Abstract
Borane–trimethylamine complex (Me3N·BH3; BTM) is the most stable of the amine–borane complexes that are commercially available, and it is cost-effective. It is a valuable reagent in organic chemistry with applications in the reduction of carbonyl groups and carbon–nitrogen double bond reduction, with considerable examples in the reduction of oximes, hydrazones and azines. The transfer hydrogenation of aromatic N-heterocycles and the selective N-monomethylation of primary anilines are further examples of recent applications, whereas the reduction of nitrobenzenes to anilines and the reductive deprotection of N-tritylamines are useful tools in the organic synthesis. Moreover, BTM is the main reagent in the regioselective cleavage of cyclic acetals, a reaction of great importance for carbohydrate chemistry. Recent innovative applications of BTM, such as CO2 utilization as feedstock and radical chemistry by photocatalysis, have extended their usefulness in new reactions. The present review is focused on the applications of borane–trimethylamine complex as a reagent in organic synthesis and has not been covered in previous reviews regarding amine–borane complexes.
1. Introduction
Boron-based reagents play an important role in modern organic synthesis and especially borane carriers have reached a predominant position in the synthesis of pharmaceutics and natural products. Boranes form complexes with Lewis bases, such as amines and pyridines, that are stable, safer and easier to handle. There are few reviews in the literature concerning the use of amine–borane complexes in organic synthesis [1,2,3,4,5,6,7,8], but in some of them, the part describing the reactivity is quite limited. An early review [2] is the most representative in the description of the reactivity of several amine–borane complexes with examples of practical application in organic synthesis covering literature up to 1984; it is also the only one that describes some applications of borane–trimethylamine complex (Me3N·BH3; BTM) while the subsequent reviews report either single reactions with this reagent or deal with different amine–boranes. A review on the chemistry of amine and phosphine–boranes was published [3] in 1999, whereas reductive amination was the topic of two reviews, one focused on amine–boranes [5] and the other with several boron reagents [4]. Amine–boranes forming frustrated Lewis pairs was the subject of a chapter in a book [6], whereas two recent reviews dealt with the reactivity of ammonia-borane complex [7,8]. This review aims to focus on the use of borane–trimethylamine complex as a reagent, mainly after 1984 and not covered in previous reviews regarding amine–borane complexes.
Of the various known complexes, BTM is the most stable [9], less sensitive to hydrolysis, even under acidic conditions [2], and does not require any special storage conditions. It is thermally stable up to 120 °C and can be purified by vacuum sublimation; conversely, ammonia–borane complex explodes when heated, and an attempted distillation of borane–pyridine complex at reduced pressure resulted in violent decomposition [1]. BTM is very soluble in a wide variety of solvents [1], and its stability significantly increases with increasing solvent polarity [10]. It is relatively inexpensive compared with the other amine–borane complexes commercially available and considering the low molecular weight. Contrarily to borane complexes of ammonia, primary and secondary alkylamines, the inert nature of trimethylamine in BTM was a further advantage in avoiding the side reactions observed with the more reactive amines. Its reactivity can be opportunely activated in the reaction medium, and it is considered a valuable reagent in organic synthesis for laboratory as well as industrial scale. Since 1937, when it was synthesized for the first time, its applications have grown steadily, with some innovative ones in recent years.
2. Reductive Transformations with Carbon–Oxygen Double Bond
2.1. Reduction of Ketones
BTM is a weak hydride donor more reactive than trialkylsilanes and trialkylgermanes, comparable with that of trialkylstannanes [11]. The greater stability of BTM in the presence of Lewis or Brønsted acid allows the activation of the substrate by acid catalysis [2]; from kinetic studies, the rate of reduction of aldehydes and ketones increases with increasing acidity of the medium suggesting the formation of a protonated carbonyl species in rapid equilibrium, followed by the rate-determining step of the reduction. BTM was the reagent of choice for the selective reduction of steroidal diones, such as 1, in the presence of wet silica gel impregnated with FeCl3 (Scheme 1): C-3 carbonyl group was reduced preferentially to the alcohol 2 [12].
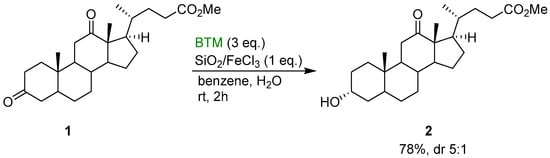
Scheme 1.
Regioselective reduction of dione 1 on a silica gel surface.
The formation of a chelate between the Lewis acid and substrates 3 and 5 proved to be effective for the completely diastereoselective reduction to the corresponding alcohols 4 and 6 (Scheme 2) [13].
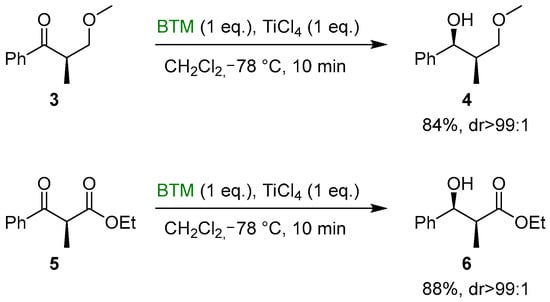
Scheme 2.
Diastereoselective reduction of ketones 3 and 5.
In a similar way, in the enantioselective total synthesis of analogs of griseusins [14], ketone 7 was reduced by BTM in good diastereoselectivity using TFA as acid (Scheme 3).
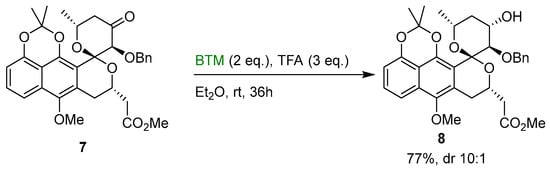
Scheme 3.
Diastereoselective reduction of ketone 7.
2.2. Reductive Bromation of Aromatic Carbonyl Compounds
Bromine was an effective activator in the reduction of both aldehydes and ketones by BTM [15], but in the case of aromatic compounds 9, the reaction proceeded beyond the reduction to alcohol, leading to bromide derivative 10 in good yields (Scheme 4) with the exception of a product obtained in low yield after recrystallization.

Scheme 4.
Reductive bromation of aromatic carbonyl compounds.
Some useful exploitations of the synthetic method were (Scheme 5) the synthesis of the 14C labeled benzylbromide 12, intermediate in the synthesis of CX3CR1 antagonist 13 [16], and the synthesis of the dibromide derivative 15, intermediate in the synthesis of crownophanes [17].
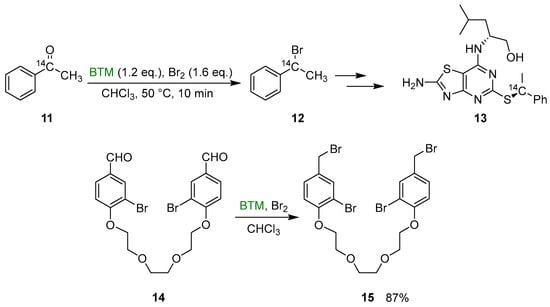
Scheme 5.
Synthesis of the 14C labeled benzylbromide 12 and the dibromide derivative 15.
2.3. Reduction of Carboxylic Acids
At room temperature, carboxylic acids are inert in presence of BTM and can be used as solvent in the reduction reactions; on the other hand, refluxing a xylene solution of BTM and carboxylic acid 16 in molar ratio of 1.5:2, ester 17 was isolated in 87% yield [18] (Scheme 6); the reaction was suitable both to aliphatic and aromatic acids with moderate to good yields (28–87%).
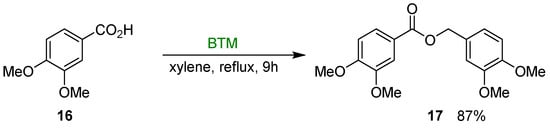
Scheme 6.
Reduction of acid 16 to ester 17.
A plausible reaction mechanism could be the presence of two concurrent reactions: the reduction of the acid 18 to alcohol 19, isolated in every reaction, and the formation of the triacyloxyborane complex 20, a known acylating compound; finally, the reaction of the two intermediates leads to the ester 21 (Scheme 7).
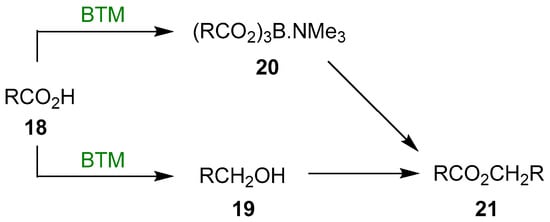
Scheme 7.
Reaction mechanism of the acid reduction.
By adding either primary or secondary amine 22 to the previous reaction mixture and changing the ratio of the reagents, a different result was obtained (Scheme 8) [19]: with the molar ratio BTM:amine 22:acid 18 = 1:1:3 led to amide 23, seemingly by the action of acylating complex 20; instead, the molar ratio 1:2:2 led to the tertiary amine 24, likely by reduction of the acid 18 to an aldehyde equivalent (boryl acetal) followed by the reductive alkylation of amine 22.
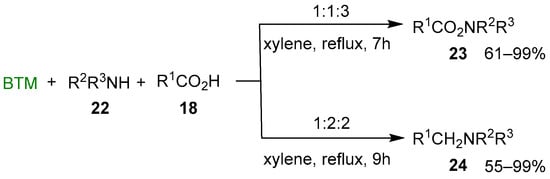
Scheme 8.
Reaction of N-acylation or N-alkylation of amines 22 by carboxylic acids 18.
2.4. Reductive Methylation with CO2
Recently, the reduction of CO2 has emerged as a topic of great interest connected with global climate change and the urgent necessity to reduce the concentration of CO2 in the atmosphere through sequestration and its utilization in the synthesis of useful compounds. An early study [20] reported the reduction of CO2 to formate by BTM weakly bonded to the bulkier Lewis acid Al(C6F5)3; then, the reaction mechanism was studied by quantum chemical calculations [21]. Subsequently, the reduction of CO2 was exploited for the selective monomethylation of 2-arylacetonitriles 25 [22]; as shown in Scheme 9, the optimized reaction conditions involved the reaction, in a sealed tube, of a DME solution of nitrile 25, tBuOK and 1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD) in an atmosphere of CO2, obtaining selectively the monomethyl derivative 26, with yields around 80% in almost all examples.
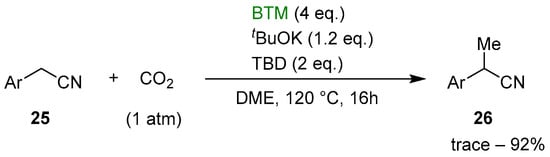
Scheme 9.
Selective monomethylation of 2-arylacetonitriles 25.
By using 13CO2, the corresponding 13C methyl derivative was obtained, confirming that the methyl group comes from CO2. In the reaction, TBD has the role of activator of the CO2 forming the adduct 27 (Scheme 10), and tBuOK of a strong base in deprotonation of 2-arylacetonitriles 25. The reaction, shown in Scheme 9, performed without nitrile 25, led to methyl borate 29 as the major product, which suggests that CO2 undergoes a six-electron reduction with the formatoborohydride 28 as an intermediate. Differently from BTM, borane–ammonia complex reduced CO2 to the formyl group; conversely, with the borane dimethylamine complex (BDM), the two-electron reduction product 28 reacted with dimethylamine, derived from BDM, leading to amide 30 and blocking further reduction (Scheme 10). In the presence of tBuOK, methyl borate 29 proved to be an effective methylating agent of nitrile 25, obtaining the methylated product 26 in 75% yield; in a similar way, two different nucleophiles, such as aniline 31 and thiophenol 33, were methylated in the same reaction condition.
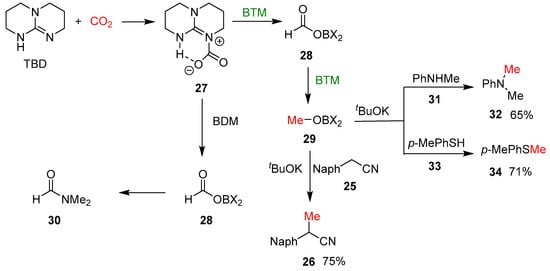
Scheme 10.
The proposed mechanism of the reaction of monomethylation.
Recently, the selective methylation of amines with CO2 was examined [23] by combining the organocatalyst 6-amino-2-picoline 36 and BTM (Scheme 11) to form a stable intramolecular frustrated Lewis pairs catalyst (Scheme 12), most of the secondary amines 35 used were N-alkyl or N-aryl anilines, with only two examples of alkyl heterocyclic amines, affording methylation products 37 in moderate to good yields.
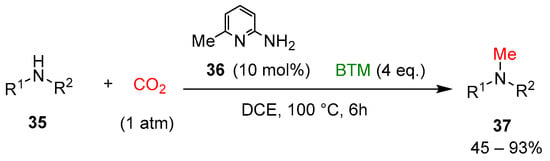
Scheme 11.
Selective methylation of amines with CO2.
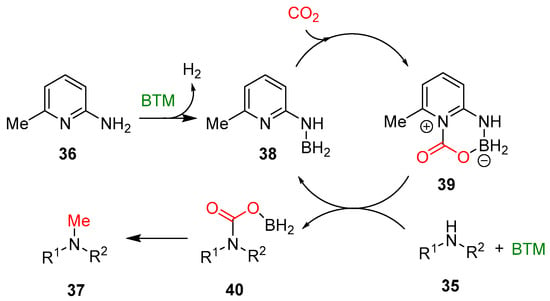
Scheme 12.
Reaction mechanism of methylation of amines with CO2.
On the basis of a series of control experiments, NMR and high-resolution mass spectrometry (HRMS) analyses, a possible reaction mechanism was proposed (Scheme 12). The first step is the reaction of 6-amino-2-picoline 36 and excess BTM leading to aminoborane 38, with the quantitative evolution of H2, followed by the CO2 activation with zwitterionic intermediate 39. The next steps are less evident, and the authors speculate the formation of intermediate 40 that is further reduced to product 37.
Without a catalyst, halving the equivalents of BTM and with DMF as solvent (Scheme 13), the reduction afforded the corresponding monoformylation products 41, an intermediate hypothesized in the reduction from 40 to product 37 (Scheme 12); most of the amines 35 used were primary anilines and yields were generally good.
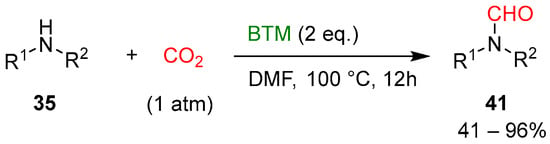
Scheme 13.
Formylation of amines with CO2.
Finally, a tandem four-component reductive methylation of primary amines 42 was realized, coupling a reductive amination to secondary amines 45, with the reduction of CO2 to a methyl group, synthesizing tertiary N-methylamines 46 [24] (Scheme 14).
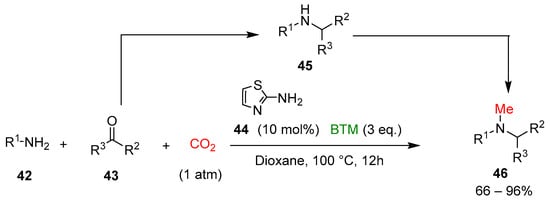
Scheme 14.
Four-component reductive methylation of primary amines with CO2.
On the contrary of catalyst 36, 2-aminothiazole 44 catalyzed the reaction with higher efficiency, whereas BTM was the best choice in the screening of different types of boron-based reducing agents. The study of the scope of the reaction involved a large number of amines 42, aldehydes and ketones 43, leading to products in mostly good yields; the gram-scale synthesis of the antifungal agent butenafine 49 was an example of the potentiality of the present synthetic method (Scheme 15).
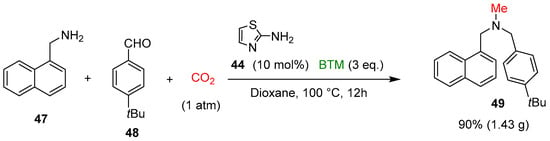
Scheme 15.
Synthesis of the antifungal agent butenafine 49.
3. Carbon–Nitrogen Double Bond Reduction
3.1. Reduction of Hydrazones and Azines
Although the reduction of hydrazones and azines is hampered by opposite conjugation effects, the presence of an acid in the reaction medium can activate the C=N bond to the attack of nucleophiles [25,26]. The improved stability of BTM in a strong acidic medium enabled the efficient reduction of both hydrazones 50 and azines 52 (Scheme 16), affording a wide range of highly functionalized mono-, di- and trialkyl hydrazines as stable and safe hydrochlorides and in excellent yields for most of the compounds.
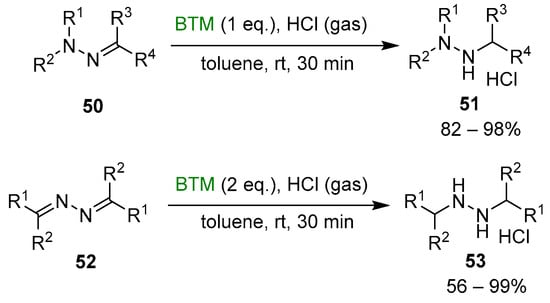
Scheme 16.
Reduction of hydrazones and azines.
The work-up operationally simple was another credit of the method, as the byproduct of reduction 56 (Scheme 17) was soluble in toluene, contrary to products 51 or 53, which are completely insoluble (except for two compounds) and easily separated by filtration.
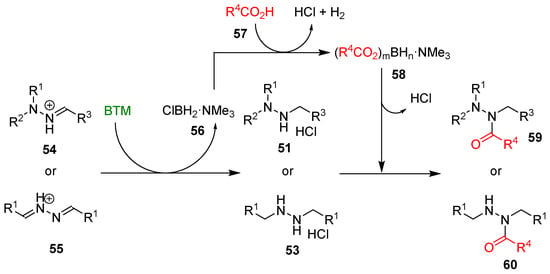
Scheme 17.
The proposed mechanism of the synthesis of hydrazides.
Byproduct 56 was exploited in the “one pot” synthesis of hydrazides by adding carboxylic acid 57 at the end of the reduction step and producing, in situ, a mixture of acyloxyboranes 58 (Scheme 17) that proved to be an effective acylating agent of both alkylhydrazine hydrochloride 51 and 53 (Scheme 18); the yields were susceptible to the bulkiness of both carboxylic acids and alkylhydrazines limiting the reaction to less hindered hydrazones and azines derived from aldehydes. The tight steric requirement for the acylation by the acyloxyboranes made the synthesis of hydrazides 60 completely selective without the formation of the related diacyl hydrazines side-product, generally observed with common acylating agents.
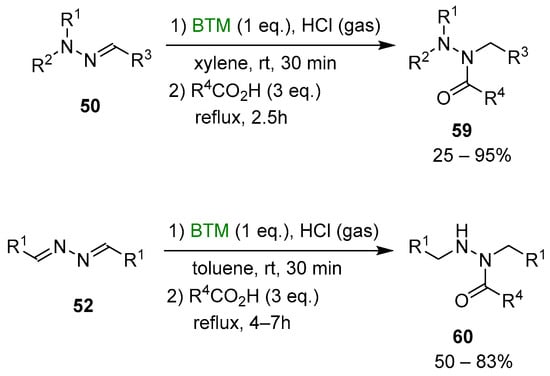
Scheme 18.
Synthesis of hydrazides.
3.2. Reduction of Oximes
Early studies on the reduction of oximes by BTM and HCl were directed to the synthesis of N-hydroxy derivatives of tryptophan 62 (Scheme 19), useful intermediates for the synthesis of β-Carbolines [27,28,29,30], fungal metabolites Neoechinulins [31,32] and recently the marine fungal metabolite raistrickindole A [33] (Scheme 20); with some oximes the reduction of indole to indoline (see Section 3.3) was observed [32].
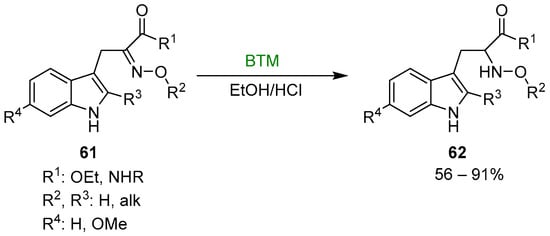
Scheme 19.
Synthesis of N-hydroxy derivatives of tryptophan 62.
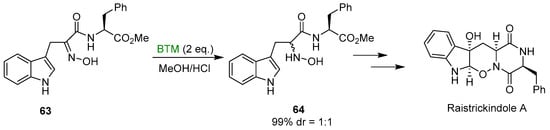
Scheme 20.
Synthesis of raistrickindole A.
Subsequently, the synthetic method was extended to the synthesis of a new type of pseudopeptides, the N-hydroxy dipeptides 66 [34], as a diastereoisomeric mixture (Scheme 21).
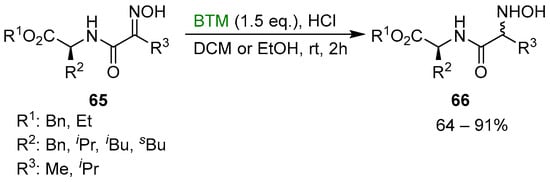
Scheme 21.
Synthesis of N-hydroxy dipeptides.
Reduction of oximes was the easiest method of synthesis of the N-alkyl-hydroxylamines 68 [35], requisite for the neoglycosylation reaction optimization in the synthesis of glycosylated 69 (Scheme 22); the yields were mediocre to moderate, likely due to the use of diluted HCl aqueous solution.
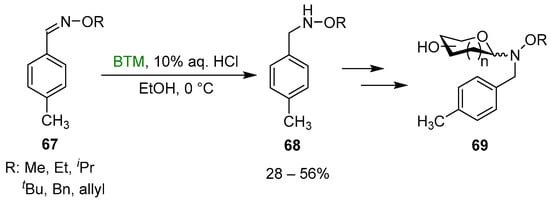
Scheme 22.
Synthesis of N-alkyl-hydroxylamines 68.
Similarly, the betulinic derivative 71 [36] and 9-amino doxycycline derivative 73 [37], suitable substrates for the neoglycosylation reaction, were synthesized from the corresponding oximes in good yields, making use of HCl in ethanol and large excess of BTM (Scheme 23).
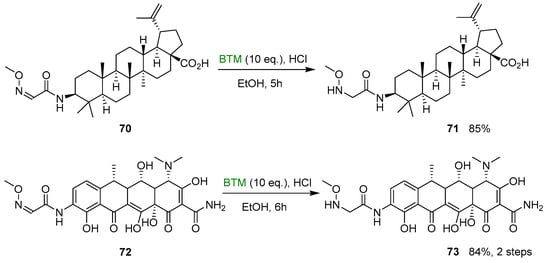
Scheme 23.
Synthesis of N-alkyl-hydroxylamines 71 and 73.
Finally, a new fluorous-tagged hydroxylamine [38], as an ammonia equivalent, was successfully exploited in the synthesis of itopride, a drug used for the treatment of functional dyspepsia; the work-up step was simplified by fluorous solid-phase extraction (F-SPE), relative with this strategy (Scheme 24).
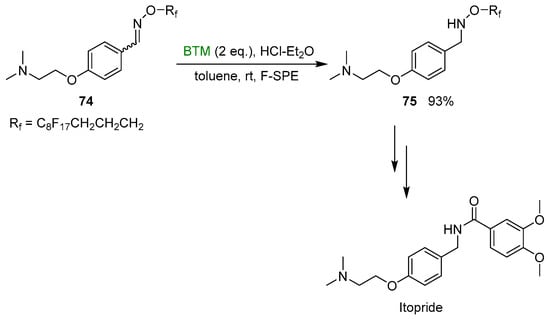
Scheme 24.
Synthesis of itopride.
3.3. Transfer Hydrogenation of Aromatic N-Heterocycles
Several aromatic N-heterocycles can react with protic acid or acylating agents, leading to salts with an immonium substructure that can be reduced by BTM similarly to the reduction of imines activated with protic acid [2]; reduction of indoles to indolines was reported in a previous review [2].
BTM reduced pyridines 76 (Scheme 25), activated by reaction with phenyl chloroformate, to 1,4 dihydropyridine 77 (majority product) and 1,2 dihydropyridine 78 (minority product) [39]; the use of triflic anhydride improved the regioselectivity toward dihydropyridine 77 reaching, in the best case, the 99:1 regioselectivity and 76:24 for the worst case. Substituents in the 4-position completely inverted the selectivity in favor of regioisomer 78. The reaction could also be applied for the synthesis of other N-heterocycles, such as dihydroquinolines 79, dihydroisoquinolines 80 and benzothiazoline 81, among others.
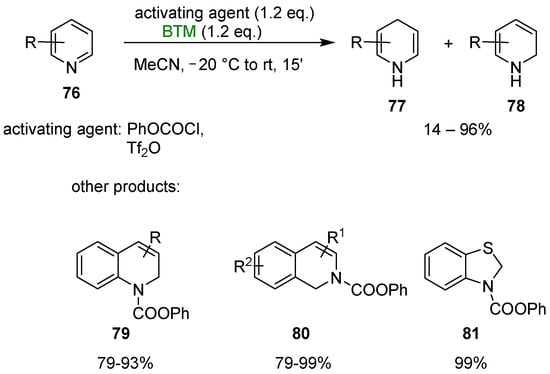
Scheme 25.
Dearomatization of N-heterocycles.
Trifluoroacetic acid (TFA), as an acidic activator, offered several opportunities; both indoles 82 and quinoxalines 84 [40] were reduced to indolines 83 and tetrahydroquinoxalines 85, respectively, in water as solvent (Scheme 26).
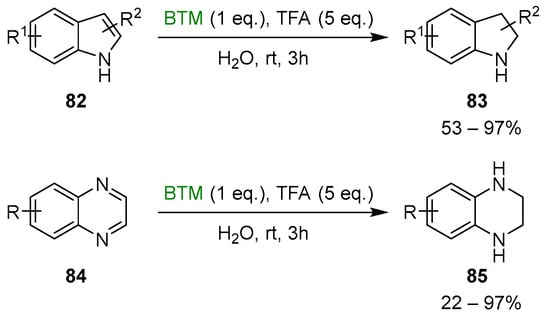
Scheme 26.
Reduction of indoles 82 and quinoxalines 84.
Tuning the reaction condition and the equivalents of BTM and TFA, as shown in Scheme 27, to go beyond the reduction, obtaining a different product [41]. A reduced amount of BTM, increased equivalents of TFA and the use of an aprotic solvent brought to the N-trifluoroacetylated indolines 86, while the increase of BTM brought to the N-trifluoroethylated indolines 87, similarly to the reduction, by BTM, of carboxylic acid in the presence of amines (Section 2.3, Scheme 8).
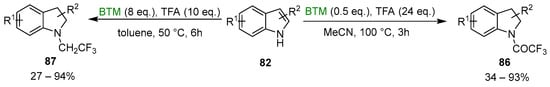
Scheme 27.
Synthesis of N-trifluoroacetylated indolines 86 and N-trifluoroethylated indolines 87.
The synthetic method was successfully extended to the synthesis of N-trifluoroethylated tetrahydroquinoline 89 and N-trifluoroethylated tetrahydroquinoxalines 91 (Scheme 28) [42].
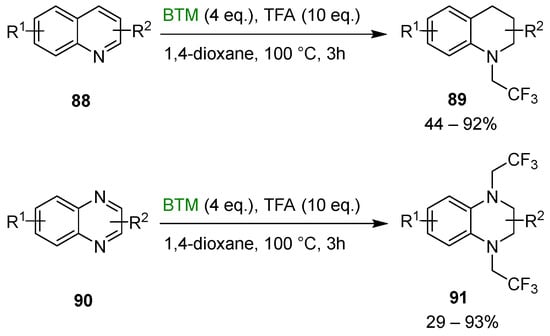
Scheme 28.
Synthesis of N-trifluoroethylated tetrahydroquinoline 89 and N-trifluoroethylated tetrahydroquinoxalines 91.
3.4. Selective N-Monomethylation of Primary Anilines
Primary anilines 92 were selectively monomethylated [43] by reaction with BTM and NaH in DMF (Scheme 29).
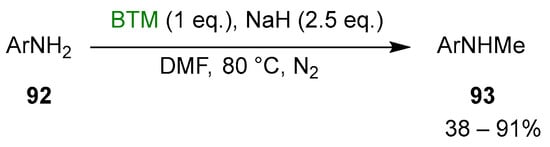
Scheme 29.
Monomethylation of anilines.
In the proposed reaction mechanism (Scheme 30), the first step is the reaction of aniline 92 with NaH and DMF obtaining amidine 94 that, likewise, the reduction of imines by BTM [2], is reduced to aminal 95; subsequent elimination of dimethylamine generated an imine that is easily reduced to the final product.
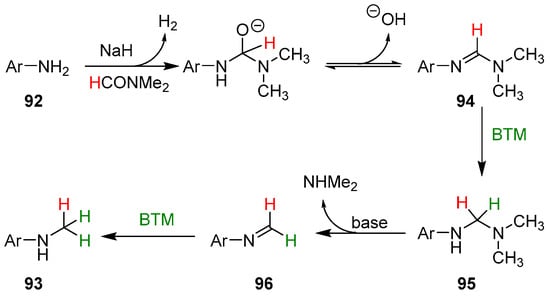
Scheme 30.
Proposed reaction mechanism of the monomethylation of anilines.
The sources of the hydrogens of the methyl group in monomethyl anilines 93 (in red and green in Scheme 30) and the easy synthesis of BTM-D3 (Me3N·BD3) by deuterium exchange in acidic D2O [2] were exploited in the synthesis of products with specific numbers of deuterium atoms into the methyl groups (Scheme 31) and excellent deuterium incorporation ratio, based on which deuterated reagent was used for the reaction.
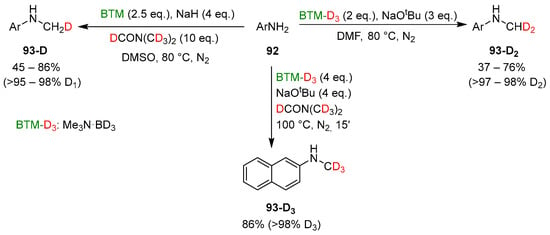
Scheme 31.
Synthesis of 93-D, 93-D2 and 93-D3 anilines.
4. Reduction of Nitrobenzenes to Anilines
BTM is hydrolytically stable in alcohols, but it can be activated in situ through palladium catalysis [44], and the reaction can be coupled with the reduction of nitroaryls 94 to anilines 95 (Scheme 32). Measuring the kinetic of the reaction pointed out that the reduction was faster than hydrogen liberation: BTM acted as hydrogen-transfer reagent and palladium hydride likely as the transient species; indeed, the reaction was an open vessel reduction, even performed at reflux, without concomitant loss of hydrogen.
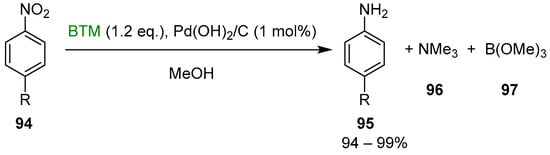
Scheme 32.
Reduction of nitroaryls 94 to anilines 95.
Credits of the reaction were excellent yield and operationally simple work-up procedure as both byproducts 96 and 97 were removed by simple concentration, and BTM surplus was completely consumed by methanolysis.
A practical use of this procedure was the “one pot” multigram synthesis of pyridine 99 (Scheme 33), a key intermediate in the synthesis of quinolone 100, a subtype-selective GABA-A receptor inverse agonist [45].
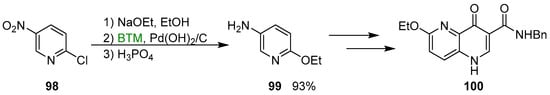
Scheme 33.
Synthesis of the pyridine 99.
5. Reductive Deprotection of N-Tritylamines
BTM reacts with some carbenium ions [11] by hydride abstraction; this reaction was exploited for the trapping of trityl cation in the deprotection of N-tritylamines, especially for sensitive substrates such as N-tritylaziridine 101 (Scheme 34) [46,47], an intermediate in the synthesis of aziridinomitosenes.
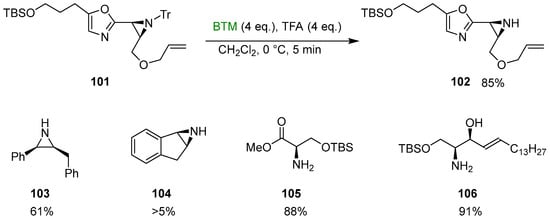
Scheme 34.
Reductive deprotection of N-tritylamines.
In addition, the method was tested for the deprotection of the aziridines 103 and 104 [48] with a low yield in the latter case due to problems with aziridine ring opening; in the absence of potential complications, the yields were excellent as for the protected serine 105 [48] and intermediate 106 in the synthesis of phytosphingosines [49].
6. Photocatalytic Difluoromethylation of Unactivated Alkenes
Ligated boryl radicals, with the general formula LB+-R2B•−, are intermediates in the activation of halogenated compounds by a reaction of halogen atom transfer (XAT) owing to the nucleophilic character of boryl radicals. Exploiting this process, BTM was utilized for the photocatalytic trifluoromethylation [50] of unactivated alkenes 107 by activation of Freon-22 108, an inexpensive feedstock, under 456 nm blue LED light irradiation (Scheme 35).
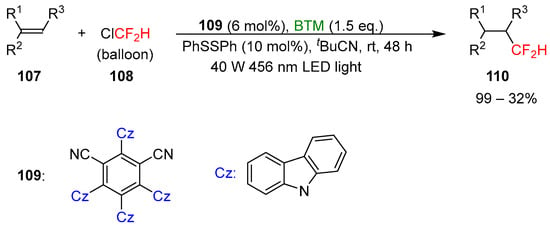
Scheme 35.
Photocatalytic difluoromethylation of unactivated alkenes.
Difluoromethylated product 110 is of enormous interest in pharmaceutical and agrochemical science owing to the properties of the group CF2H, bioisostere of hydroxyl, thiol, and amine groups. Alkenes suitable for the reaction protocol were extremely broad with the limitation of sterically encumbered tetra-substituted alkenes, unreactive, styrenes and electron-deficient alkenes where the reduction was a competitive reaction. The tolerance of various functional groups was also proved in the late-stage functionalization of complex pharmaceutical molecules and natural products; the yields were generally good, with limited examples with low yields. Supported by experimental and calculation results, the proposed mechanism of the reaction is shown in Scheme 36.
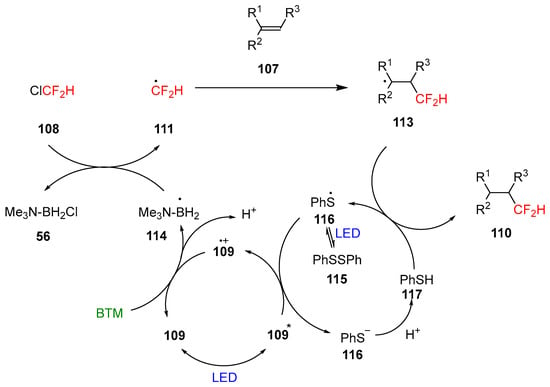
Scheme 36.
Proposed mechanism of the reaction.
Aryl thiyl radical 116 is generated from disulfide homolysis under blue light irradiation and subsequently reduced to thiolate 116 by the excited state of 109; the produced oxidized form 109.+ is reduced back by BTM generating the amine-boryl radical 114 that undergoes a XAT reaction with Freon-22 108 to generate the transient radical 111. Subsequently, the intermolecular radical addition with the alkene substrate 107 and the quench of the radical adduct 113 by thiol 117 completed the reaction.
7. Reductive Cleavage of Acetals
Regioselective cleavage of cyclic acetals in the presence of a Lewis acid is the main application of BTM in the field of organic synthesis, and it is almost completely related to carbohydrate chemistry. Carbohydrates protected as 4,6-O-benzylidene acetals were regioselectively reduced by BTM in the presence of AlCl3 [51,52,53,54,55,56,57,58,59,60,61,62,63,64,65,66,67,68,69,70,71,72,73,74,75,76,77,78,79,80,81,82,83,84,85,86,87,88,89,90,91,92,93,94,95,96,97,98,99,100,101,102], forming a free alcohol at one position and a benzyl ether protection at the other, useful for further modifications; Scheme 37 shows an example [93].
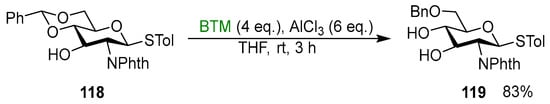
Scheme 37.
Regioselective cleavage of benzylidene acetal 118.
Alternative acidic activators were BF3·Et2O [103,104,105,106,107,108,109,110,111,112,113,114,115,116,117,118], Me2BBr [119] and methanesulfonic acid [120]. Five-membered cyclic benzylidene acetals were suitable reactants for the synthetic protocol as well [121,122,123,124,125,126,127,128,129,130,131,132,133,134]; Scheme 38 shows a recent example [134].
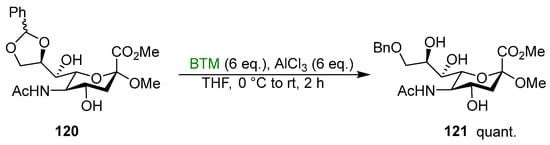
Scheme 38.
Regioselective cleavage of benzylidene acetal 121.
Acetals 122 (Scheme 39) with R different from the phenyl group allowed us to obtain the reduced product 123 with a hydroxyl protected with a different protecting group instead of the benzyl group, allowing more flexibility in the synthesis. Several examples were reported in the literature for R = pMeOPh (for six [135,136,137,138] and five [139] membered cyclic acetals), 2-naphthyl (for six- [140,141,142,143,144,145] and five- [146,147,148] membered cyclic acetals) and vinyl (five-membered cyclic acetals [149]).
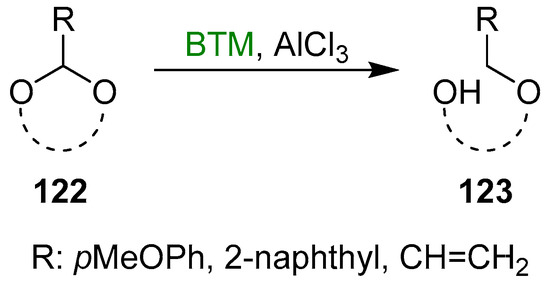
Scheme 39.
Cleavage of cyclic acetals 122.
The reaction was extensively investigated, and an early study [150] showed that the addition of two equivalents of water to the reaction mixture (four equivalents of BTM and six equivalents of AlCl3) improved the efficiency of benzylidene reductive cleavage without the observation of products of benzylidene acetal hydrolysis and rate enhancement of approximately four times. In order to decipher the mechanistic details of the reaction, several model compounds, kinetic experiments, 11B NMR spectroscopy, computational calculations, deuterium labeling, alternative reducing reagents and solvents were used [151,152,153,154], bringing to the proposed mechanism for the reaction in THF, where the Lewis acid is complexed to the solvent (Scheme 40). In the first step, BTM is activated by AlCl3, making the borane the most electrophilic species and leading to the interaction with the most electron-rich oxygen in intermediate 126, whilst the driving force is the formation of the highly stabilized AlCl3·NMe3 125; the opening of the acetal is obtained by the action of a second Lewis acid molecule with the formation of an oxocarbenium ion 127 as the rate-controlling step; finally, oxocarbenium ion is then reduced with low stereoselectivity to give product 128.
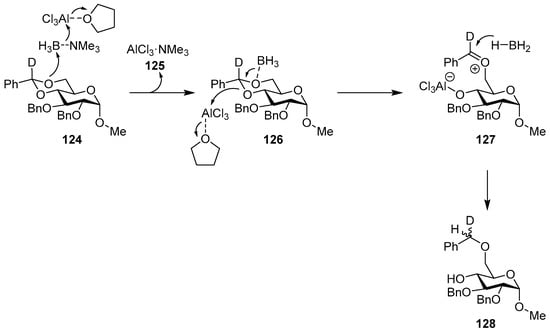
Scheme 40.
Proposed mechanism for the reaction in THF.
The reaction in toluene had different regioselectivity and usually gave low yields due to degradation. In this case, in the proposed mechanism (Scheme 41), the strongest Lewis acid is AlCl3, which reacts very fast to give the oxocarbenium ion 130 that is then reduced by BTM, with low stereoselectivity to give product 131.
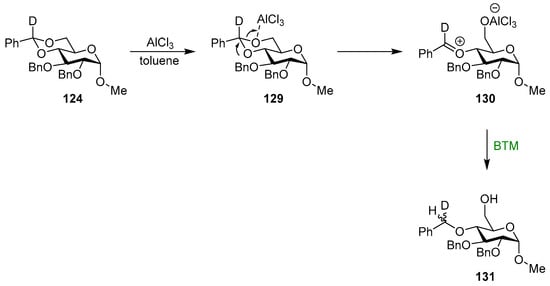
Scheme 41.
Proposed mechanism for the reaction in toluene.
8. Conclusions
In conclusion, BTM has several credits as a reagent in modern organic synthesis. It is relatively inexpensive, and considering its low molecular weight, it has a low price per mole. It is a stable solid with a good safety profile linked with its relative inertness. Its reactivity can be opportunely activated in the reaction medium, generally in the presence of Lewis or Brønsted acids. BTM undergoes rapid deuterium exchange in acidic D2O, allowing easy conversion to BTM-D3, an effective reagent for the synthesis of deuterium-labeled compounds. BTM is very soluble in a wide variety of solvents, offering more versatility in the reaction options. The tolerance of various functional groups was a well-substantiated feature of this reagent. The main application of BTM is the regioselective cleavage of cyclic acetals, a reaction of great importance for carbohydrate chemistry. Carbon–nitrogen double bond reduction is another class of reactions where the activation by acids plays an important role in the BTM reactivity. Finally, recent findings in organocatalysis have contributed to develop some innovative applications of BTM, such as the CO2 utilization as feedstock and the radical chemistry by photocatalysis.
Funding
This research received no external funding.
Data Availability Statement
Not applicable.
Conflicts of Interest
The author declares no conflicts of interest.
References
- Lane, C.F. The borane.amine complexes. Aldrichimica Acta 1973, 6, 51–58. [Google Scholar]
- Hutchins, R.O.; Learn, K.; Nazer, B.; Pytlewski, D.; Pelter, A. Amine boranes as selective reducing and hydroborating agents. A review. Org. Prep. Proced. Int. 1984, 16, 335–372. [Google Scholar] [CrossRef]
- Carboni, B.; Monnier, B. Recent developments in the chemistry of amine- and phosphine-boranes. Tetrahedron 1999, 55, 1197–1248. [Google Scholar] [CrossRef]
- Matos, K.; Pichlmair, S.; Burkhardt, E.R. Boron reagents for reductive amination. Chim. Oggi 2007, 25, 17–20. [Google Scholar]
- Matos, K.; Burkhardt, E.R. Direct reductive amination with amine boranes. In Pharmaceutical Process Chemistry, 1st ed.; Shioiri, T., Izawa, K., Konoike, T., Eds.; Wiley: New York, NY, USA, 2010; pp. 127–143. [Google Scholar] [CrossRef]
- Sumerin, V.; Chernichenko, K.; Schulz, F.; Lesleka, M.; Rieger, B.; Repo, T. Amine-borane mediated metal-free hydrogen activation and catalytic hydrogenation. In Frustrated Lewis Pairs I; Erker, G., Douglas, W.S., Eds.; Topics in Current Chemistry; Springer: Berlin/Heidelberg, Germany, 2012; Volume 332, pp. 111–155. [Google Scholar] [CrossRef]
- Lau, S.; Gasperini, D.; Webster, R.L. Amine–boranes as transfer hydrogenation and hydrogenation reagents: A mechanistic perspective». Angew. Chem. Int. Ed. 2021, 60, 14272–14294. [Google Scholar] [CrossRef]
- Faverio, C.; Boselli, M.F.; Medici, F.; Benaglia, M. Ammonia borane as a reducing agent in organic synthesis. Bioinorg. Chem. Appl. 2020, 18, 7789–7813. [Google Scholar] [CrossRef]
- Karthikeyan, S.; Sedlak, R.; Hobza, P. On the nature of stabilization in weak, medium, and strong charge-transfer complexes: CCSD(T)/CBS and SAPT calculations. J. Phys. Chem. A 2011, 115, 9422–9428. [Google Scholar] [CrossRef]
- Lo, R.; Manna, D.; Lamanec, M.; Dračínský, M.; Bouř, P.; Wu, T.; Bastien, G.; Kaleta, J.; Miriyala, M.V.; VŠpirko, V.; et al. The stability of covalent dative bond significantly increases with increasing solvent polarity. Nat. Commun. 2022, 13, 2107. [Google Scholar] [CrossRef]
- Funke, M.-A.; Mayr, H. Kinetics and mechanism of the reactions of amine boranes with carbenium ions. Chem. Eur. J. 1997, 3, 1214–1222. [Google Scholar] [CrossRef]
- Gohzu, S.; Tada, M. Regioselective reduction of polyketones on silica gel surface with borane–trimethylamine complex. Chem. Lett. 1986, 15, 61–64. [Google Scholar] [CrossRef]
- Sarko, C.R.; Guch, I.C.; DiMare, M. Chelation-controlled protocol for the diastereoselective reduction of ketones. J. Org. Chem. 1994, 59, 705–706. [Google Scholar] [CrossRef]
- Zhang, Y.; Ye, Q.; Ponomareva, L.V.; Cao, Y.; Liu, Y.; Cui, Z.; Van Lanen, S.G.; Voss, S.R.; She, Q.-B.; Thorson, J.S. Total synthesis of griseusins and elucidation of the griseusin mechanism of action. Chem. Sci. 2019, 10, 7641–7648. [Google Scholar] [CrossRef]
- Le Corre, M.; Gheerbrant, E.; Le Deit, H. Trimethylamine–borane bromide as alternative reagent for reductive bromation of aromatic carbonyl compounds. J. Chem. Soc. Chem. Commun. 1989, 5, 313–314. [Google Scholar] [CrossRef]
- Malmquist, J.; Ström, P. Multiple labeling of a potent CX3CR1 antagonist for the treatment of multiple sclerosis. J. Label. Compd. Radiopharm. 2012, 55, 387–392. [Google Scholar] [CrossRef]
- Stengel, I.; Götz, G.; Weil, M.; Bäuerle, P. A dinuclear (Bpy)Pt II -decorated crownophane. Eur. J. Org. Chem. 2015, 18, 3887–3893. [Google Scholar] [CrossRef]
- Trapani, G.; Reho, A.; Latrofa, A.; Liso, G. Trimethylamine-borane a useful reagent in the one-pot preparation of carboxylic esters from carboxylic acids. Synthesis 1990, 9, 853–854. [Google Scholar] [CrossRef]
- Trapani, G.; Reho, A.; Latrofa, A. Trimethylamine-borane as useful reagent in the N-acylation or N-alkylation of amines by carboxylic acids. Synthesis 1983, 12, 1013–1014. [Google Scholar] [CrossRef]
- Ménard, G.; Stephan, D.W. CO2 reduction via aluminum complexes of ammonia boranes. Dalton Trans. 2013, 42, 5447–5453. [Google Scholar] [CrossRef]
- Roy, L.; Ghosh, B.; Paul, A. Lewis acid promoted hydrogenation of CO2 and HCOO—By amine boranes: Mechanistic insight from a computational approach. J. Phys. Chem. A 2017, 121, 5204–5216. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, S.; Xi, C. α-Methylation of 2-arylacetonitrile by a trimethylamine-borane-CO2 system. J. Org. Chem. 2019, 84, 9744–9749. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, H.; Gao, K. Borane–trimethylamine complex as a reducing agent for selective methylation and formylation of amines with CO2. Org. Lett. 2021, 23, 8282–8286. [Google Scholar] [CrossRef]
- Xiong, F.; Cheng, Q.; Dang, Y.; Gao, K. A tandem reduction of primary amines, carbonyl compounds, CO2, and boranes catalyzed by in situ formed frustrated lewis pairs. Org. Chem. Front. 2022, 9, 4882–4889. [Google Scholar] [CrossRef]
- Perdicchia, D.; Licandro, E.; Maiorana, S.; Baldoli, C.; Giannini, C. A new ‘one-pot’ synthesis of hydrazides by reduction of hydrazones. Tetrahedron 2003, 59, 7733–7742. [Google Scholar] [CrossRef]
- Perdicchia, D. Ionic hydrogenation of azines: An efficient synthesis of 1,2-dialkylhydrazines. Tetrahedron 2023, 139, 133432. [Google Scholar] [CrossRef]
- Plate, R.; Hermkens, P.H.H.; Smits, J.M.M.; Ottenheijm, H.C.G. Nitrone cycloaddition in the stereoselective synthesis of β-carbolines from N-hydroxytryptophan. J. Org. Chem. 1986, 51, 309–314. [Google Scholar] [CrossRef]
- Plate, R.; Hermkens, P.H.H.; Smits, J.M.M.; Nivard, R.J.F.; Ottenheijm, H.C.G. Employment of nitriles in the stereoselective cycloaddition to nitrones. J. Org. Chem. 1987, 52, 1047–1051. [Google Scholar] [CrossRef]
- Hermkens, P.H.H.; Van Maarseveen, J.H.; Berens, H.W.; Smits, J.M.M.; Kruse, C.G.; Scheeren, H.W. Intramolecular Pictet-Spengler reaction of N-alkoxytryptophans and tryptamines. 2. Synthesis of corynanthe alkaloid derivatives containing a tetrahydro-1,2-oxazine as the D ring. J. Org. Chem. 1990, 55, 2200–2206. [Google Scholar] [CrossRef]
- Hermkens, P.H.H.; Van Maarseveen, J.H.; Cobben, P.L.H.M.; Ottenheijm, H.C.G.; Kruse, C.G.; Scheeren, H.W. Syntheses of 1,3-disubstituted N-oxy-β-carbolines by the Pictet-Spengler reactions of N-oxy-tryptophan and -tryptamine derivatives. Tetrahedron 1990, 46, 833–846. [Google Scholar] [CrossRef]
- Plate, R.; Ottenheijm, H.C.G. Synthesis of 2-(dimethylallyl)-N-hydroxytryptophans from indole. Tetrahedron Lett. 1986, 27, 3755–3758. [Google Scholar] [CrossRef]
- Plate, R.; Nivard, R.J.F.; Ottenheijm, H.C.G. Conversion of N-hydroxytryptophans into α,β-dehydrotryptophan. An approach to the neoechinulin series. J. Chem. Soc. Perkin Trans. 1 1987, 2473–2480. [Google Scholar] [CrossRef]
- Pham, T.L.; Sae-Lao, P.; Toh, H.H.M.; Csókás, D.; Bates, R.W. The total synthesis of Raistrickindole A. J. Org. Chem. 2022, 87, 16111–16114. [Google Scholar] [CrossRef]
- Lawrence, J.; Cointeaux, L.; Maire, P.; Vallée, Y.; Blandin, V. N-Hydroxy and N-acyloxy peptides: Synthesis and chemical modifications. Org. Biomol. Chem. 2006, 4, 3125–3141. [Google Scholar] [CrossRef]
- Langenhan, J.M.; Endo, M.M.; Engle, J.M.; Fukumoto, L.L.; Rogalsky, D.R.; Slevin, L.K.; Fay, L.R.; Lucker, R.W.; Rohlfing, J.R.; Smith, K.R.; et al. Synthesis and biological evaluation of RON-neoglycosides as tumor cytotoxins. Carbohydr. Res. 2011, 346, 2663–2676. [Google Scholar] [CrossRef]
- Goff, R.D.; Thorson, J.S. Enhancing the divergent activities of betulinic acid via neoglycosylation. Org. Lett. 2009, 11, 461–464. [Google Scholar] [CrossRef]
- Zhang, J.; Ponomareva, L.V.; Marchillo, K.; Zhou, M.; Andes, D.R.; Thorson, J.S. The synthesis and antibacterial activity of doxycycline neoglycosides. J. Nat. Prod. 2013, 76, 1627–1636. [Google Scholar] [CrossRef]
- Nielsen, S.D.; Smith, G.; Begtrup, M.; Kristensen, J.L. Synthesis and application of a new fluorous-tagged ammonia equivalent. Chem. Eur. J. 2010, 16, 4557–4566. [Google Scholar] [CrossRef]
- Heusler, A.; Fliege, J.; Wagener, T.; Glorius, F. Substituted dihydropyridine synthesis by dearomatization of pyridines. Angew. Chem. Int. Ed. 2021, 60, 13793–13797. [Google Scholar] [CrossRef]
- Zeng, Y.-F.; Li, Y.-N.; Zhou, M.-X.; Han, S.; Guo, Y.; Wang, Z. Metal-free hydrogenation of N-heterocycles with trimethylamine borane and TFA in aqueous solution. Adv. Synth. Catal. 2022, 364, 3664–3669. [Google Scholar] [CrossRef]
- Zeng, Y.-F.; Zhou, M.-X.; Li, Y.-N.; Wu, X.; Guo, Y.; Wang, Z. Switchable reductive N-trifluoroethylation and N-trifluoroacetylation of indoles with trifluoroacetic acid and trimethylamine borane. Org. Lett. 2022, 24, 7440–7445. [Google Scholar] [CrossRef]
- Li, Y.-N.; Zhou, M.-X.; Wu, J.-B.; Wang, Z.; Zeng, Y.-F. Tandem reduction and trifluoroethylation of quinolines and quinoxalines with trifluoroacetic acid and trimethylamine borane. Org. Biomol. Chem. 2022, 20, 9613–9617. [Google Scholar] [CrossRef]
- Meng, J.; Xia, H.M.; Xu, A.-Q.; Wang, Y.-F.; Wang, Z.; Zhang, F.-L. Selective N-monomethylation of primary anilines with the controllable installation of N-CH2D, N-CHD2, and N-CD3 units. Org. Biomol. Chem. 2020, 18, 4922–4926. [Google Scholar] [CrossRef]
- Couturier, M.; Tucker, J.L.; Andresen, B.M.; Dubé, P.; Brenek, S.J.; Negri, J.T. Palladium catalyzed activation of borane–amine adducts: Rate enhancement of amine–borane methanolysis in the reduction of nitrobenzenes to anilines. Tetrahedron Lett. 2001, 42, 2285–2288. [Google Scholar] [CrossRef]
- Beaudin, J.; Bourassa, D.E.; Bowles, P.; Castaldi, M.J.; Clay, R.; Couturier, M.A.; Karrick, G.; Makowski, T.W.; McDermott, R.E.; Meltz, C.N.; et al. Synthesis and purification of 6-ethoxy-4-oxo-1,4-dihydro-[1,5]naphthyridine-3-carboxylic acid benzylamide. Org Process Res Dev 2003, 7, 873–878. [Google Scholar] [CrossRef]
- Klapars, E.A.V.; Naidu, B.N.; Piotrowski, D.W.; Tucci, F.C. Enantiocontrolled synthesis of (1S,2S)-6-desmethyl-(methylaziridino)mitosene. J. Am. Chem. Soc. 2000, 122, 5401–5402. [Google Scholar] [CrossRef]
- Vedejs, E.; Naidu, B.N.; Klapars, A.; Warner, D.L.; Li, V.-S.; Na, Y.; Kohn, H. Synthetic enantiopure aziridinomitosenes: Preparation, reactivity, and DNA alkylation studies. J. Am. Chem. Soc. 2003, 125, 15796–157806. [Google Scholar] [CrossRef]
- Vedejs, E.; Klapars, A.; Warner, D.L.; Weiss, A.H. Reductive deprotection of N-tritylaziridines. J. Org. Chem. 2001, 66, 7542–7546. [Google Scholar] [CrossRef]
- Park, J.-J.; Lee, J.H.; Li, Q.; Diaz, K.; Chang, Y.-T.; Chung, S.-K. Divergent syntheses of all stereoisomers of phytosphingosine and their use in the construction of a ceramide library. Bioorg. Chem. 2008, 36, 220–228. [Google Scholar] [CrossRef]
- Zhang, Z.-Q.; Sang, Y.-Q.; Wang, C.-Q.; Dai, P.; Xue, X.-S.; Piper, J.L.; Peng, Z.-H.; Ma, J.-A.; Zhang, F.-G.; Wu, J. Difluoromethylation of unactivated alkenes using freon-22 through tertiary amine-borane-triggered halogen atom transfer. J. Am. Chem. Soc. 2022, 144, 14288–14296. [Google Scholar] [CrossRef]
- Ek, M.; Garegg, P.J.; Hultberg, H.; Oscarson, S. Reductive ring openings of carbohydrate benzylidene acetals using borane-trimethylamine and aluminium chloride. Regioselectivity and solvent dependance. J. Carbohydr. Chem. 1983, 2, 305–311. [Google Scholar] [CrossRef]
- Fügedi, P.; Birberg, W.; Garegg, P.J.; Pilotti, A. Syntheses of a branched heptasaccharide having phytoalexin-elicitor activity. Carbohydr. Res. 1987, 164, 297–312. [Google Scholar] [CrossRef]
- Sato, S.; Nunomura, S.; Nakano, T.; Ito, Y.; Ogawa, T. An efficient approach to stereoselective glycosylation of ceramide derivatives: Use of pivaloyl group as a stereocontrolling auxiliary. Tetrahedron Lett. 1988, 29, 4097–4100. [Google Scholar] [CrossRef]
- Garegg, P.J. Saccharides of biological importance: Challenges and opportunities for organic synthesis. Acc. Chem. Res. 1992, 25, 575–580. [Google Scholar] [CrossRef]
- Garegg, P.J.; Olsson, L.; Oscarson, S. Synthesis of methyl (ethyl 2-O-acyl-3,4-di-O-benzyl-1-thio-β-D-glucopyranosid)uronates and evaluation of their use as reactive β-selective glucuronic acid donors. J. Org. Chem. 1995, 60, 2200–2204. [Google Scholar] [CrossRef]
- Karst, N.; Jacquinet, J.-C. Chemical synthesis of β-D-GlcpA(2SO4)-(1→3)-D-GalpNAc(6SO4), the disaccharide repeating unit of shark cartilage chondroitin sulfate D, and of its methyl β-D-glycoside derivative. J. Chem. Soc. Perkin Trans. 1 2000, 16, 2709–2717. [Google Scholar] [CrossRef]
- Svansson, L.; Johnston, B.D.; Gu, J.-H.; Patrick, B.; Pinto, B.M. Synthesis and conformational analysis of a sulfonium-ion analogue of the glycosidase inhibitor castanospermine. J. Am. Chem. Soc. 2000, 122, 10769–10775. [Google Scholar] [CrossRef]
- Li, X.; Ohtake, H.; Takahashi, H.; Ikegami, S. Direct methylenation of partially benzyl-protected sugar lactones by dimethyltitanocene. Synlett 2001, 12, 1885–1888. [Google Scholar] [CrossRef]
- Hoffmann, B.; Zanini, D.; Ripoche, I.; Bürli, R.; Vasella, A. Oligosaccharide analogues of polysaccharides, part 22, synthesis of cyclodextrin analogues containing a buta-1,3-diyne-1,4-diyl or a butane-1,4-diyl unit. Helv. Chim. Acta 2001, 84, 1862–1888. [Google Scholar] [CrossRef]
- Sherman, A.A.; Yudina, O.N.; Mironov, Y.V.; Sukhova, E.V.; Shashkov, A.S.; Menshov, V.M.; Nifantiev, N.E. Study of glycosylation with N-trichloroacetyl-D-glucosamine derivatives in the syntheses of the spacer-armed pentasaccharides sialyl lacto-N-neotetraose and sialyl lacto-N-tetraose, their fragments, and analogues. Carbohydr. Res. 2001, 336, 13–46. [Google Scholar] [CrossRef]
- Kitov, P.I.; Bundle, D.R. Synthesis and structure–activity relationships of di- and trisaccharide inhibitors for Shiga-like toxin type 1. J. Chem. Soc. Perkin Trans. 1 2001, 8, 838–853. [Google Scholar] [CrossRef]
- Sherman, A.A.; Yudina, O.N.; Shashkov, A.S.; Menshov, V.M.; Nifant’ev, N.E. Synthesis of Neu5Ac- and Neu5Gc-α-(2→6′)-lactosamine 3-aminopropyl glycosides. Carbohydr. Res. 2001, 330, 445–458. [Google Scholar] [CrossRef] [PubMed]
- Amaya, T.; Tanaka, H.; Yamaguchi, T.; Naoto Shibuya, N.; Takahashi, T. The first synthesis of tetraglucosyl glucitol having phytoalexin-elicitor activity in rice cells based on a sequential glycosylation strategy. Tetrahedron Lett. 2001, 42, 9191–9194. [Google Scholar] [CrossRef]
- Chowdhury, A.R.; Siriwardena, A.; Boons, G.-J. A highly convergent approach for the synthesis of disaccharide repeating units of peptidoglycan. Tetrahedron Lett. 2002, 43, 7805–7807. [Google Scholar] [CrossRef]
- Elsayed, G.A.; Zhu, T.; Boons, G.-J. Demixing libraries of saccharides using a multi-linker approach in combination with a soluble polymeric support. Tetrahedron Lett. 2002, 43, 4691–4694. [Google Scholar] [CrossRef]
- Dohi, H.; Nishida, Y.; Furuta, Y.; Uzawa, H.; Yokoyama, S.-I.; Ito, S.; Mori, H.; Kobayashi, K. Molecular design and biological potential of galacto-type trehalose as a nonnatural ligand of Shiga toxins. Org. Lett. 2002, 4, 355–357. [Google Scholar] [CrossRef]
- Manabe, S.; Ito, Y. On-resin real-time reaction monitoring of solid-phase oligosaccharide synthesis. J. Am. Chem. Soc. 2002, 124, 12638–12639. [Google Scholar] [CrossRef]
- Takahashi, T.; Okano, A.; Amaya, T.; Tanaka, H.; Doi, T. Solid-Phase Synthesis of a phytoalexin elicitor-active tetraglucosyl glucitol. Synlett 2002, 6, 911–914. [Google Scholar] [CrossRef]
- Kanemitsu, T.; Wong, C.-H.; Kanie, O. Solid-phase synthesis of oligosaccharides and on-resin quantitative monitoring using gated decoupling 13C NMR. J. Am. Chem. Soc. 2002, 124, 3591–3599. [Google Scholar] [CrossRef]
- Ágoston, K.; Kerékgyártó, J.; Hajkó, J.; Batta, G.; Lefeber, D.J.; Kamerling, J.P.; Vliegenthart, J.F.G. Synthesis of fragments of the glycocalyx glycan of the Parasiteschistosoma mansoni. Chem. Eur. J. 2002, 8, 151–161. [Google Scholar] [CrossRef]
- Tanaka, H.; Amaya, T.; Takahashi, T. Parallel synthesis of multi-branched oligosaccharides related to elicitor active pentasaccharide in rice cell based on orthogonal deprotection and glycosylation strategy. Tetrahedron Lett. 2003, 44, 3053–3057. [Google Scholar] [CrossRef]
- Alpe, M.; Oscarson, S.; Svahnberg, P. Synthesis of Cryptococcus neoformans capsular polysaccharide structures. IV. Construction of thioglycoside donor blocks and their subsequent assembly. J. Carbohydr. Chem. 2003, 22, 565–577. [Google Scholar] [CrossRef]
- Tarling, C.A.; Withers, S.G. The synthesis of a series of modified mannotrisaccharides as probes of the enzymes involved in the early stages of mammalian complex N-glycan formation. Carbohydr. Res. 2004, 339, 2487–2497. [Google Scholar] [CrossRef]
- Tsvetkov, Y.E.; Nifantiev, N.E. Enhanced sialylating activity of O-chloroacetylated 2-thioethyl sialosides. Synlett 2005, 9, 1375–1380. [Google Scholar] [CrossRef]
- Wang, J.; Li, J.; Chen, H.-N.; Chang, H.; Tanifum, C.T.; Liu, H.-H.; Czyryca, P.G.; Chang, C.-W.T. Glycodiversification for the optimization of the Kanamycin class aminoglycosides. J. Med. Chem. 2005, 48, 6271–6285. [Google Scholar] [CrossRef]
- Veselý, J.; Rohlenová, A.; Džoganová, M.; Trnka, T.; Tišlerová, I.; Šaman, D.; Ledvina, M. Preparation of ethyl 2-azido-2-deoxy-1-thio-β-D-mannopyranosides, and their rearrangement to 2-S-ethyl-2-thio-β-D-mannopyranosylamines. Synthesis 2006, 4, 699–705. [Google Scholar] [CrossRef]
- Khatuntseva, E.A.; Tsvetkov, Y.E.; Grachev, A.A.; Nifant’ev, N.E. Synthesis of aminoethyl glycosides of type 2 chain A tetrasaccharide and related trisaccharides. Russ. J. Org. Chem. 2005, 41, 1814–1823. [Google Scholar] [CrossRef]
- Matsuoka, K.; Goshu, Y.; Takezawa, Y.; Mori, T.; Sakamoto, J.-I.; Yamada, A.; Onaga, T.; Koyama, T.; Hatano, K.; Snyder, P.W.; et al. Practical synthesis of fully protected globotriaose and its glycopolymers. Carbohydr. Polym. 2007, 69, 326–335. [Google Scholar] [CrossRef]
- Yamada, A.; Hatano, K.; Matsuoka, K.; Koyama, T.; Esumi, Y.; Koshino, H.; Hino, K.; Nishikawa, K.; Natori, Y.; Terunuma, D. Syntheses and Vero toxin-binding activities of carbosilane dendrimers periphery-functionalized with galabiose. Tetrahedron 2006, 62, 5074–5083. [Google Scholar] [CrossRef]
- Komarova, B.S.; Tsvetkov, Y.E.; Knirel, Y.A.; Zähringer, U.; Pier, G.B.; Nifantiev, N.E. Synthesis of a common trisaccharide fragment of glycoforms of the outer core region of the Pseudomonas aeruginosa lipopolysaccharide. Tetrahedron Lett. 2006, 47, 3583–3587. [Google Scholar] [CrossRef]
- Nakano, J.; Akihiro Ishiwata, A.; Ohta, H.; Ito, Y. Synthesis of complex-type glycans derived from parasitic helminths. Carbohydr. Res. 2007, 342, 675–695. [Google Scholar] [CrossRef]
- Sukhova, E.V.; Dubrovskii, A.V.; Tsvetkov, Y.E.; Nifantiev, N.E. Synthesis of oligosaccharides related to the HNK-1 antigen. 5. Synthesis of a sulfo-mimetic of the HNK-1 antigenic trisaccharide. Russ. Chem. Bull. 2007, 56, 1655–1670. [Google Scholar] [CrossRef]
- Tatai, J.; Osztrovszky, G.; Kajtár-Peredy, M.; Fügedi, P. An efficient synthesis of L-idose and L-iduronic acid thioglycosides and their use for the synthesis of heparin oligosaccharides. Carbohydr. Res. 2008, 343, 596–606. [Google Scholar] [CrossRef]
- Komarova, B.S.; Tsvetkov, Y.E.; Pier, G.B.; Nifantiev, N.E. First Synthesis of pentasaccharide glycoform I of the outer core region of the Pseudomonas aeruginosa lipopolysaccharide. J. Org. Chem. 2008, 73, 8411–8421. [Google Scholar] [CrossRef]
- Lin, Y.-S.; Tungpradit, R.; Sinchaikul, S.; An, F.-M.; Liu, D.-Z.; Phutrakul, S.; Chen, S.-T. Targeting the delivery of glycan-based paclitaxel prodrugs to cancer cells via glucose transporters. J. Med. Chem. 2008, 51, 7428–7441. [Google Scholar] [CrossRef]
- Schwardt, O.; Gäthje, H.; Vedani, A.; Mesch, S.; Gao, G.-P.; Spreafico, M.; von Orelli, J.; Kelm, S.; Ernst, B.J. Examination of the Biological Role of the α (2→ 6)-Linked Sialic Acid in Gangliosides Binding to the Myelin-Associated Glycoprotein (MAG). Med. Chem. 2009, 52, 989–1004. [Google Scholar] [CrossRef]
- Rasmussen, T.S.; Jensen, H.H. Chiral pool synthesis of calystegine A3 from 2-deoxyglucose via a Brown allylation. Carbohydr. Res. 2011, 346, 2855–2861. [Google Scholar] [CrossRef]
- Titz, A.; Marra, A.; Cutting, B.; Smieško, M.; Papandreou, G.; Dondoni, A.; Ernst, B. Conformational constraints: Nature does it best with sialyl Lewisx. Eur. J. Org. Chem. 2012, 28, 5534–5539. [Google Scholar] [CrossRef]
- Xia, L.; Zheng, R.B.; Lowary, T.L. Revisiting the specificity of an α-(1→4)-mannosyltransferase involved in mycobacterial methylmannose polysaccharide biosynthesis. ChemBioChem 2012, 13, 1139–1151. [Google Scholar] [CrossRef]
- Tamura, J.-I.; Tsutsumishita-Nakai, N.; Nakao, Y.; Kawano, M.; Kato, S.; Takeda, N.; Nadanaka, S.; Kitagawa, H. Synthesis and interaction with Midkine of biotinylated chondroitin sulfate tetrasaccharides. Bioorg. Med. Chem. Lett. 2012, 22, 1371–1374. [Google Scholar] [CrossRef]
- Takeda, N.; Ikeda-Matsumi, R.; Ebara-Nagahara, K.; Otaki-Nanjo, M.; Taniguchi-Morita, K.; Nanjo, M.; Tamura, J.-I. Synthesis of heparan sulfate tetrasaccharide as a substrate for human heparanase. Carbohydr. Res. 2012, 353, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Komarova, B.S.; Tsvetkov, Y.E.; Pier, G.P.; Nifantiev, N.E. Synthesis of pentasaccharides corresponding to the glycoform II of the outer core region of the Pseudomonas aeruginosa lipopolysaccharide. Carbohydr. Res. 2012, 360, 56–68. [Google Scholar] [CrossRef]
- Zhang, J.; Zou, L.; Lowary, T.L. Synthesis of the tolerance-inducing oligosaccharide lacto-N-fucopentaose III bearing an activated linker. ChemistryOpen 2013, 2, 156–163. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, Y.; Koshino, H.; Nakamura, T.; Tsuchida, A.; Nitoda, T.; Kanzaki, H.; Matsuoka, K.; Takahashi, S. Total synthesis of the proposed structure for pochonicine and determination of its absolute configuration. Tetrahedron Lett. 2013, 54, 1456–1459. [Google Scholar] [CrossRef]
- Yudina, O.N.; Tsvetkov, Y.E.; Nifantiev, N.E. Synthesis of 2-aminoethyl glycosides of chitooligosaccharides. Russ. Chem. Bull. 2015, 64, 2932–2941. [Google Scholar] [CrossRef]
- Fan, Q.-H.; Pickens, J.B.; Striegler, S.; Gervaise, C.D. Illuminating the binding interactions of galactonoamidines during the inhibition of β-galactosidase (E. coli). Bioorg. Med. Chem. 2016, 24, 661–671. [Google Scholar] [CrossRef] [PubMed]
- d’Ortoli, T.A.; Widmalm, G. Synthesis of the tetrasaccharide glycoside moiety of Solaradixine and rapid NMR-based structure verification using the program CASPER. Tetrahedron 2016, 72, 912–927. [Google Scholar] [CrossRef]
- Vinnitskiy, D.Z.; Ustyuzhanina, N.E.; Andrey, S.D.; Shashkov, A.S.; Nifantiev, N.E. Synthesis and NMR analysis of model compounds related to fucosylated chondroitin sulfates: GalNAc and Fuc(1 → 6)GalNAc derivatives. Carbohydr. Res. 2017, 438, 9–17. [Google Scholar] [CrossRef]
- Tsvetkov, Y.E.; Yashunsky, D.V.; Sukhova, E.V.; Kurbatova, E.A.; Nifantiev, N.E. Synthesis of oligosaccharides structurally related to fragments of Streptococcus pneumoniae type 3 capsular polysaccharide. Russ. Chem. Bull. 2017, 66, 111–122. [Google Scholar] [CrossRef]
- Demeter, F.; Veres, F.; Herczeg, M.; Borbás, A. Short synthesis of Idraparinux by applying a 2-O-Methyl-4,6-O-arylmethylene thioidoside as a 1,2-trans-α-selective glycosyl donor. Eur. J. Org. Chem. 2018, 48, 6901–6912. [Google Scholar] [CrossRef]
- Tateda, N.; Ajisaka, K.; Ishiguro, M.; Miyazaki, T. Synthesis of 5a,5a′-dicarba-D-glucobioses from conformationally restricted carbaglucosyl triflates using SN2-type inversion with carbaglucosyl nucleophiles. Bioorg. Med. Chem. 2019, 27, 2345–2367. [Google Scholar] [CrossRef]
- Dhaene, S.; Van Der Eycken, J.; Beerens, K.; Franceus, J.; Desmet, T.; Caroen, J. Synthesis, trehalase hydrolytic resistance and inhibition properties of 4- and 6-substituted trehalose derivatives. J. Enzyme Inhib. Med. Chem. 2020, 35, 1964–1989. [Google Scholar] [CrossRef]
- Inamura, S.; Fukase, K.; Kusumoto, S. Synthetic study of peptidoglycan partial structures. synthesis of tetrasaccharide and octasaccharide fragments. Tetrahedron Lett. 2001, 42, 7613–7616. [Google Scholar] [CrossRef]
- Hesek, D.; Lee, M.; Morio, K.-I.; Mobashery, S. Synthesis of a fragment of bacterial cell wall. J. Org. Chem. 2004, 69, 2137–2146. [Google Scholar] [CrossRef]
- Inamura, S.; Fujimoto, Y.; Kawasaki, A.; Shiokawa, Z.; Woelk, E.; Heine, H.; Lindner, B.; Inohara, N.; Kusumoto, S.; Fukase, K. Synthesis of peptidoglycan fragments and evaluation of their biological activity. Org. Biomol. Chem. 2006, 4, 232–242. [Google Scholar] [CrossRef] [PubMed]
- Bohn, M.L.; Colombo, M.I.; Stortz, C.A.; Rúveda, E.A. A comparative study of the influence of some protecting groups on the reactivity of D-glucosamine acceptors with a galactofuranosyl donor. Carbohydr. Res. 2006, 341, 1096–1104. [Google Scholar] [CrossRef]
- Bohn, M.L.; Colombo, M.I.; Pisano, P.L.; Stortz, C.A.; Rúveda, E.A. Differential O-3/O-4 regioselectivity in the glycosylation of α and β anomers of 6-O-substituted N-dimethylmaleoyl-protected D-glucosamine acceptors. Carbohydr. Res. 2007, 342, 2522–2536. [Google Scholar] [CrossRef]
- Fujimoto, Y.; Konishi, Y.; Kubo, O.; Hasegawa, M.; Inohara, N.; Fukase, K. Synthesis of crosslinked peptidoglycan fragments for investigation of their immunobiological functions. Tetrahedron Lett. 2009, 50, 3631–3634. [Google Scholar] [CrossRef]
- Danieli, E.; Lay, L.; Proietti, D.; Berti, F.; Costantino, P.; Adamo, R. First synthesis of C. difficile PS-II cell wall polysaccharide repeating unit. Org. Lett. 2011, 13, 378–381. [Google Scholar] [CrossRef] [PubMed]
- Colombo, M.I.; Rúveda, E.A.; Stortz, C.A. Regioselectivity of the glycosylation of N-dimethylmaleoyl-protected hexosamine acceptors. An experimental and DFT approach. Org. Biomol. Chem. 2011, 9, 3020–3025. [Google Scholar] [CrossRef]
- Adamo, R.; Romano, M.R.; Berti, F.; Leuzzi, R.; Tontini, M.; Danieli, E.; Cappelletti, E.; Cakici, O.S.; Swennen, E.; Pinto, V.; et al. Phosphorylation of the synthetic hexasaccharide repeating unit is essential for the induction of antibodies to Clostridium difficile PSII cell wall polysaccharide. ACS Chem. Biol. 2012, 7, 1420–1428. [Google Scholar] [CrossRef]
- Wang, N.; Huang, C.-Y.; Hasegawa, M.; Inohara, N.; Fujimoto, Y.; Fukase, K. Glycan sequence-dependent Nod2 activation investigated by using a chemically synthesized bacterial peptidoglycan fragment library. ChemBioChem 2013, 14, 482–488. [Google Scholar] [CrossRef]
- Adamo, R.; Micoli, F.; Proietti, D.; Berti, F. Efficient synthesis of Meningococcal X polysaccharide repeating unit (N-acetylglucosamine-4-phosphate) as analytical standard for polysaccharide determination. Synth. Commun. 2014, 44, 1266–1273. [Google Scholar] [CrossRef]
- Enugala, R.; Pires, M.J.D.; Marques, M.M.B. Synthesis of the NAG–NAM disaccharide via a versatile intermediate. Carbohydr. Res. 2014, 384, 112–118. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Matsuo, Y.; Pradipta, A.R.; Inohara, N.; Fujimoto, Y.; Fukase, K. Synthesis of characteristic mycobacterium peptidoglycan (PGN) fragments utilizing with chemoenzymatic preparation of meso-diaminopimelic acid (DAP), and their modulation of innate immune responses. Org. Biomol. Chem. 2016, 14, 1013–1023. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Hasegawa, H.; Huang, C.-Y.; Fukase, K.; Fujimoto, Y. Synthesis of peptidoglycan fragments from Enterococcus faecalis with Fmoc-strategy for glycan elongation. Chem. Asian J. 2017, 12, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Del Bino, L.; Calloni, I.; Oldrini, D.; Raso, M.M.; Cuffaro, R.; Ardá, A.; Codée, J.D.C.; Jiménez-Barbero, J.; Adamo, R. Regioselective glycosylation strategies for the synthesis of group Ia and Ib Streptococcus related glycans enable elucidating unique conformations of the capsular polysaccharides. Chem. Eur. J. 2019, 25, 16277–16287. [Google Scholar] [CrossRef] [PubMed]
- Dallabernardina, P.; Benazzi, V.; Laman, J.D.; Seeberger, P.H.; Loeffler, F.F. Automated glycan assembly of peptidoglycan backbone fragments. Org. Biomol. Chem. 2021, 19, 9829–9832. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, M.; Dulina, R.G.; Kakarla, R.; Sofia, M.J. Efficient synthesis of a stereochemically defined carbohydrate scaffold: Carboxymethyl 2-acetamido-6-azido-4-O-benzyl-2-deoxy-α-D-glucopyranoside. J. Org. Chem. 2000, 65, 8387–8390. [Google Scholar] [CrossRef] [PubMed]
- Périon, R.; Lemée, L.; Ferrières, V.; Duval, R.; Plusquellec, D. A new synthesis of the oligosaccharide domain of acarbose. Carbohydr. Res. 2003, 338, 2779–2792. [Google Scholar] [CrossRef] [PubMed]
- Ando, H.; Koike, Y.; Koizumi, S.; Ishida, H.; Kiso, M. 1,5-Lactamized sialyl acceptors for various disialoside syntheses: Novel method for the synthesis of glycan portions of Hp-s6 and HLG-2 gangliosides. Angew. Chem. Int. Ed. 2005, 44, 6759–6763. [Google Scholar] [CrossRef]
- Ando, H.; Shimizu, H.; Katano, Y.; Koike, Y.; Koizumi, S.; Ishida, H.; Kiso, M. Studies on the α-(1→4)- and α-(1→8)-fucosylation of sialic acid for the total assembly of the glycan portions of complex HPG-series gangliosides». Carbohydr. Res. 2006, 341, 1522–1532. [Google Scholar] [CrossRef]
- Tanaka, H.; Nishiura, Y.; Adachi, M.; Takahashi, T. Synthetic study of α(2,8) oligosialoside using N-Troc sialyl N-phenyltrifluoroimidate. Heterocycles 2006, 67, 107–112. [Google Scholar] [CrossRef]
- Shelke, S.V.; Gao, G.-P.; Mesch, S.; Gäthje, H.; Kelm, S.; Schwardt, O.; Ernst, B. Synthesis of sialic acid derivatives as ligands for the myelin-associated glycoprotein (MAG). Bioorg. Med. Chem. 2007, 15, 4951–4965. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, H.; Nishiura, Y.; Takahashi, T. Stereoselective synthesis of α(2,9) di- to tetrasialic acids, using a 5,4-N,O-carbonyl protected thiosialoside. J. Org. Chem. 2009, 74, 4383–4386. [Google Scholar] [CrossRef]
- Hanashima, S.; Ishikawa, D.; Akai, S.; Sato, K.-I. Synthesis of the starfish ganglioside LLG-3 tetrasaccharide. Carbohydr. Res. 2009, 344, 747–752. [Google Scholar] [CrossRef]
- Meinke, S.; Schroven, A.; Thiem, J. Sialic acid C-glycosides with aromatic residues: Investigating enzyme binding and inhibition of Trypanosoma Cruzi trans-sialidase. Org. Biomol. Chem. 2011, 9, 4487–4497. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, H.; Iwayama, Y.; Imamura, A.; Ando, H.; Ishida, H.; Kiso, M. Synthesis of the disialic acid-embedded glycan part of ganglioside HPG-1. Biosci. Biotechnol. Biochem. 2011, 75, 2079–2082. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Schumann, B.; Pragani, R.; Anish, C.; Pereira, C.L.; Seeberger, P.H. Synthesis of conjugation-ready zwitterionic oligosaccharides by chemoselective thioglycoside activation. Chem. Sci. 2014, 5, 1992–2002. [Google Scholar] [CrossRef]
- Cheallaigh, N.A.; Oscarson, S. Synthesis of building blocks for an iterative approach towards oligomers of the Streptococcus Pneumoniae type 1 zwitterionic capsular polysaccharide repeating unit. Can. J. Chem. 2016, 94, 940–960. [Google Scholar] [CrossRef]
- Podvalnyy, N.M.; Malysheva, N.N.; Panova, M.V.; Zinin, A.I.; Chizhov, A.O.; Orlova, A.V.; Kononov, L.O. Stereoselective sialylation with O-trifluoroacetylated thiosialosides: Hydrogen bonding involved? Carbohydr. Res. 2017, 451, 12–28. [Google Scholar] [CrossRef]
- Shirasaki, J.; Tanaka, H.-N.; Konishi, M.; Hirose, Y.; Imamura, A.; Ishida, H.; Kiso, M.; Ando, H. Systematic strategy utilizing 1,5-lactamization for the synthesis of the trisialylated galactose unit of c-series gangliosides. Tetrahedron Lett. 2020, 61, 151759. [Google Scholar] [CrossRef]
- Wu, Y.-F.; Tsai, Y.-F.; Huang, Y.-S.; Shih, J.-F. Total synthesis of the echinodermatous ganglioside LLG-3 possessing the biological function of promoting the neurite outgrowth. Org. Lett. 2020, 22, 7491–7495. [Google Scholar] [CrossRef] [PubMed]
- Johansson, E.; Caraballo, R.; Elofsson, M. Synthesis of 4-O-alkylated N-acetylneuraminic acid derivatives. J. Org. Chem. 2021, 86, 9145–9154. [Google Scholar] [CrossRef]
- Hirschmann, R.; Ducry, L.; Smith, A.B. Development of an efficient, regio- and stereoselective route to libraries based on the β-D-glucose scaffold. J. Org. Chem. 2000, 65, 8307–8316. [Google Scholar] [CrossRef] [PubMed]
- Morii, Y.; Matsuda, H.; Ohara, K.; Hashimoto, M.; Miyairi, K.; Okuno, T. Synthetic studies on oligosaccharides composed of 5-thioglucopyranose units. Bioorg. Med. Chem. 2005, 13, 5113–5144. [Google Scholar] [CrossRef]
- Danieli, E.; Lalot, J.; Murphy, P.V. Selective protecting group manipulations on the 1-Deoxynojirimycin scaffold. Tetrahedron 2007, 63, 6827–6834. [Google Scholar] [CrossRef]
- Noguchi, S.; Takemoto, S.; Kidokoro, S.-I.; Yamamoto, K.; Hashimoto, M. Syntheses of cellotriose and cellotetraose analogues as transition state mimics for mechanistic studies of cellulases. Bioorg. Med. Chem. 2011, 19, 3812–3830. [Google Scholar] [CrossRef]
- Weïwer, M.; Chen, C.-C.; Kemp, M.M.; Linhardt, R.J. Synthesis and biological evaluation of non-hydrolyzable 1,2,3-triazole-linked sialic acid derivatives as neuraminidase inhibitors. Eur. J. Org. Chem. 2009, 16, 2611–2620. [Google Scholar] [CrossRef] [PubMed]
- Borbás, A.; Szabó, Z.B.; Szilágyi, L.; Bényei, A.; Lipták, A. Dioxane-type (2-naphthyl)methylene acetals of glycosides and their hydrogenolytic transformation into 6-O- and 4-O-(2-naphthyl)methyl (NAP) ethers. Tetrahedron 2002, 58, 5723–5732. [Google Scholar] [CrossRef]
- Tanaka, H.; Tateno, Y.; Takahashi, T. Convergent stereoselective synthesis of multiple sulfated GlcNα(1,4)GlcAβ(1,4) dodecasaccharides. Org. Biomol. Chem. 2012, 10, 9570–9582. [Google Scholar] [CrossRef]
- Oka, H.; Koyama, T.; Hatano, K.; Matsuoka, K. Synthetic studies of bi-fluorescence-labeled maltooligosaccharides as substrates for α-amylase on the basis of fluorescence resonance energy transfer (FRET). Bioorg. Med. Chem. 2012, 20, 435–445. [Google Scholar] [CrossRef]
- Takeda, N.; Tamura, J.-I. Synthesis of biotinylated keratan sulfate repeating disaccharides. Biosci. Biotechnol. Biochem. 2014, 78, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Takeda-Okuda, N.; Yamaguchi, Y.; Uzawa, J.; Tamura, J.-I. Synthesis of a biotinylated keratan sulfate tetrasaccharide composed of dimeric Galβ1-4GlcNAc6Sβ. Carbohydr. Res. 2017, 452, 97–107. [Google Scholar] [CrossRef]
- Herczeg, M.; Demeter, F.; Balogh, T.; Kelemen, V.; Borbás, A. Rapid synthesis ofL-idosyl glycosyl donors from α-thioglucosides for the preparation of heparin disaccharides. Eur. J. Org. Chem. 2018, 25, 3312–3316. [Google Scholar] [CrossRef]
- Lipták, A.; Borbás, A.; Jánossy, L.; Szilágyi, L. Preparation of (2-naphthyl)methylene acetals of glycosides and their hydrogenolytic transformation into 2-naphthylmethyl (NAP) ethers. Tetrahedron Lett. 2000, 41, 4949–4953. [Google Scholar] [CrossRef]
- Borbás, A.; Szabó, Z.B.; Szilágyi, L.; Bényei, A.; Lipták, A. Stereoselective (2-naphthyl)methylation of sugar hydroxyls by the hydrogenolysis of diastereoisomeric dioxolane-type (2-naphthyl)methylene acetals. Carbohydr. Res. 2002, 337, 1941–1951. [Google Scholar] [CrossRef]
- Aoyagi, T.; Ohira, S.; Fuse, S.; Uzawa, J.; Yamaguchi, Y.; Tanaka, H. The α-glycosidation of partially unprotected N-acetyl and N-glycolyl sialyl donors in the absence of a nitrile solvent effect. Chem. Eur. J. 2016, 22, 6968–6973. [Google Scholar] [CrossRef]
- Matsushita, K.; Sato, Y.; Funamoto, S.; Tamura, J.-I. Side reactions with 2,2,2-trichloroethoxysulfates during the synthesis of glycans. Carbohydr. Res. 2014, 396, 14–24. [Google Scholar] [CrossRef]
- Sherman, A.A.; Mironov, Y.V.; Yudina, O.N.; Nifantiev, N.E. The presence of water improves reductive openings of benzylidene acetals with trimethylaminoborane and aluminium chloride. Carbohydr. Res. 2003, 338, 697–703. [Google Scholar] [CrossRef] [PubMed]
- Johnsson, R.; Mani, K.; Cheng, F.; Ellervik, U. Regioselective reductive openings of acetals; mechanistic details and synthesis of fluorescently labeled compounds. J. Org. Chem. 2006, 71, 3444–3451. [Google Scholar] [CrossRef]
- Johnsson, R.; Olsson, D.; Ellervik, U. Reductive openings of acetals: Explanation of regioselectivity in borane reductions by mechanistic studies. J. Org. Chem. 2008, 73, 5226–5232. [Google Scholar] [CrossRef]
- Johnsson, R.; Cukalevski, R.; Dragén, F.; Ivanisevic, D.; Johansson, I.; Petersson, L.; Wettergren, E.E.; Yam, K.B.; Yang, B.; Ellervik, U. Reductive openings of benzylidene acetals. kinetic studies of borane and alane activation by lewis acids. Carbohydr. Res. 2008, 343, 2997–3000. [Google Scholar] [CrossRef] [PubMed]
- Johnsson, R.; Ohlin, M.; Ellervik, U. Reductive openings of benzylidene acetals revisited: A mechanistic scheme for regio- and stereoselectivity. J. Org. Chem. 2010, 75, 8003–8011. [Google Scholar] [CrossRef] [PubMed]









































Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).