


tert-Butyl Hypochlorite: A Reagent for the Synthesis of Chlorinated Oxindole and Indole Derivatives


Abstract
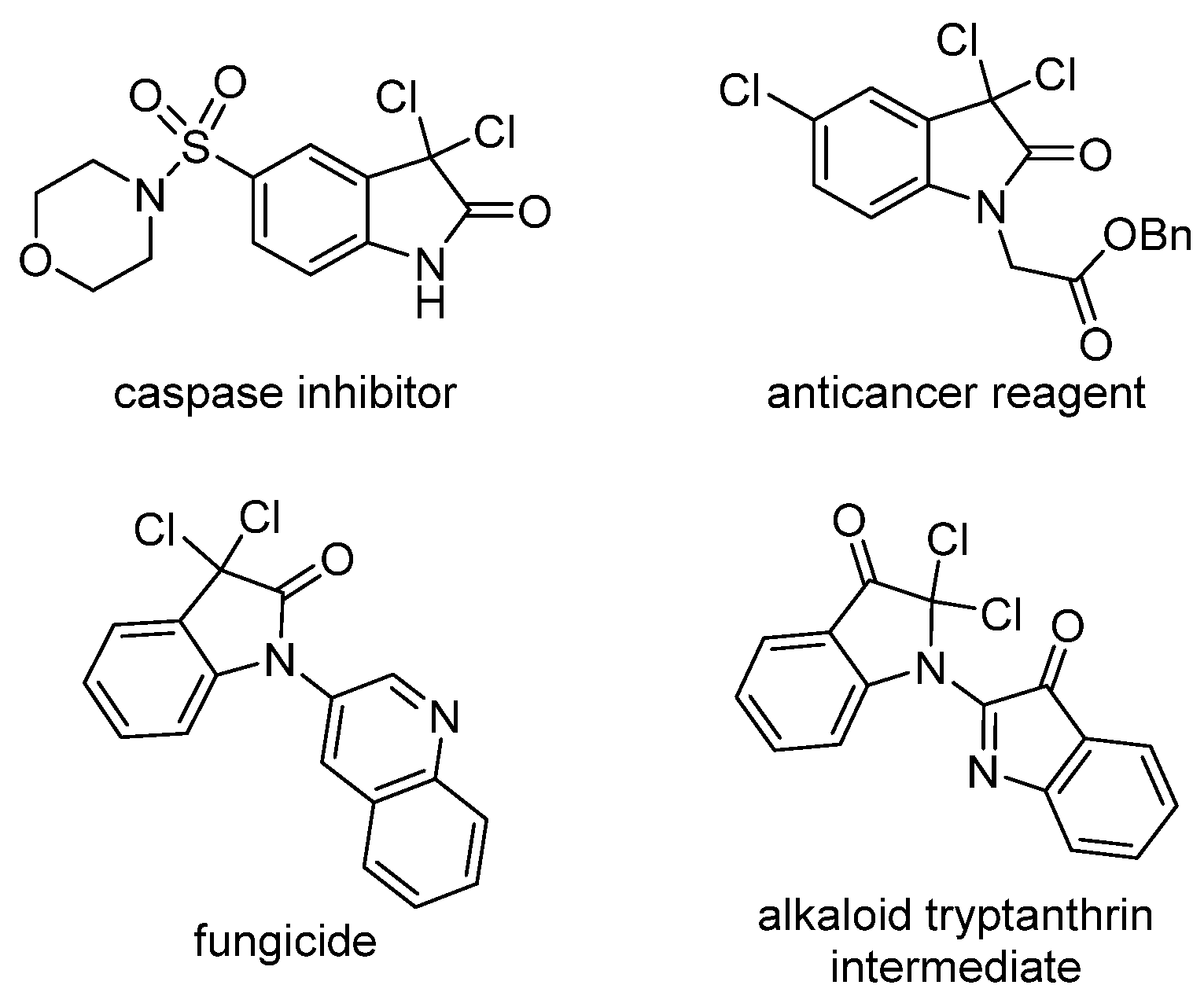
1. Introduction
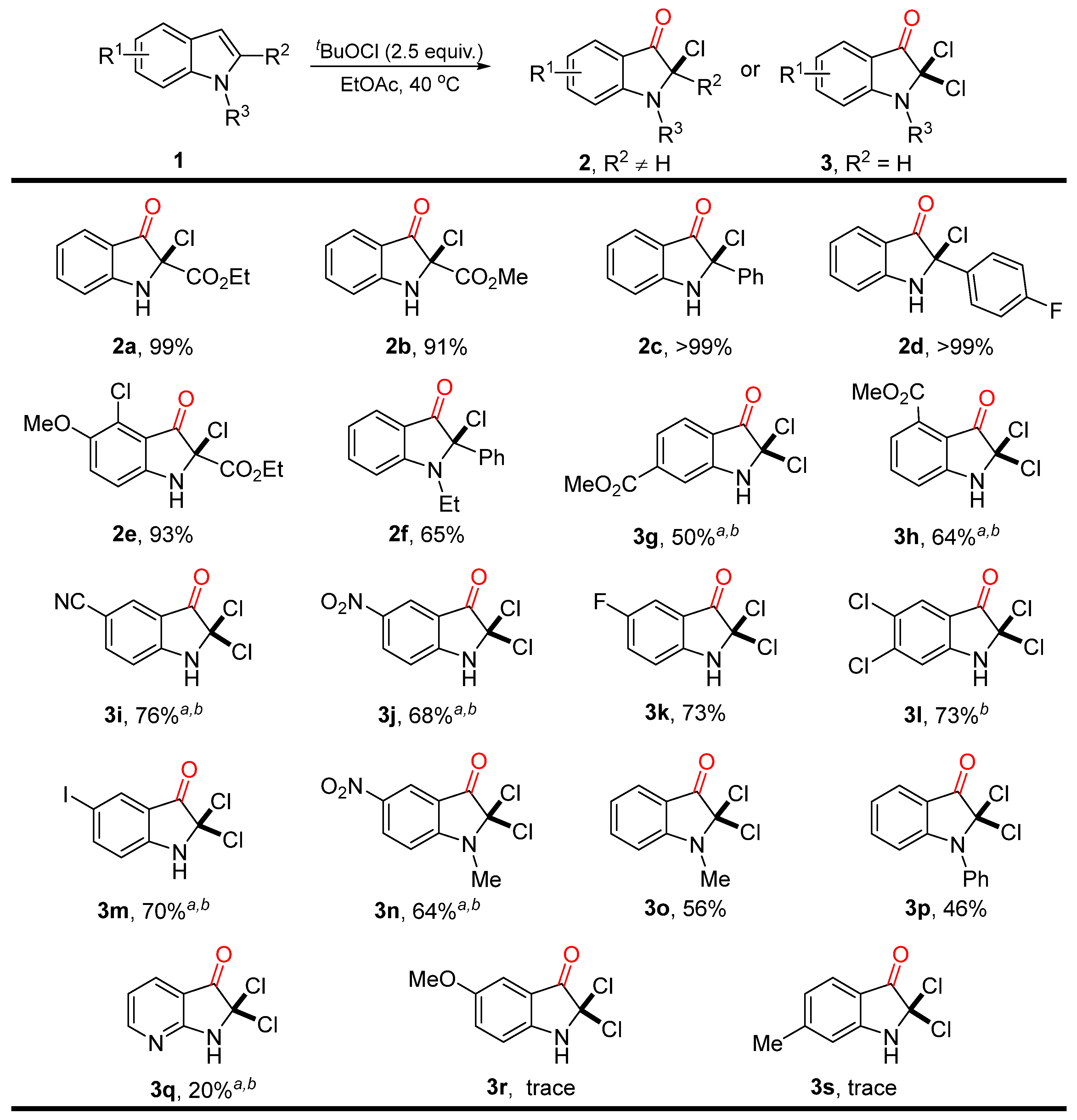
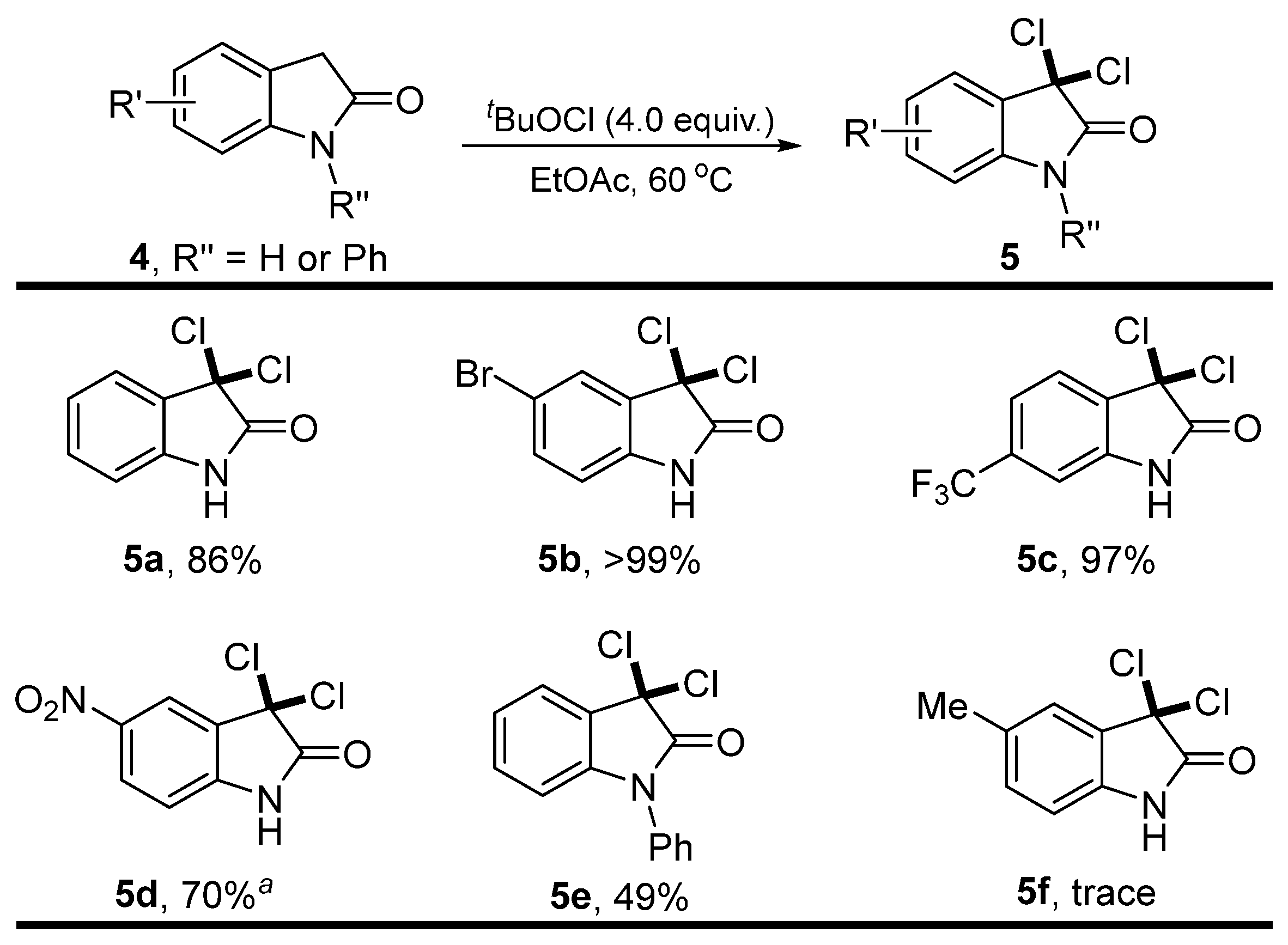
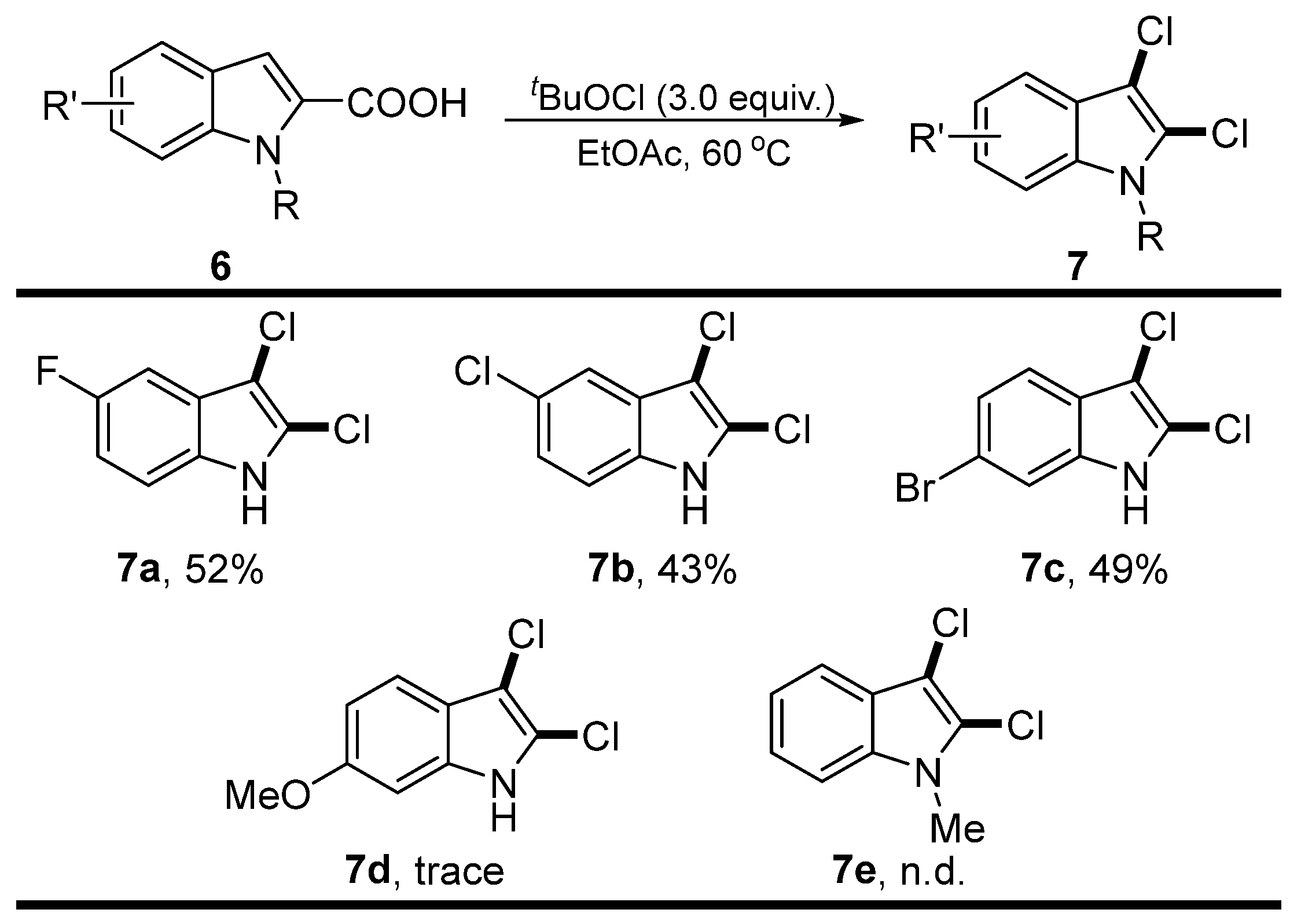
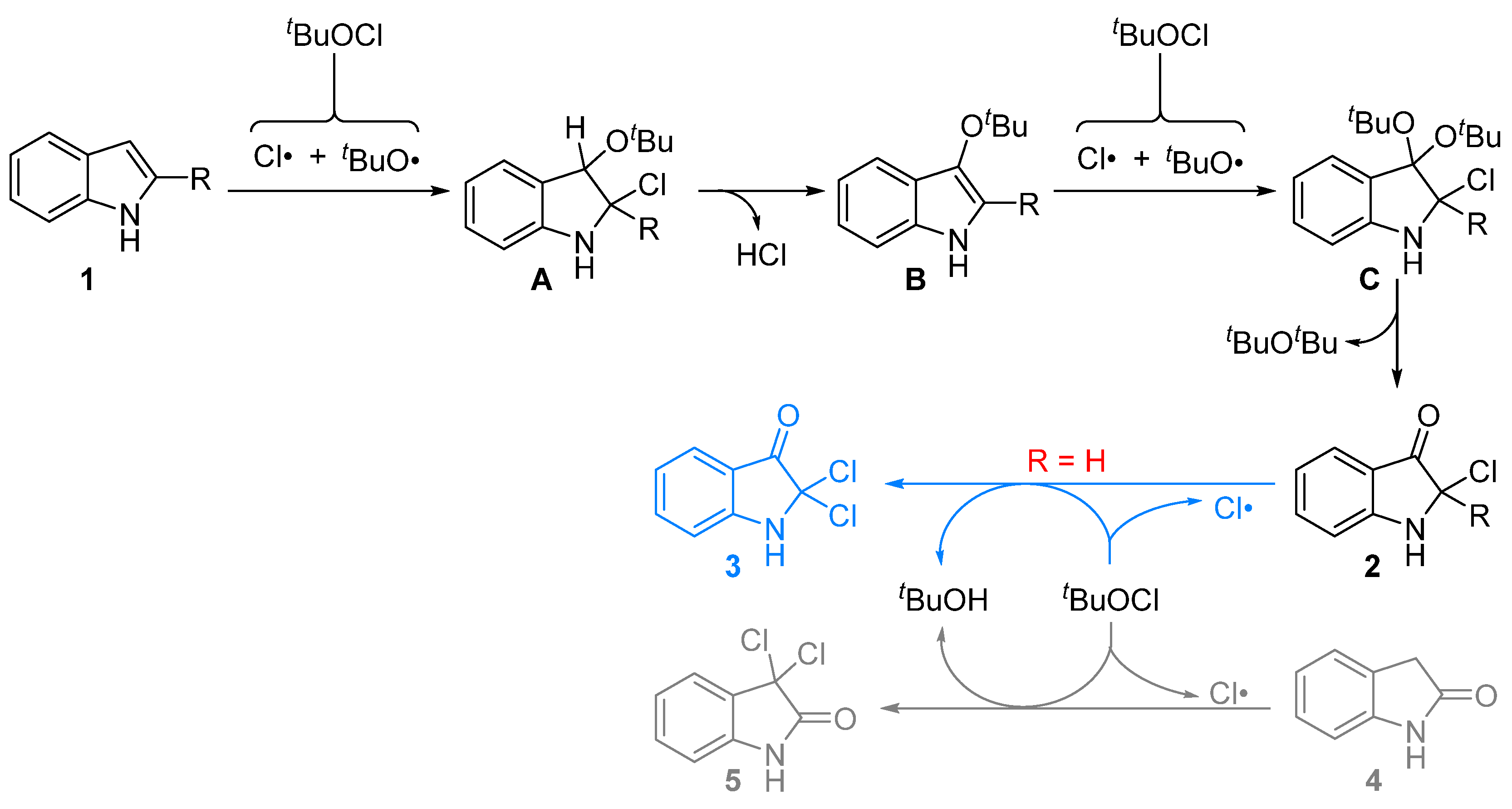
2. Results
3. Conclusions
4. Materials and Methods
4.1. General Information
4.2. Synthetic Procedures
4.2.1. The Typical Procedure for Chlorooxidation of Indoles with tBuOCl
4.2.2. The Typical Procedure for Chlorination of 2-Oxindoles with tBuOCl
4.2.3. The Typical Procedure for Decarboxylative Chlorination of Indole-2-Carboxylic Acids
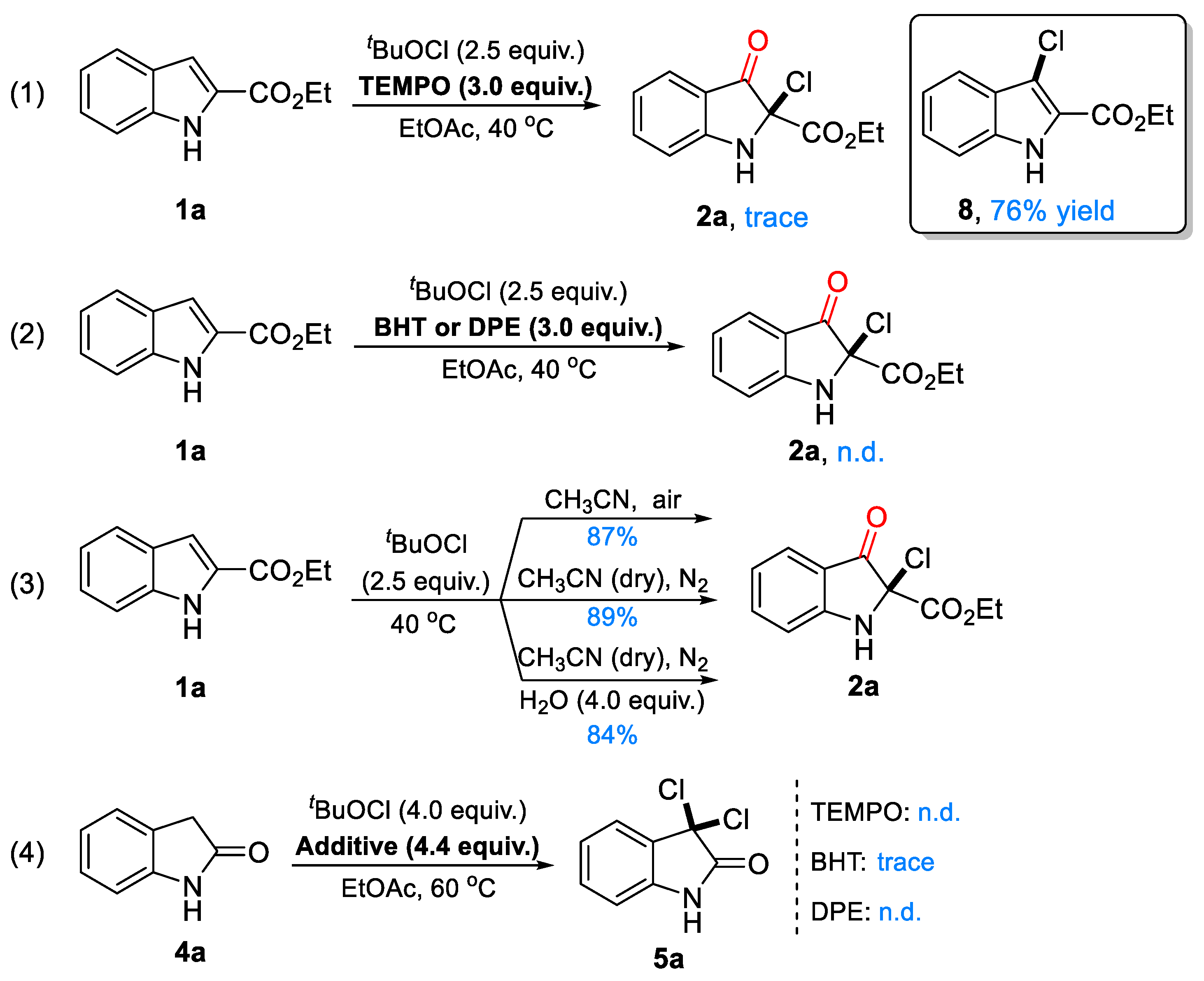
4.3. Control Experiments
4.3.1. The Effect of Radical Scavenger TEMPO on the Reaction
4.3.2. The Effect of Radical Scavenger BHT and DPE on the Reaction
4.3.3. The Effect of Water and Air on the Reaction
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Nakamura, I.; Yamamoto, Y. Transition-Metal-Catalyzed Reactions in Heterocyclic Synthesis. Chem. Rev. 2004, 104, 2127–2198. [Google Scholar] [CrossRef]
- Bariwal, J.; Voskressensky, L.G.; der Eycken, E.V.V. Recent advances in spirocyclization of indole derivatives. Chem. Soc. Rev. 2018, 47, 3831–3848. [Google Scholar] [CrossRef] [PubMed]
- Sakla, A.P.; Kansal, P.; Shankaraiah, N. Syntheses and Applications of Spirocyclopropyl Oxindoles: A Decade Review. Eur. J. Org. Chem. 2021, 2021, 757–772. [Google Scholar] [CrossRef]
- Dong, Y.; Hu, F.; Wu, H.; Guo, F.-W.; Wang, L.; Du, F.-Y.; Li, S.-S. Controllable Synthesis of N-Heterocycles via Hydride Transfer Strategy-Enabled Formal [5 + 1] and [5 + 2] Cyclizations. Org. Lett. 2024, 26, 332–337. [Google Scholar] [CrossRef]
- Xiao, J.; Westbroek, W.; Motabar, O.; Lea, W.A.; Hu, X.; Velayati, A.; Zheng, W.; Southall, N.; Gustafson, A.M.; Goldin, E.; et al. Discovery of a Novel Noniminosugar Acid α Glucosidase Chaperone Series. J. Med. Chem. 2012, 55, 7546–7559. [Google Scholar] [CrossRef]
- Liu, L.; Li, Y.; Huang, T.; Kong, D.; Wu, M. A novel methodology for the efficient synthesis of 3-monohalooxindoles by acidolysis of 3-phosphate-substituted oxindoles with haloid acids. Beilstein J. Org. Chem. 2021, 17, 2321–2328. [Google Scholar] [CrossRef]
- Li, R.-L.; Fang, Q.-Y.; Li, M.-Y.; Wang, X.-S.; Zhao, L.-M. A rearrangement of saccharin-derived cyclic ketimines with 3-chlorooxindoles leading to spiro-1,3-benzothiazine oxindoles. Chem. Commun. 2021, 57, 11322–11325. [Google Scholar] [CrossRef] [PubMed]
- Meesin, J.; Chotsaeng, N.; Kuhakarn, C. Dimerization of 3-Chlorooxindoles Mediated by Potassium Ethyl-xanthate: Synthesis of Isoindigos. Synlett 2022, 33, 1317–1322. [Google Scholar] [CrossRef]
- Ošeka, M.; Noole, A.; Žari, S.; Öeren, M.; Järving, I.; Lopp, M.; Kanger, T. Asymmetric Diastereoselective Synthesis of Spirocyclopropane Derivatives of Oxindole. Eur. J. Org. Chem. 2014, 2014, 3599–3606. [Google Scholar] [CrossRef]
- Liu, X.-L.; Yue, J.; Chen, S.; Liu, H.-H.; Yang, K.-M.; Feng, T.-T.; Zhou, Y. Thermal-mediated catalyst-free heterolytic cleavage of 3-halooxindoles: Rapid access to 3-functionalized-2-oxindoles. Org. Chem. Front. 2019, 6, 256–262. [Google Scholar] [CrossRef]
- Liu, R.-R.; Ye, S.-C.; Lu, C.-J.; Zhuang, G.-L.; Gao, J.-R.; Jia, Y.-X. Dual Catalysis for the Redox Annulation of Nitroalkynes with Indoles: Enantioselective Construction of Indolin-3-ones Bearing Quaternary Stereocenters. Angew. Chem. Int. Ed. 2015, 54, 11205–11208. [Google Scholar] [CrossRef] [PubMed]
- Shu, C.; Li, L.; Xiao, X.-Y.; Yu, Y.-F.; Ping, Y.-F.; Zhou, J.-M.; Ye, L.-W. Flexible and practical synthesis of 3-oxyindoles through gold-catalyzed intermolecular oxidation of o-ethynylanilines. Chem. Commun. 2014, 50, 8689–8692. [Google Scholar] [CrossRef] [PubMed]
- Mothe, S.R.; Novianti, M.L.; Ayers, B.J.; Chan, P.W.H. Silver-Catalyzed Tandem Hydroamination/Hydroarylation of 1-(2-Allylamino)phenyl-4-hydroxy-but-2-yn-1-ones to 1′-Allylspiro[indene-1,2′-indolin]-3′-ones. Org. Lett. 2014, 16, 4110–4113. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, M.; Takao, Y.; Katsurayama, H.; Mizota, I. Synthesis of Indolin-3-ones and Tetrahydro-4-quinolones from α-Imino Esters. Asian J. Org. Chem. 2013, 2, 130–134. [Google Scholar] [CrossRef]
- Kothandaraman, P.; Koh, B.Q.; Limpanuparb, T.; Hirao, H.; Chan, P.W.H. 1-(2′-Anilinyl)prop-2-yn-1-ol Rearrangement for Oxindole Synthesis. Chem. Eur. J. 2013, 19, 1978–1985. [Google Scholar] [CrossRef] [PubMed]
- Wetzel, A.; Gagosz, F. Gold-Catalyzed Transformation of 2-Alkynyl Arylazides: Efficient Access to the Valuable Pseudoindoxyl and Indolyl Frameworks. Angew. Chem. Int. Ed. 2011, 50, 7354–7358. [Google Scholar] [CrossRef] [PubMed]
- Okuma, K.; Matsunaga, N.; Nagahora, N.; Shioji, K.; Yokomori, Y. Reaction of arynes with amino acid esters. Chem. Commun. 2011, 47, 5822–5824. [Google Scholar] [CrossRef]
- Sun, Y.; Fan, R. Construction of 3-oxyindoles via hypervalent iodine mediated tandem cyclization–acetoxylation of o-acyl anilines. Chem. Commun. 2010, 46, 6834–6836. [Google Scholar] [CrossRef]
- Kiraz, C.I.A.; Emge, T.J.; Jimenaz, L.S. Oxidation of Indole Substrates by Oxodiperoxomolybdenum·Trialkyl(aryl)- phosphine Oxide Complexes. J. Org. Chem. 2004, 69, 2200–2202. [Google Scholar] [CrossRef]
- Higuchi, K.; Sato, Y.; Kojima, S.; Tsuchimochi, M.; Sugiura, K.; Hatori, M.; Kawasaki, T. Preparation of 2,2-disubstituted 1,2-dihydro-3H-indol-3-ones via oxidation of 2-substituted indoles and Mannich-type reaction. Tetrahedron 2010, 66, 1236–1243. [Google Scholar] [CrossRef]
- Zhang, X.; Foote, C.S. Dimethyldioxirane oxidation of indole derivatives. Formation of novel indole-2,3-epoxides and a versatile synthetic route to indolinones and indulines. J. Am. Chem. Soc. 1993, 115, 8867–8868. [Google Scholar] [CrossRef]
- Colandrea, V.J.; Rajaraman, S.; Jimenez, L.S. Synthesis of the Mitomycin and FR900482 Ring Systems via Dimethyldioxirane Oxidation. Org. Lett. 2003, 5, 785–787. [Google Scholar] [CrossRef] [PubMed]
- Guchhait, S.K.; Chaudhary, V.; Rana, V.A.; Priyadarshani, G.; Kandekar, S.; Kashyap, M. Oxidative Dearomatization of Indoles via Pd-Catalyzed C-H Oxygenation: An Entry to C2-Quaternary Indolin-3-ones. Org. Lett. 2016, 18, 1534–1537. [Google Scholar] [CrossRef]
- Xu, D.; Sun, W.-W.; Xie, Y.; Liu, J.-K.; Liu, B.; Zhou, Y.; Wu, B. Metal-Free Regioselective Hypervalent Iodine-Mediated C-2 and C-3 Difunctionalization of N-Substituted Indoles. J. Org. Chem. 2016, 81, 11081–11094. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Zhou, X.-Y.; Feng, X.-J.; Bao, M. Ruthenium-Catalyzed Oxidative Dearomatization of N-Boc Indoles. Synthesis 2021, 53, 1121–1126. [Google Scholar]
- Jaegli, S.; Dufour, J.; Wei, H.-L.; Piou, T.; Duan, X.-H.; Vors, J.-P.; Neuville, L.; Zhu, J. Palladium-Catalyzed Carbo-Heterofunctionalization of Alkenes for the Synthesis of Oxindoles and Spirooxindoles. Org. Lett. 2010, 12, 4498–4501. [Google Scholar] [CrossRef]
- Wei, H.-L.; Piou, T.; Dufour, J.; Neuville, L.; Zhu, J. Iodo-Carbocyclization of Electron-Deficient Alkenes: Synthesis of Oxindoles and Spirooxindoles. Org. Lett. 2011, 13, 2244–2247. [Google Scholar] [CrossRef] [PubMed]
- Mu, X.; Wu, T.; Wang, H.-Y.; Guo, Y.-L.; Liu, G. Palladium-Catalyzed Oxidative Aryltrifluoromethylation of Activated Alkenes at Room Temperature. J. Am. Chem. Soc. 2012, 134, 878–881. [Google Scholar] [CrossRef] [PubMed]
- Singh, J.; Sharma, A. Visible Light Mediated Synthesis of Oxindoles. Adv. Synth. Catal. 2021, 363, 4284–4308. [Google Scholar] [CrossRef]
- Dou, X.; Zhou, B.; Yao, W.; Zhong, F.; Jiang, C.; Lu, Y. A Facile Approach for the Asymmetric Synthesis of Oxindoles with a 3-Sulfenyl-Substituted Quaternary Stereocenter. Org. Lett. 2013, 15, 4920–4923. [Google Scholar] [CrossRef]
- He, Y.; Guo, C.; Sun, B.; Quinn, J.; Li, Y. (3E,7E)-3,7-Bis(2-oxoindolin-3-ylidene)-5,7-dihydropyrrolo[2,3-f]indole-2,6(1H,3H)-dione based polymers for ambipolar organic thin film transistors. Chem. Commun. 2015, 51, 8093–8096. [Google Scholar] [CrossRef]
- Hepples, C.; Murphy, G.K. Synthesis of 3,3-dichloro-2-oxindoles from isatin-3-p-tosylhydrazones and (dichloroiodo)benzene. Tetrahedron Lett. 2015, 56, 4971–4974. [Google Scholar] [CrossRef]
- Lötter, A.N.C.; Pathak, R.; Sello, T.S.; Fernandes, M.A.; van Otterlo, W.A.L.; de Koning, C.B. Synthesis of the dibenzopyrrocoline alkaloid skeleton: Indolo[2,1-a]isoquinolines and related analogues. Tetrahedron 2007, 63, 2263–2274. [Google Scholar] [CrossRef]
- Kravchenkoa, D.V.; Kuzovkova, J.A.; Kysil, V.M.; Tkachenko, S.E.; Malarchuk, S.; Okun, I.M.; Ivachtchenko, A.V. Pyrrolo[3,4-c]Quinoline-1,3-Diones as Potent Caspase-3 Inhibitors: Synthesis and SAR of 8-Sulfamoyl-1,3-Dioxo-2,3-Dihydro-1H-Pyrrolo[3,4-c]Quinolines. Lett. Drug Des. Discov. 2006, 3, 61–70. [Google Scholar] [CrossRef]
- Jiang, Y.; Hansen, T.V. Isatin 1,2,3-triazoles as potent inhibitors against caspase-3. Bioorg. Med. Chem. Lett. 2011, 21, 1626–1629. [Google Scholar] [CrossRef] [PubMed]
- Dolci, S.; Marchetti, F.; Pampaloni, G.; Zacchini, S. A crystallographic and spectroscopic study on the reactions of WCl6 with carbonyl compounds. Dalton Trans. 2013, 42, 5635–5648. [Google Scholar] [CrossRef]
- Foglia, T.A.; Swern, D. Pseudohalogens. XII. Reaction of N,N-dichlorourethane with indole and derivatives. J. Org. Chem. 1968, 33, 4440–4442. [Google Scholar] [CrossRef]
- Bass, R.J. Chlorination of 5-methoxyindole derivatives. Tetrahedron 1971, 27, 3263–3270. [Google Scholar] [CrossRef]
- Hamamoto, H.; Umemoto, H.; Umemoto, M.; Ohta, C.; Miki, Y. Hypervalent Iodine(III) Mediated Decarboxylative Halogenation of Indolecarboxylic Acids for the Synthesis of Haloindole Derivatives. Synlett 2010, 2010, 2593–2596. [Google Scholar] [CrossRef]
- Hamamoto, H.; Umemoto, H.; Umemoto, M.; Ohta, C.; Fujita, E.; Nakamura, A.; Maegawa, T.; Miki, Y. Decarboxylative halogenation of indolecarboxylic acids using hypervalent iodine (III) reagent and its application to the synthesis of polybromoindoles. Heterocycles 2015, 91, 561–572. [Google Scholar] [CrossRef]
- Valiulin, R.A.; Mamidyala, S.; Finn, M.G. Taming chlorine azide: Access to 1,2-azidochlorides from alkenes. J. Org. Chem. 2015, 80, 2740–2755. [Google Scholar] [CrossRef]
- Reddy, V.L.; Prathima, P.S.; Rao, V.J.; Bikshapathi, R. An efficient synthesis of versatile synthon 3-chlorooxindoles with NaCl/oxone. New J. Chem. 2018, 42, 20152–20155. [Google Scholar] [CrossRef]
- Ma, T.; Zheng, Y.; Huang, S. SO2ClF: A Reagent for Controllable Chlorination and Chlorooxidation of Simple Unprotected Indoles. J. Org. Chem. 2023, 88, 4839–4847. [Google Scholar] [CrossRef]
- Jiang, X.; Yang, L.; Yang, W.; Zhu, Y.; Fang, L.; Yu, C. Controllable synthesis of 3-chloro- and 3,3-dichloro-2-oxindoles via hypervalent iodine-mediated chlorooxidation. Org. Biomol. Chem. 2019, 17, 6920–6924. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Reintjens, N.R.M.; Dhineshkumar, J.; Witte, M.D.; Adriaan, J.M. Site-Selective Dehydroxy-Chlorination of Secondary Alcohols in Unprotected Glycosides. Org. Lett. 2022, 24, 5339–5344. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Shi, J.; Yu, W. Photoinduced Site-Selective C(sp3)-H Chlorination of AliphaticAmides. Org. Lett. 2020, 22, 8899–8903. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, S.; Li, S.; Feng, X.; Yamamoto, Y.; Bao, M. Three-component addition of terminal alkynes, carboxylic acids, and tert-butyl hypochlorite. Chem. Commun. 2022, 58, 2670–2673. [Google Scholar] [CrossRef]
- Shen, D.; Sun, C.; Han, Y.; Luo, Z.; Ren, T.; Zhang, Q.; Huang, W.; Xie, J.; Jia, Y.; Chao, M. Additive-free oxychlorination of unsaturated C-C bonds with tert-butyl hypochlorite and water. Org. Biomol. Chem. 2024, 22, 3080–3085. [Google Scholar] [CrossRef] [PubMed]
- Qiu, K.; Wang, R. tert-Butyl Hypochlorite Mediated Oxidative Chlorination of S-Alkylisothiourea Salts: Synthesis of Sulfonyl Chlorides. Synthesis 2015, 47, 3186–3190. [Google Scholar] [CrossRef]
- Li, X.; Wang, B.; Li, D.; Zhao, J.; Qu, J.; Zhou, Y. KI-Mediated Chlorine Gas-Free Synthesis of 3,3-Dichloro-2-oxindole Derivatives. Eur. J. Org. Chem. 2023, 26, e202201452. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | ||||
|---|---|---|---|---|
| Entry | tBuOCl (x Equiv.) | Solvent | Temp. (°C) | Yield (%) b |
| 1 | 3.0 | THF | 60 | 77 |
| 2 | 3.0 | EtOAc | 60 | 91 |
| 3 | 3.0 | 1,4-dioxane | 60 | 61 |
| 4 | 3.0 | CH3CN | 60 | 87 |
| 5 | 3.0 | toluene | 60 | 53 |
| 6 | 2.5 | EtOAc | 60 | 94 |
| 7 | 2.0 | EtOAc | 60 | 85 |
| 8 | 1.5 | EtOAc | 60 | 56 |
| 9 | 2.5 | EtOAc | 50 | 97 |
| 10 | 2.5 | EtOAc | 40 | 99 |
| 11 | 2.5 | EtOAc | 30 | 92 |
| 12 c | 2.5 | EtOAc | 40 | 99 |
| 13 d | 2.5 | EtOAc | 40 | 88 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, C.-Y.; Chen, X.; Liu, H.-L.; Wang, N.; Zhou, X.-Y. tert-Butyl Hypochlorite: A Reagent for the Synthesis of Chlorinated Oxindole and Indole Derivatives. Molecules 2025, 30, 102. https://doi.org/10.3390/molecules30010102
Liu C-Y, Chen X, Liu H-L, Wang N, Zhou X-Y. tert-Butyl Hypochlorite: A Reagent for the Synthesis of Chlorinated Oxindole and Indole Derivatives. Molecules. 2025; 30(1):102. https://doi.org/10.3390/molecules30010102
Chicago/Turabian StyleLiu, Chun-Yan, Xia Chen, Hai-Long Liu, Nan Wang, and Xiao-Yu Zhou. 2025. "tert-Butyl Hypochlorite: A Reagent for the Synthesis of Chlorinated Oxindole and Indole Derivatives" Molecules 30, no. 1: 102. https://doi.org/10.3390/molecules30010102
APA StyleLiu, C.-Y., Chen, X., Liu, H.-L., Wang, N., & Zhou, X.-Y. (2025). tert-Butyl Hypochlorite: A Reagent for the Synthesis of Chlorinated Oxindole and Indole Derivatives. Molecules, 30(1), 102. https://doi.org/10.3390/molecules30010102