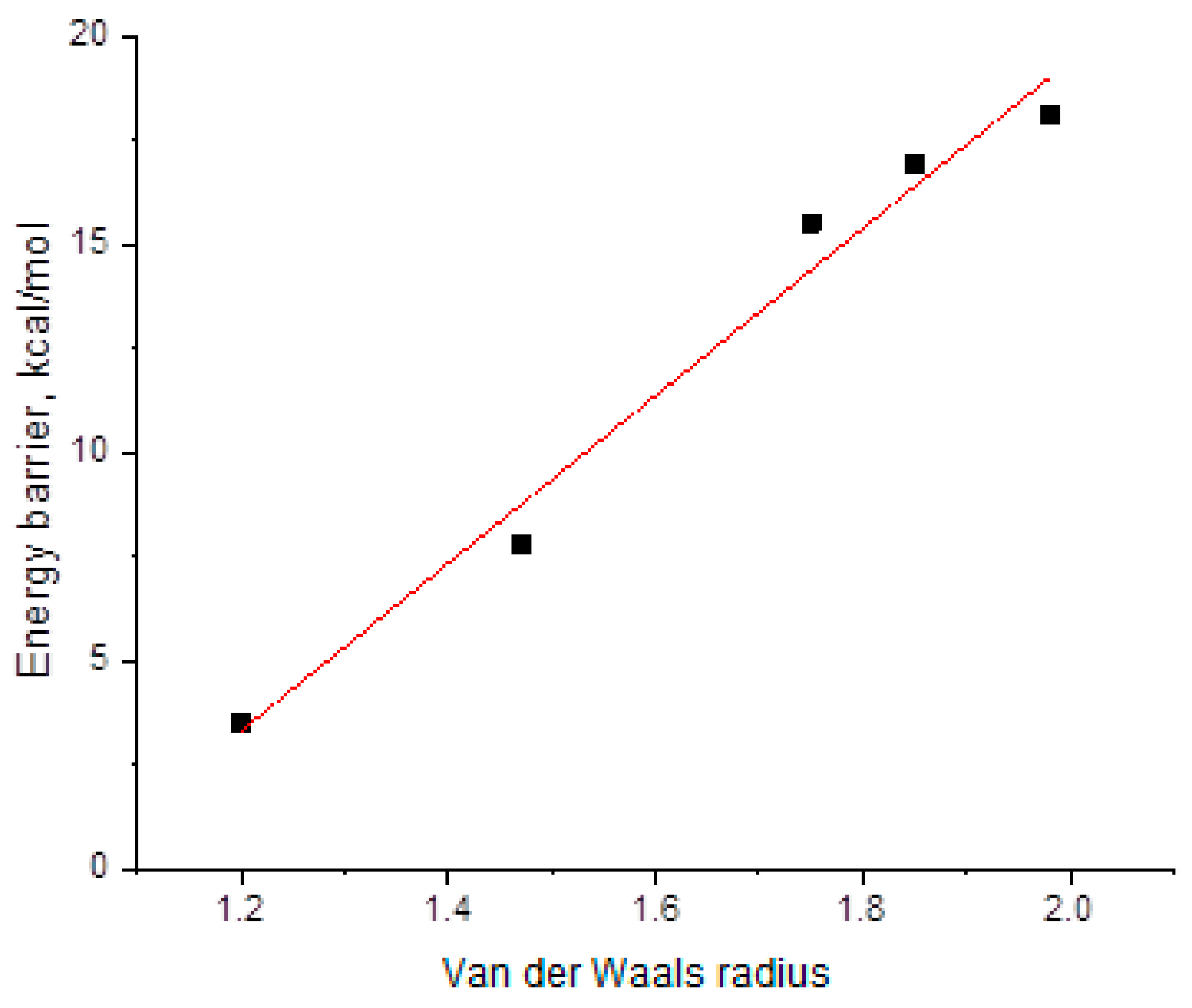
2.4. DFT Studies
3. Materials and Methods
3.1. General Information
3.2. GC and GC-MS Analyses
3.3. Initial NMR Measurements at Room Temperature
3.4. General Procedure for Preparation
3.4.1. Chlorination of Carvacrol
3.4.2. Bromination of Carvacrol
3.4.3. Iodination of Carvacrol
3.4.4. Etherification of Compounds 2 and 3
3.4.5. Esterification of Compounds 2 and 3
3.5. Dynamic NMR Studies
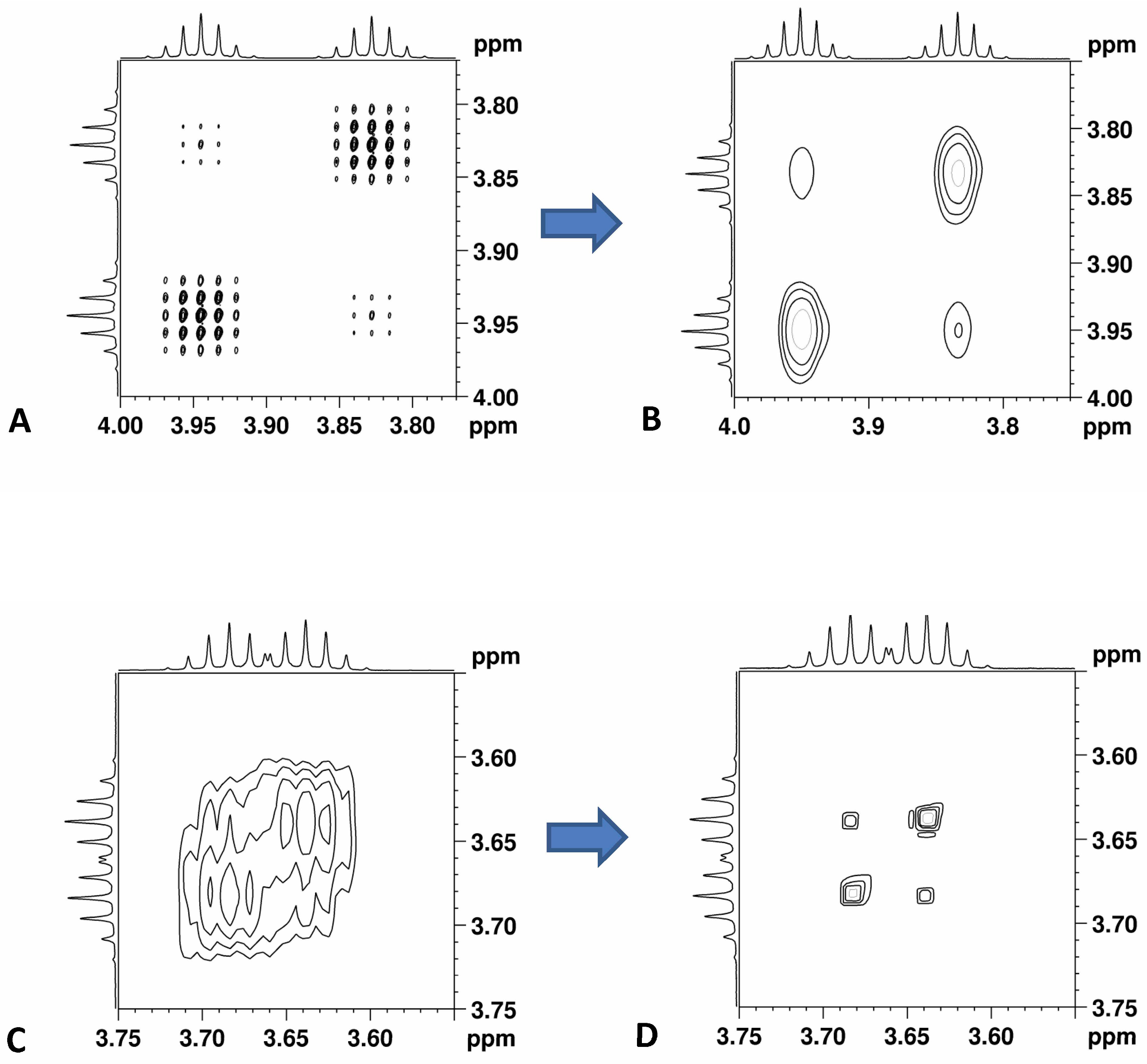
3.6. HOmodecoupled Band-Selective NMR Experiments (HOBS)
3.7. NOE Experiments
3.8. DFT Calculations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cheng, J.K.; Xiang, S.-H.; Li, S.; Ye, L.; Tan, B. Recent Advances in Catalytic Asymmetric Construction of Atropisomers. Chem. Rev. 2021, 121, 4805–4902. [Google Scholar] [CrossRef] [PubMed]
- Kumarasamy, E.; Raghunathan, R.; Sibi, M.P.; Sivaguru, J. Nonbiaryl and Heterobiaryl Atropisomers: Molecular Templates with Promise for Atropselective Chemical Transformations. Chem. Rev. 2015, 115, 11239–11300. [Google Scholar] [CrossRef] [PubMed]
- Smyth, J.E.; Butler, N.M.; Keller, P.A. A Twist of Nature—The Significance of Atropisomers in Biological Systems. Nat. Prod. Rep. 2015, 32, 1562–1583. [Google Scholar] [CrossRef] [PubMed]
- Lanman, B.A.; Parsons, A.T.; Zech, S.G. Addressing Atropisomerism in the Development of Sotorasib, a Covalent Inhibitor of KRAS G12C: Structural, Analytical, and Synthetic Considerations. Acc. Chem. Res. 2022, 55, 2892–2903. [Google Scholar] [CrossRef]
- LaPlante, S.R.; Fader, L.D.; Fandrick, K.R.; Fandrick, D.R.; Hucke, O.; Kemper, R.; Miller, S.P.F.; Edwards, P.J. Assessing Atropisomer Axial Chirality in Drug Discovery and Development. J. Med. Chem. 2011, 54, 7005–7022. [Google Scholar] [CrossRef]
- LaPlante, S.R.; Edwards, P.J.; Fader, L.D.; Jakalian, A.; Hucke, O. Revealing atropisomer axial chirality in drug discovery. ChemMedChem 2011, 6, 505–513. [Google Scholar] [CrossRef] [PubMed]
- Toenjes, S.T.; Gustafson, J.L. Atropisomerism in medicinal chemistry: Challenges and opportunities. Future Med. Chem. 2018, 10, 409–422. [Google Scholar] [CrossRef]
- Chandross, E.A.; Sheley, C.F., Jr. Some 9-aryl fluorenes. Ring-current effects on nuclear magnetic resonance spectra, carbonium ions, and the 9-mesitylfluorenyl radical. J. Am. Chem. Soc. 1968, 90, 4345–4354. [Google Scholar] [CrossRef]
- Lomas, J.S.; Dubois, J.-E. Conformational isomerism in o-tolyldi-tert-butylcarbinol. J. Org. Chem. 1976, 41, 3033–3034. [Google Scholar] [CrossRef]
- Lomas, J.S.; Luong, P.K.; Dubois, J.-E. Nucleophilic addition of o-tolyllithium compounds to di-tert-butyl ketone. Thermal and organolithium-catalyzed isomerization of o-tolyldi-tert-butylcarbinol rotamers. J. Org. Chem. 1977, 42, 3394–3399. [Google Scholar] [CrossRef]
- Casarini, D.; Lunazzi, L.; Mazzanti, A. Conformational Studies by Dynamic Nuclear Magnetic Resonance. 59. (1) Stereodynamics of Conformational Enantiomers in the Atropisomers of Hindered Naphthylcarbinols. J. Org. Chem. 1997, 62, 3315–3323. [Google Scholar] [CrossRef] [PubMed]
- Casarini, D.; Lunazzi, L.; Mancinelli, M.; Mazzanti, A. Conformation and Stereodynamics of Symmetrically Ortho-Disubstituted Aryl Carbinols and Aryl Ethers. J. Org. Chem. 2007, 72, 998–1004. [Google Scholar] [CrossRef] [PubMed]
- Mori, T.; Ōki, M. Restricted Rotation Involving the Tetrahedral Carbon. XXXVIII. Barriers to Rotation and Population Distributions of 9-(8-Methyl-1-naphthyl)fluorene and Its 1-Methyl Derivative. Bull. Chem. Soc. Jpn. 1981, 54, 1199–1202. [Google Scholar] [CrossRef]
- Nakamura, M.; Ōki, M. Restricted Rotation Involving the Tetrahedral Carbon. XXXIII. Restricted Rotation about a Csp3–Csp2 Bond in 10,10-Disubstituted 9-(2,6-Xylyl)-9,10-dihydroanthracene Derivatives. Bull. Chem. Soc. Jpn. 1980, 53, 2977–2980. [Google Scholar] [CrossRef]
- Berber, H.; Lameiras, P.; Denhez, C.; Antheaume, C.; Clayden, J. Atropisomerism about Aryl–Csp3 Bonds: The Electronic and Steric Influence of ortho-Substituents on Conformational Exchange in Cannabidiol and Linderatin Derivatives. J. Org. Chem. 2014, 79, 6015–6027. [Google Scholar] [CrossRef]
- Flos, M.; Lameiras, P.; Denhez, C.; Mirand, C.; Berber, H. Atropisomerism about Aryl–C(sp3) Bonds: Conformational Behavior of Substituted Phenylcyclohexanes in Solution. J. Org. Chem. 2016, 81, 2372–2382. [Google Scholar] [CrossRef] [PubMed]
- Eto, M.; Yamaguchi, K.; Shinohara, I.; Ito, F.; Yoshitake, Y. Conformation of aromatic rings in isolable atropisomers of 2-arylindoline derivatives and kinetic evidences for π–π interaction. Tetrahedron 2010, 66, 898–903. [Google Scholar] [CrossRef]
- Toda, Y.; Kooguchi, A.; Sukegawa, K.; Kikuchi, A.; Suga, H. Ring-fused hexahydro-1,2,4,5-tetrazines: Synthesis, structure, and mechanistic studies on isolable rotational isomers. Chem. Commun. 2023, 59, 700–703. [Google Scholar] [CrossRef] [PubMed]
- Li, S.-G.; Wang, Y.-T.; Zhang, Q.; Wang, K.-B.; Xue, J.-J.; Li, D.-H.; Jing, Y.-K.; Lin, B.; Hua, H.-M. Pegaharmols A–B, Axially Chiral β-Carboline-quinazoline Dimers from the Roots of Peganum harmala. Org. Lett. 2020, 22, 7522–7525. [Google Scholar] [CrossRef]
- Isaka, M.; Tanticharoen, M.; Kongsaeree, P.; Thebtaranonth, Y. Structures of cordypyridones A-D, antimalarial N-hydroxy- and N-methoxy-2-pyridones from the insect pathogenic fungus Cordyceps nipponica. J. Org. Chem. 2001, 66, 4803–4808. [Google Scholar] [CrossRef]
- Jones, I.L.; Moore, F.K.; Chai, C.L.L. Total Synthesis of (±)-Cordypyridones A and B and Related Epimers. Org. Lett. 2009, 11, 5526–5529. [Google Scholar] [CrossRef] [PubMed]
- Delaye, P.-O.; Lameiras, P.; Kervarec, N.; Mirand, C.; Berber, H. Improved Enantioselective Synthesis of (−)-Linderol A: Hindered Rotation about Aryl−Csp3 Bond. J. Org. Chem. 2010, 75, 2501–2509. [Google Scholar] [CrossRef] [PubMed]
- He, Q.-F.; Wu, Z.-L.; Huang, X.-J.; Xia, T.-Q.; Tang, G.; Tang, W.; Shi, L.; Ye, W.-C.; Wang, Y. Cajanusoids A–D, Unusual Atropisomeric Stilbene Dimers with PTP1B Inhibitory Activities from the Leaves of Cajanus cajan. J. Org. Chem. 2021, 86, 5870–5882. [Google Scholar] [CrossRef]
- He, Q.-F.; Wu, Z.-L.; Huang, X.-J.; Zhong, Y.-L.; Li, M.-M.; Jiang, R.-W.; Li, Y.-L.; Ye, W.-C.; Wang, Y. Cajanusflavanols A–C, Three Pairs of Flavonostilbene Enantiomers from Cajanus cajan. Org. Lett. 2018, 20, 876–879. [Google Scholar] [CrossRef] [PubMed]
- EAFUS. A Food Additive Database Centre for Food Safety Applied Nutrition; Food, and Drug Administration: Washington, DC, USA, 2006.
- European Parliament and Council. Regulation (EC) No 2232/96 the European Parliament, and of the Council on 28 October 1996, Commission Decision of 23 February 1999 adopting a register of flavouring substances used in or on foodstuffs. Off. J. Eur. Commun. 1996, 39, 1–37. [Google Scholar]
- Hyldgaard, M.; Mygind, T.; Meyer, R.L. Essential Oils in Food Preservation: Mode of Action, Synergies, and Interactions with Food Matrix Components. Front. Microbiol. 2012, 3, 12. [Google Scholar] [CrossRef] [PubMed]
- Langeveld, W.T.; Veldhuizen, E.J.A.; Burt, S.A. Synergy between essential oil components and antibiotics: A review. Crit. Rev. Microbiol. 2014, 40, 76–94. [Google Scholar] [CrossRef] [PubMed]
- De Oliveira, A.S.; Llanes, L.C.; Brighente, I.M.C.; Nunes, R.J.; Yunes, R.A.; Máximo, N., Jr.; Baumgart, A.M.K.; Aust, A.N.; Cruz, A.B. New Sulfonamides Derived from Carvacrol: Compounds with High Antibacterial Activity against Resistant Staphylococcus aureus Strains. J. Biosci. Med. 2016, 4, 105–114. [Google Scholar] [CrossRef]
- Bansal, A.; Saleh-E-In, M.; Kar, P.; Roy, A.; Sharma, N.R. Synthesis of Carvacrol Derivatives as Potential New Anticancer Agent against Lung Cancer. Molecules 2022, 27, 4597. [Google Scholar] [CrossRef]
- Cocolas, A.H.; Parks, E.L.; Ressler, A.J.; Havasi, M.H.; Seeram, N.P.; Henry, G.E. Heterocyclic β-keto sulfide derivatives of carvacrol: Synthesis and copper (II) ion reducing capacity. Bioorg. Med. Chem. Lett. 2019, 29, 126636. [Google Scholar] [CrossRef]
- Cocker, W.; Shannon, P.V.R.; Dowsett, M. The chemistry of terpenes. Part 27. The halogenation of (+)-thujone and of (−)-carvotanacetone, and the stereochemistry and mechanism of formation of ‘tribromothujone’. J. Chem. Soc. Perkin Trans. 1988, 1, 1527–1535. [Google Scholar] [CrossRef]
- Miller, B.; Haggerty, J.G. Effects of halogen substitution on reactions of o-quinol acetates with isopropylmagnesium bromide and diisopropylmagnesium. Competition between unimolecular decomposition and bimolecular reactions of radical anions. J. Org. Chem. 1986, 51, 174–179. [Google Scholar] [CrossRef]
- Soderberg, B.C.; Fields, S.L. Expedient syntheses of espintanol, p-methoxycarvacrol and thymoqutnol dimethyl ether. Org. Prep. Proced. Int. 1996, 28, 221–225. [Google Scholar] [CrossRef]
- Majetich, G.; Grove, J.L. Total Synthesis of (+)-19-Deoxyicetexone, (−)-Icetexone, and (+)-5-Epi-icetexone. Org. Lett. 2009, 11, 2904–2907. [Google Scholar] [CrossRef] [PubMed]
- Radulović, N.S.; Đorđević, M.R.; Blagojević, P.D. Structural revision of aristol: A fresh look at the oxidative coupling of thymol under iodination conditions. RSC Adv. 2016, 6, 69067–69082. [Google Scholar] [CrossRef]
- Enevoldsen, T.; Oddershede, J.; Sauer, S.P.A. Correlated calculations of indirect nuclear spin-spin coupling constants using second-order polarization propagator approximations: SOPPA and SOPPA (CCSD). Theor. Chem. Acc. 1998, 100, 275–284. [Google Scholar] [CrossRef]
- Provasi, P.F.; Aucar, G.A.; Sauer, S.P.A. The effect of lone pairs and electronegativity on the indirect nuclear spin–spin coupling constants in CH2X (X=CH2, NH, O, S): Ab initio calculations using optimized contracted basis sets. J. Chem. Phys. 2001, 115, 1324–1334. [Google Scholar] [CrossRef]
- Kjær, H.; Sauer, S.P.A. Pople Style Basis Sets for the Calculation of NMR Spin-Spin Coupling Constants: The 6-31G-J and 6-311G-J Basis Sets. J. Chem. Theory Comput. 2011, 7, 4070–4076. [Google Scholar] [CrossRef] [PubMed]
- Castañar, L.; Parella, T. Broadband 1H homodecoupled NMR experiments: Recent developments, methods and applications. Magn. Reson. Chem. 2015, 53, 399–426. [Google Scholar] [CrossRef]
- Zangger, K. Pure shift NMR. Prog. Nucl. Magn. Reson. Spectrosc. 2015, 86–87, 1–20. [Google Scholar] [CrossRef]
- McKenna, J.M.; Parkinson, J.A. HOBS methods for enhancing resolution and sensitivity in small DNA oligonucleotide NMR studies. Magn. Reson. Chem. 2015, 53, 249–255. [Google Scholar] [CrossRef] [PubMed]
- Ying, J.; Roche, J.; Bax, A. Homonuclear decoupling for enhancing resolution and sensitivity in NOE and RDC measurements of peptides and proteins. J. Magn. Reson. 2014, 241, 97–102. [Google Scholar] [CrossRef] [PubMed]
- Ilgen, J.; Nowag, J.; Kaltschnee, L.; Schmidts, V.; Thiele, C.M. Gradient selected pure shift EASY-ROESY techniques facilitate the quantitative measurement of 1H,1H-distance restraints in congested spectral regions. J. Magn. Reson. 2021, 324, 106900. [Google Scholar] [CrossRef] [PubMed]
- Castañar, L.; Nolis, P.; Virgili, A.; Parella, T. Full Sensitivity and Enhanced Resolution in Homodecoupled Band-Selective NMR Experiments. Chem.—Eur. J. 2013, 19, 17283–17286. [Google Scholar] [CrossRef] [PubMed]
- Dangalov, M.; Stoyanova, M.; Petrov, P.; Putala, M.; Vassilev, N.G. Fluxional Pd (II) NHC complexes–Synthesis, structure elucidation and catalytic studies. J. Organomet. Chem. 2016, 817, 1–14. [Google Scholar] [CrossRef]
- Philipova, I.; Stavrakov, G.; Vassilev, N.; Nikolova, R.; Shivachev, B.; Dimitrov, V. Cytisine as a scaffold for ortho-diphenylphosphinobenzenecarboxamide ligands for Pd-catalyzed asymmetric allylic alkylation. J. Organomet. Chem. 2015, 778, 10–20. [Google Scholar] [CrossRef]
- Philipova, I.; Stavrakov, G.; Dimitrov, V.; Vassilev, N. Galantamine derivatives: Synthesis, NMR study, DFT calculations and application in asymmetric catalysis. J. Mol. Struct. 2020, 1219, 128568. [Google Scholar] [CrossRef]
- Smith, M.B.; March, J. March’s Advanced Organic Chemistry: Reactions, Mechanisms, and Structure; Wiley: Hoboken, NJ, USA, 2007. [Google Scholar]
- Eliel, E.L.; Wilen, S.H.; Mander, L.N. Stereochemistry of Organic Compounds; Wiley: Hoboken, NJ, USA, 1994. [Google Scholar]
- Adams, R.P. Identification of Essential Oil Components by Gas Chromatography/Mass Spectrometry; Allured Publishing Corporation: Carol Stream, IL, USA, 2007. [Google Scholar]
- Binsch, G. Dynamic NMR Spectroscopy; Jackman, L.M., Cotton, F.A., Eds.; Academic Press: New York, NY, USA, 1975; pp. 45–82. [Google Scholar]
- Heinzer, J.; Oth, J. Iterative Least-Squares Lineshape Fitting of 1H-decoupled 13C-DNMR. Spectra. Helv. Chim. Acta 1981, 64, 258. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian; Gaussian, Inc.: Wallingford, CT, USA, 2013. [Google Scholar]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Hariharan, P.C.; Pople, J.A. The influence of polarization functions on molecular orbital hydrogenation energies. Theoret. Chim. Acta 1973, 28, 213–222. [Google Scholar] [CrossRef]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef] [PubMed]
- Wolinski, K.; Hinton, J.F.; Pulay, P. Efficient implementation of the gauge-independent atomic orbital method for NMR chemical shift calculations. J. Am. Chem. Soc. 1990, 112, 8251–8260. [Google Scholar] [CrossRef]
- Cheeseman, J.R.; Trucks, G.W.; Keith, T.A.; Frisch, M.J. A comparison of models for calculating nuclear magnetic resonance shielding tensors. J. Chem. Phys. 1996, 104, 5497–5509. [Google Scholar] [CrossRef]
- Helgaker, T.; Watson, M.; Handy, N.C. Analytical calculation of nuclear magnetic resonance indirect spin–spin coupling constants at the generalized gradient approximation and hybrid levels of density-functional theory. J. Chem. Phys. 2000, 113, 9402–9409. [Google Scholar] [CrossRef]
- Sychrovský, V.; Gräfenstein, J.; Cremer, D. Nuclear magnetic resonance spin–spin coupling constants from coupled perturbed density functional theory. J. Chem. Phys. 2000, 113, 3530–3547. [Google Scholar] [CrossRef]
- Peralta, J.E.; Scuseria, G.E.; Cheeseman, J.R.; Frisch, M.J. Basis set dependence of NMR spin–spin couplings in density functional theory calculations: First row and hydrogen atoms. Chem. Phys. Lett. 2003, 375, 452–458. [Google Scholar] [CrossRef]
- Deng, W.; Cheeseman, J.R.; Frisch, M.J. Calculation of nuclear spin−spin coupling constants of molecules with first and second row atoms in study of basis set dependence. J. Chem. Theory Comput. 2006, 2, 1028–1037. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound a | X | R1 | H b | T Range, K | H c | T Range, K |
|---|---|---|---|---|---|---|
| 1 | Cl | H | H-5 | 233–258 | H-8 | 233–268 |
| 2 | Br | H | H-8 | 253–273 | H-8 | 253–283 |
| 3 | I | H | OH | 253–293 | H-8 | 263–293 |
| 4 | Br | CH3 | H-5 | 253–278 | H-8 | 253–273 |
| 5 | I | CH3 | H-8 | 253–273 | H-8 | 263–293 |
| 6 | Br | Allyl | H-8 | 243–283 | H-8 | 253–283 |
| 7 | I | Allyl | H-8 | 253–273 | H-8 | 263–293 |
| 8 | Br | C(=O)i-Pr | H-5 | 253–273 | H-8 | 263–288 |
| 9 | I | C(=O)i-Pr | H-8 | 263–283 | H-8 | 263–293 |
| 10 | Br | C(=O)Et | H-5 | 253–293 | H-8 | 263–293 |
| 11 | I | C(=O)Et | H-8 | 273–313 | H-8 | 273–313 |
| 12 | F | H | ||||
| 13 | H | H |
| Compound | Process | X | R1 | ∆H≠(298 K), kcal/mol | ∆S≠(298 K), e.u. | ∆G≠(298 K), kcal/mol | T Range, K |
|---|---|---|---|---|---|---|---|
| 1 | anti to syn | Cl | H | 15.4 ± 1.0 | 4.2 ± 3.6 | 14.1 ± 0.1 | 233–268 |
| syn to anti | 15.5 ± 0.7 | 5.0 ± 2.7 | 14.0 ± 0.1 | ||||
| 2 | anti to syn | Br | H | 16.2 ± 1.1 | 1.9 ± 3.2 | 15.6 ± 0.1 | 253–283 |
| syn to anti | 17.5 ± 0.8 | 7.4 ± 3.0 | 15.3 ± 0.1 | ||||
| 3 | anti to syn | I | H | 12.3 ± 1.2 | −15.2 ± 4.4 | 16.9 ± 0.1 | 263–293 |
| syn to anti | 13.0 ± 1.0 | −13.0 ± 3.5 | 16.9 ± 0.1 | ||||
| 4 | anti to syn | Br | CH3 | 18.7 ± 1.2 | 10.9 ± 4.4 | 15.5 ± 0.1 | 253–273 |
| syn to anti | 16.8 ± 1.6 | 3.9 ± 12.6 | 15.6 ± 0.1 | ||||
| 5 | anti to syn | I | CH3 | 16.7 ± 0.9 | 0.0 ± 2.7 | 16.7 ± 0.1 | 263–293 |
| syn to anti | 16.6 ± 0.9 | 0.0 ± 1.2 | 16.6 ± 0.1 | ||||
| 6 | anti to syn | Br | Allyl | 15.2 ± 0.8 | −1.9 ± 2.4 | 15.8 ± 0.1 | 253–283 |
| syn to anti | 16.3 ± 0.8 | 1.9 ± 2.5 | 15.7 ± 0.1 | ||||
| 7 | anti to syn | I | Allyl | 16.4 ± 0.9 | −0.1 ± 1.4 | 16.4 ± 0.1 | 263–293 |
| syn to anti | 16.3 ± 0.8 | −1.1 ± 3.9 | 16.7 ± 0.1 | ||||
| 8 | anti to syn | Br | C(=O)i-Pr | 17.4 ± 1.1 | 7.2 ± 4.5 | 15.3 ± 0.1 | 263–288 |
| syn to anti | 17.5 ± 1.4 | 7.6 ± 6.4 | 15.2 ± 0.1 | ||||
| 9 | anti to syn | I | C(=O)i-Pr | 18.1 ± 1.2 | 5.3 ± 3.7 | 16.6 ± 0.1 | 263–293 |
| syn to anti | 17.0 ± 0.8 | 0.6 ± 3.2 | 16.8 ± 0.1 | ||||
| 10 | anti to syn | Br | C(=O)Et | 14.6 ± 0.9 | −3.4 ± 2.9 | 15.6 ± 0.1 | 263–293 |
| syn to anti | 14.8 ± 1.0 | −2.9 ± 2.8 | 15.6 ± 0.1 | ||||
| 11 | anti to syn | I | C(=O)Et | 15.9 ± 0.8 | −2.8 ± 3.1 | 16.7 ± 0.1 | 273–313 |
| syn to anti | 15.4 ± 0.8 | −4.8 ± 2.4 | 16.8 ± 0.1 |
| Compound | Barrier | ΔH≠(298 K) | ΔS≠(298 K) | ΔG≠(298 K) | ΔG≠(298 K)eff |
|---|---|---|---|---|---|
| 1 | TS1—anti-GS | 14.4 | −4.9 | 15.9 | 15.5 |
| TS2—anti-GS | 14.4 | −4.9 | 15.9 | ||
| 2 | TS1—anti-GS | 16.0 | −4.8 | 17.4 | 17.0 |
| TS2—anti-GS | 16.0 | −4.8 | 17.4 | ||
| 3 | TS1—anti-GS | 17.1 | −4.9 | 18.6 | 18.1 |
| TS2—anti-GS | 17.1 | −4.9 | 18.6 | ||
| 4 | TS1—anti-GS | 15.6 | −4.3 | 16.9 | 16.7 |
| TS2—anti-GS | 16.2 | −4.5 | 17.5 | ||
| 5 | TS1—anti-GS | 16.3 | −3.5 | 17.3 | 17.3 |
| TS2—anti-GS | 17.1 | −4.5 | 18.4 | ||
| 6 | TS1—anti-GS | 16.1 | −3.8 | 17.2 | 16.0 |
| TS2—anti-GS | 15.5 | −2.2 | 16.1 | ||
| 7 | TS1—anti-GS | 17.2 | −4.7 | 18.6 | 17.6 |
| TS2—anti-GS | 16.4 | −4.4 | 17.7 | ||
| 8 | TS1—anti-GS | 15.5 | −4.2 | 16.7 | 16.7 |
| TS2—anti-GS | 16.3 | −5.7 | 18.0 | ||
| 9 | TS1—anti-GS | 16.0 | −2.8 | 16.9 | 17.0 |
| TS2—anti-GS | 17.4 | −2.5 | 18.2 | ||
| 10 | TS1—anti-GS | 15.6 | −4.2 | 16.9 | 16.7 |
| TS2—anti-GS | 16.2 | −4.3 | 17.4 | ||
| 11 | TS1—anti-GS | 16.5 | −3.3 | 17.5 | 17.4 |
| TS2—anti-GS | 17.3 | −4.5 | 18.7 | ||
| 12 | TS1—anti-GS | 6.2 | −5.8 | 8.0 | 7.6 |
| TS2—anti-GS | 6.2 | −5.8 | 8.0 | ||
| 13 | TS1—anti-GS | 1.9 | −7.1 | 4.0 | 3.6 |
| TS2—anti-GS | 1.9 | −7.1 | 4.0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Đorđević Zlatković, M.R.; Radulović, N.S.; Dangalov, M.; Vassilev, N.G. Conformation Analysis and Stereodynamics of Symmetrically ortho-Disubstituted Carvacrol Derivatives. Molecules 2024, 29, 1962. https://doi.org/10.3390/molecules29091962
Đorđević Zlatković MR, Radulović NS, Dangalov M, Vassilev NG. Conformation Analysis and Stereodynamics of Symmetrically ortho-Disubstituted Carvacrol Derivatives. Molecules. 2024; 29(9):1962. https://doi.org/10.3390/molecules29091962
Chicago/Turabian StyleĐorđević Zlatković, Miljana R., Niko S. Radulović, Miroslav Dangalov, and Nikolay G. Vassilev. 2024. "Conformation Analysis and Stereodynamics of Symmetrically ortho-Disubstituted Carvacrol Derivatives" Molecules 29, no. 9: 1962. https://doi.org/10.3390/molecules29091962
APA StyleĐorđević Zlatković, M. R., Radulović, N. S., Dangalov, M., & Vassilev, N. G. (2024). Conformation Analysis and Stereodynamics of Symmetrically ortho-Disubstituted Carvacrol Derivatives. Molecules, 29(9), 1962. https://doi.org/10.3390/molecules29091962