
Crystal Structures of d-Lyxono-1,4-lactone and Its O-Tosyl Derivative


 , ,
, ,  and
and
Abstract
:1. Introduction
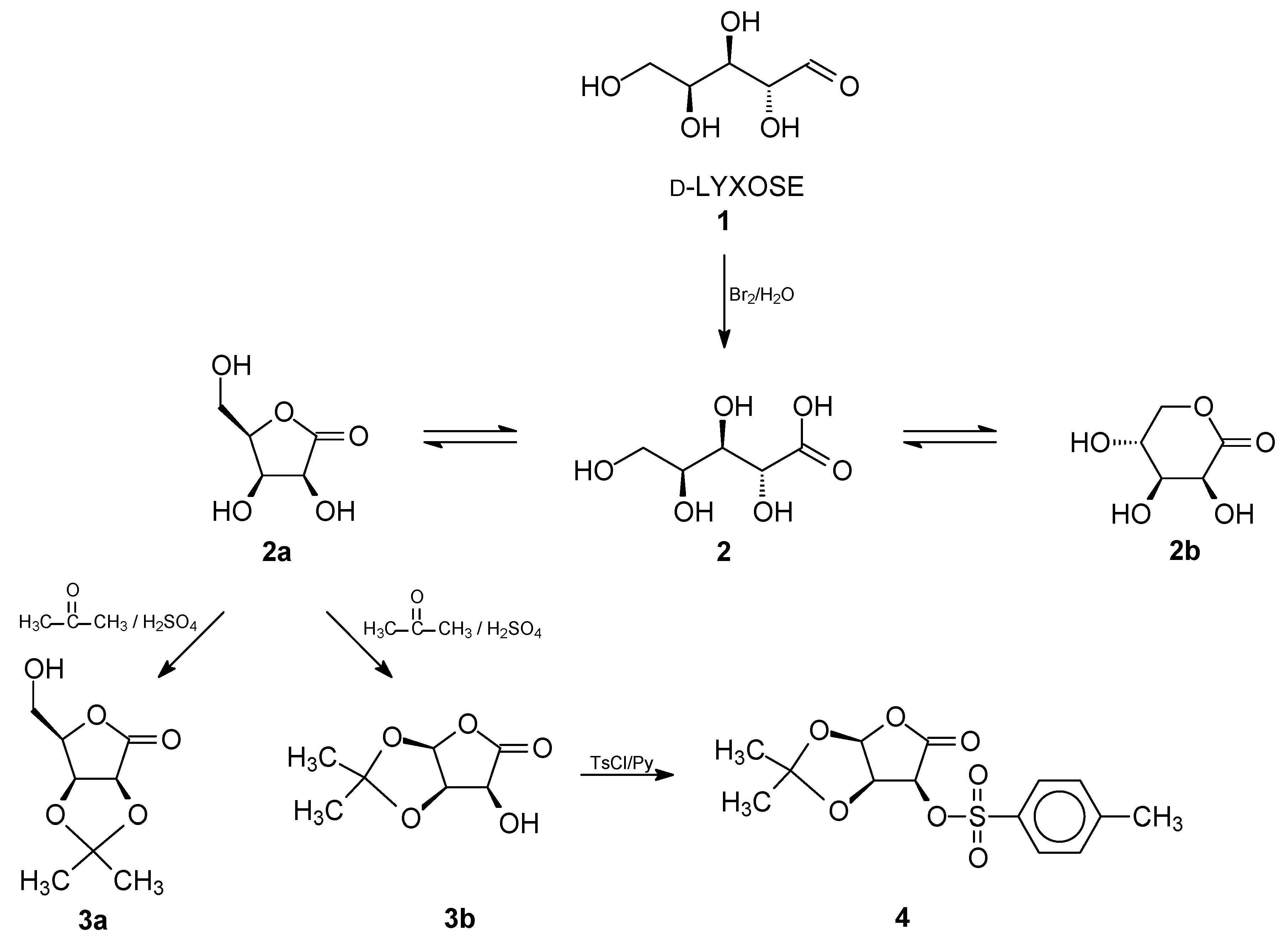
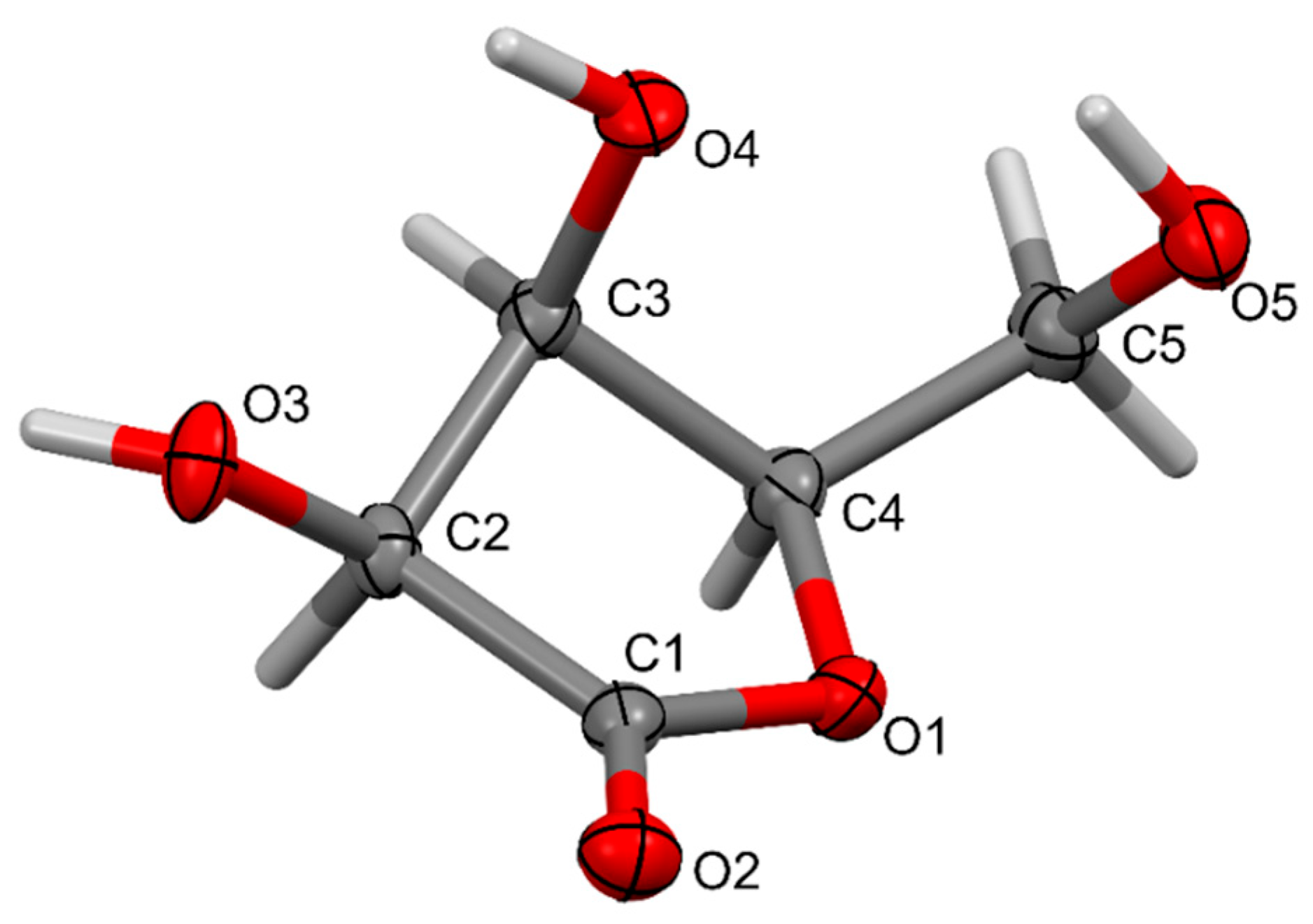
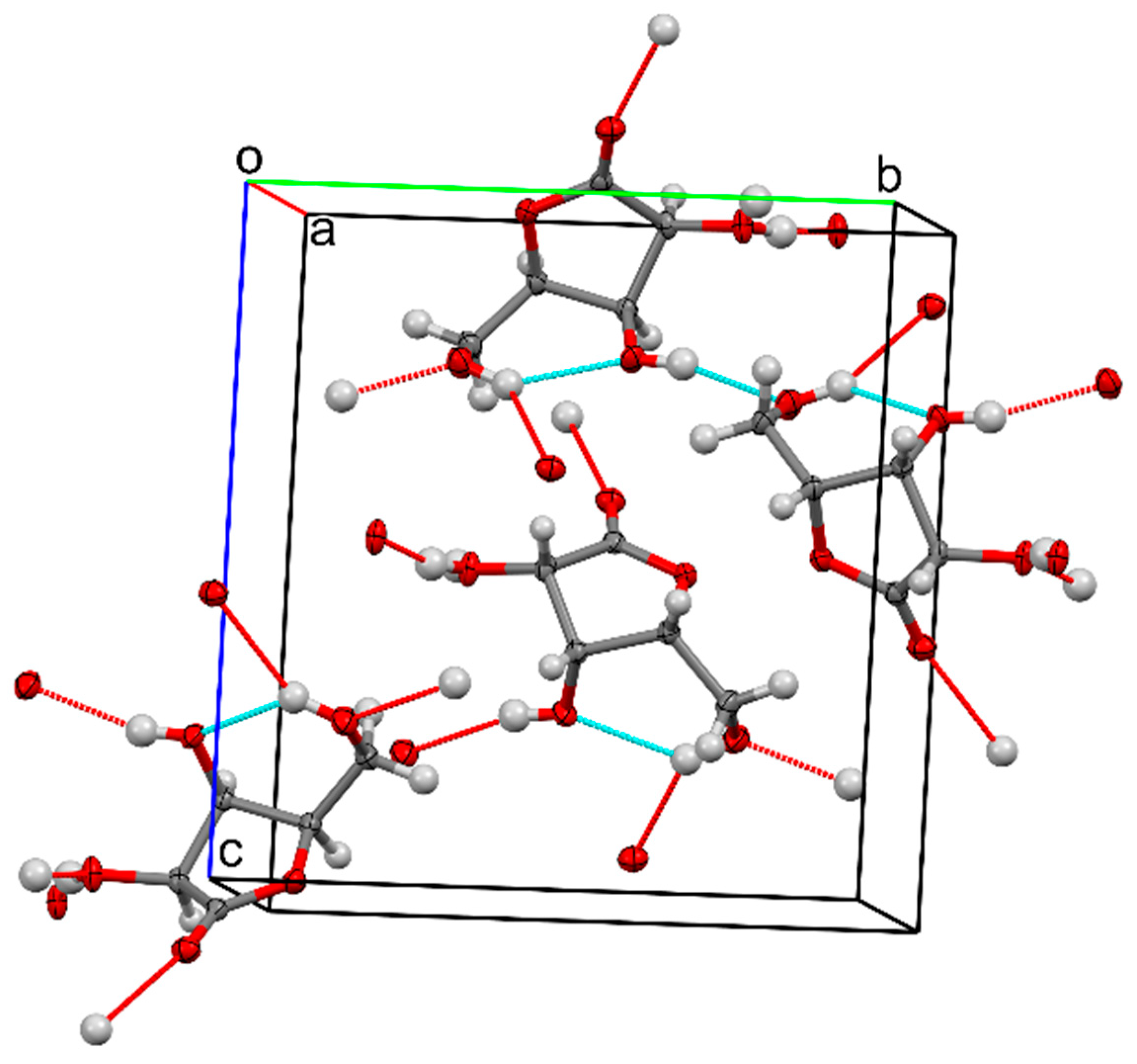
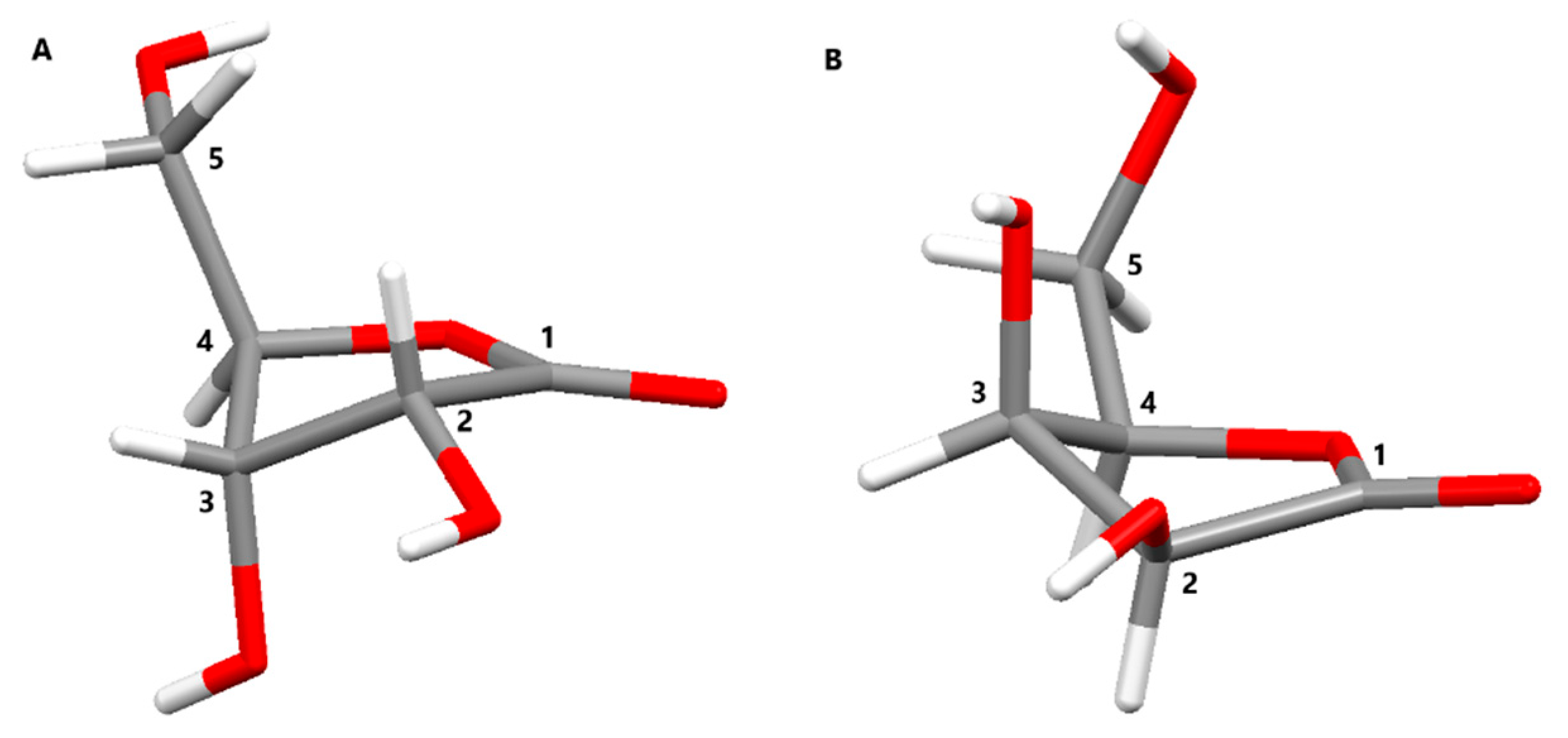
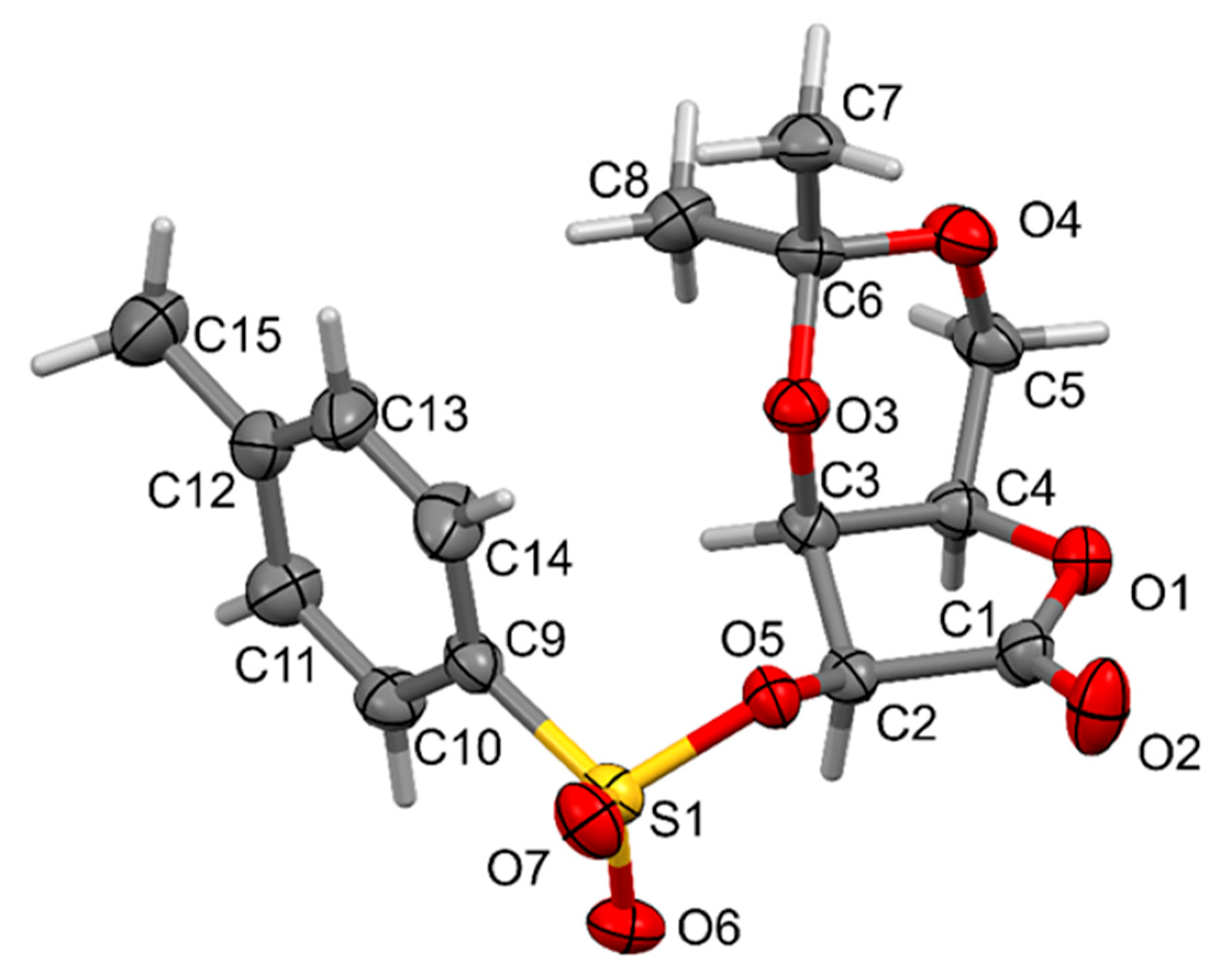
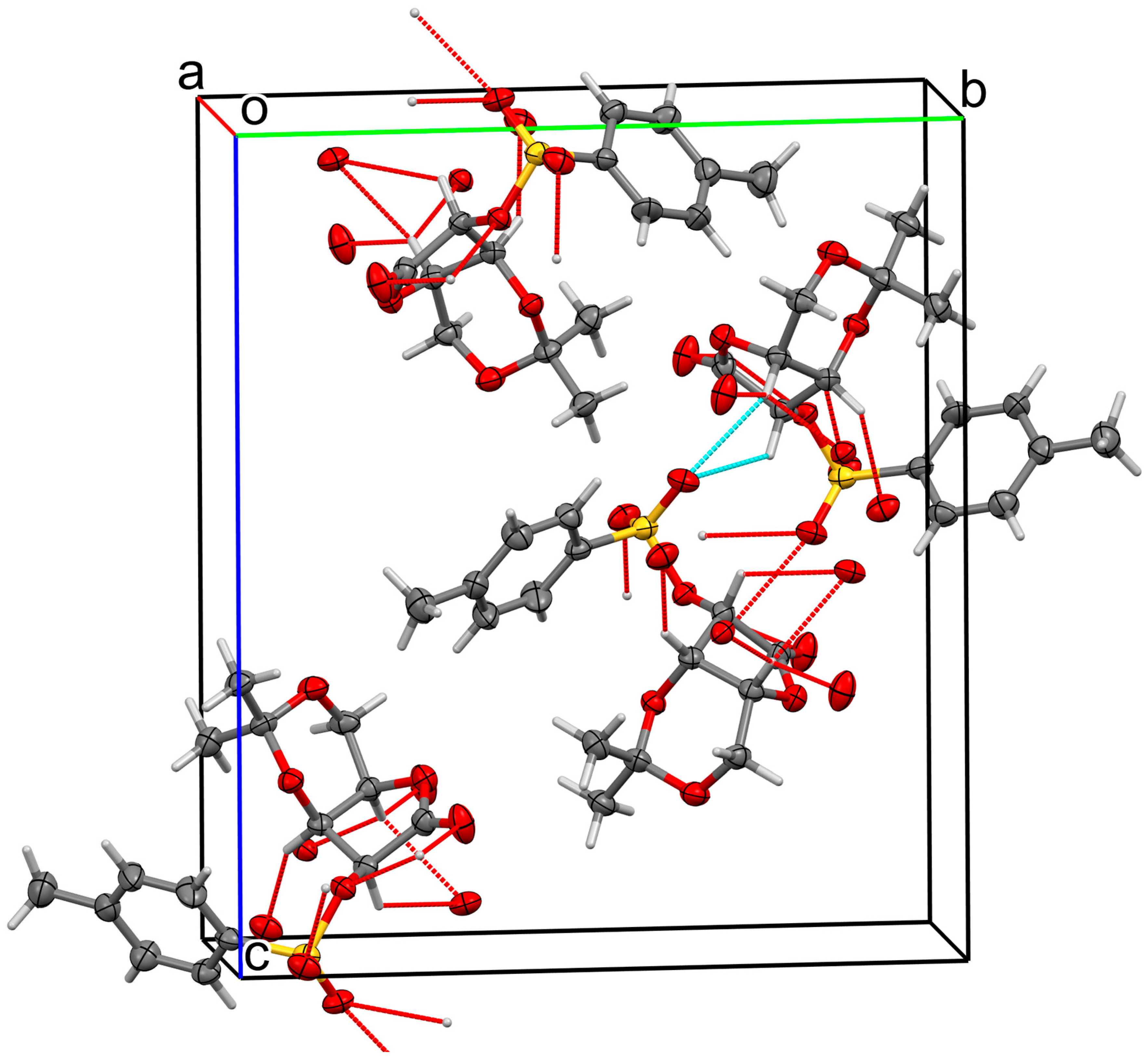
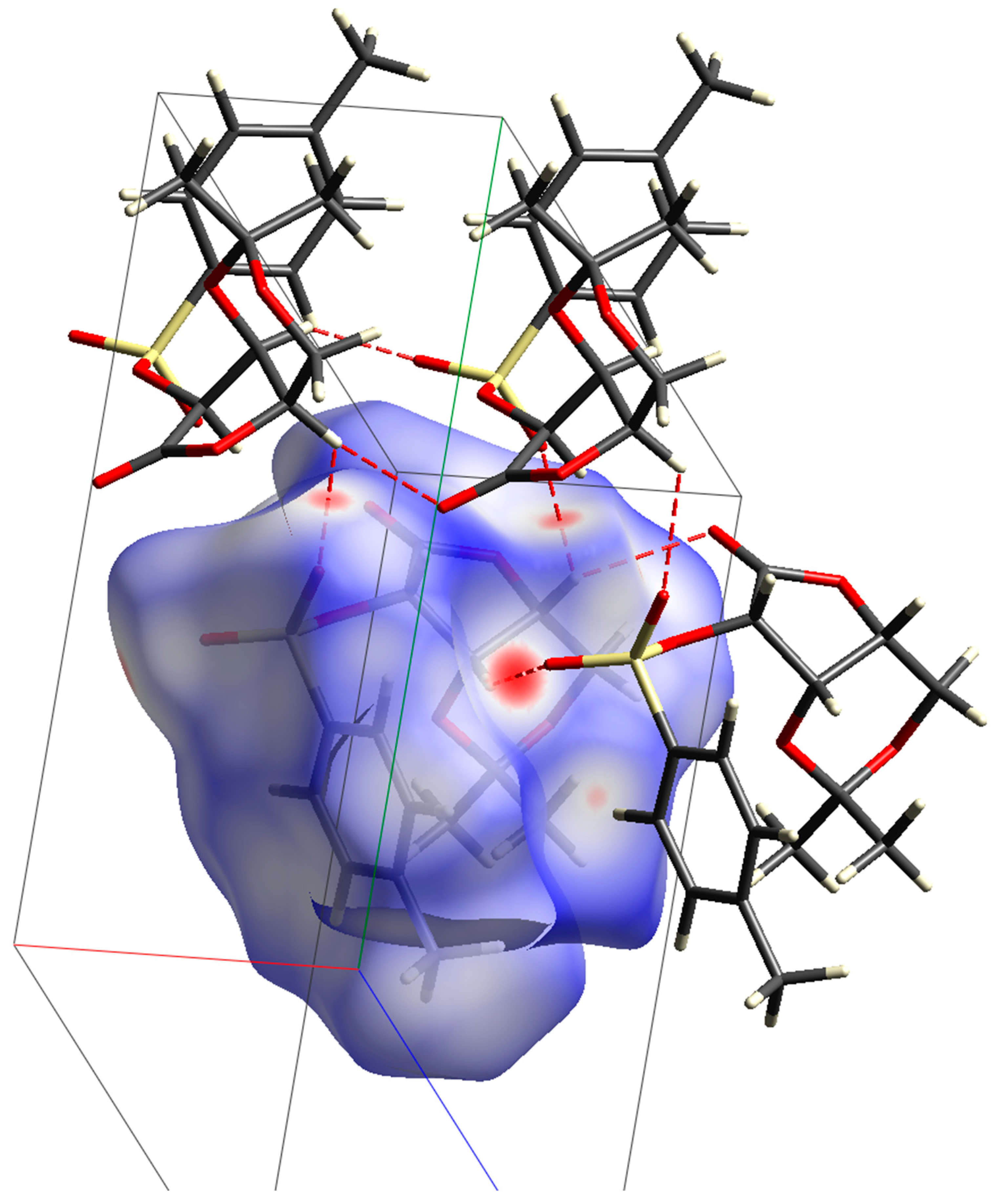
2. Results and Discussion
3. Materials and Methods
3.1. General Section
3.2. NMR Measurements
3.3. Mass Spectrometry
3.4. Infrared Spestroscopy
3.5. Single-Crystal X-Ray Diffraction
3.6. d-Lyxono-1,4-Lactone (2a)
3.7. 2,3-O-Isopropylidene-d-Lyxono-1,4-Lactone (3a)
3.7.1. Procedure A
3.7.2. Procedure B
3.8. 2,3-O-Isopropylidene-5-O-tosyl-D-Lyxono-1,4-Lactone (4)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Moss, G.P.; Smith, P.A.S.; Tavernier, D. Glossary of class names of organic compounds and reactivity intermediates based on structure (IUPAC Recommendations 1995). Pure Appl. Chem. 1995, 67, 1307–1375. [Google Scholar] [CrossRef]
- Koszelewski, D.; Borys, F.; Brodzka, A.; Ostaszewski, R. Synthesis of Enantiomerically Pure 5,6-Dihydropyran-2-ones via Chemoenzymatic Sequential DKR-RCM Reaction. Eur. J. Org. Chem. 2019, 2019, 1653–1658. [Google Scholar] [CrossRef]
- Sardan, M.; Sezer, S.; Günel, A.; Akkaya, M.; Tanyeli, C. Synthesis and biological evaluation of optically active conjugated γ- and δ-lactone derivatives. Bioorg. Med. Chem. Lett. 2012, 22, 5814–5818. [Google Scholar] [CrossRef] [PubMed]
- Kozioł, A.; Mroczko, L.; Niewiadomska, M.; Lochyński, S. γ-Lactones with potential biological activity. Pol. J. Nat. Sci. 2017, 32, 495–511. [Google Scholar]
- Ozçelik, B.; Gurbuz, I.; Karaaoglu, T.; Yesilada, E. Antiviral and antimicrobial activities of three sesquiterpene lactones from Centaurea solstitialis L. ssp. Solstitialis. Microbiol. Res. 2009, 164, 545–552. [Google Scholar] [CrossRef] [PubMed]
- Muthukumar, C.; Ignacimuthu, S.; Al-Harbi, N.A.; Duraipandiyan, V. Antimicrobial activity of sesquiterpene lactones isolated from traditional medicinal plant, Costus speciosus (Koen ex. Retz.) Sm. BMC Complement. Altern. Med. 2012, 12, 12–23. [Google Scholar] [CrossRef]
- Toyang, N.J.; Krause, M.A.; Fairhurst, R.M.; Tane, P.; Bryant, J.; Verpoorte, R. Antiplasmodial activity of sesquiterpene lactones and a sucrose ester from Vernonia guineensis Benth. (Asteraceae). J. Ethnopharmacol. 2013, 147, 618–621. [Google Scholar] [CrossRef]
- Fortuna, A.M.; Juárez, Z.N.; Bach, H.; Nematallah, A.; Av-Gay, Y.; Sánchez-Arreola, E.; Catalán, C.A.N.; Turbay, S.; Hernández, L.R. Antimicrobial activities of sesquiterpene lactones and inositol derivatives from Hymenoxys robusta. Phytochemistry 2011, 72, 2413–2418. [Google Scholar] [CrossRef] [PubMed]
- Djeddi, S.; Karioti, A.; Sokovic, M.; Stojkovic, D.; Seridi, R.; Skaltsa, H. Minor Sesquiterpene Lactones from Centaurea pullata and their antimicrobial activity. J. Nat. Prod. 2007, 70, 1796–1799. [Google Scholar] [CrossRef]
- Hamann, H.J.; Abutaleb, N.S.; Pal, R.; Seleem, M.N.; Ramachandran, P.V. β,γ-Diaryl α-methylene-γ-butyrolactones as potent antibacterials against methicillin-resistant Staphylococcus aureus. Bioorg. Chem. 2020, 104, 104183. [Google Scholar] [CrossRef]
- Gładkowski, W.; Skrobiszewski, A.; Mazur, M.; Siepka, M.; Pawlak, A.; Obmińska-Mrukowicz, B.; Białońska, A.; Poradowski, D.; Drynda, A.; Urbaniak, M. Synthesis and anticancer activity of novel halolactones with β-aryl substituents from simple aromatic aldehydes. Tetrahedron 2013, 69, 10414–10423. [Google Scholar] [CrossRef]
- Włoch, A.; Stygar, D.; Bahri, F.; Bazanów, B.; Kuropka, P.; Chełmecka, E.; Pruchnik, H.; Gładkowski, W. Antiproliferative, antimicrobial and antiviral activity of β-aryl-δ-iodo-γ-lactones, their effect on cellular oxidative stress markers and biological membranes. Biomolecules 2020, 10, 1594–1615. [Google Scholar] [CrossRef]
- Herkommer, D.; Thiede, S.; Wosniok, P.R.; Dreisigacker, S.; Tian, M.; Debnar, T.; Irschik, H.; Menche, D. Stereochemical determination of the leupyrrins and total synthesis of leupyrrin A1. J. Am. Chem. Soc. 2015, 137, 4086–4089. [Google Scholar] [CrossRef] [PubMed]
- Wosniok, P.R.; Knopf, C.; Dreisigacker, S.; Orozco-Rodriguez, J.M.; Hinkelmann, B.; Mueller, P.P.; Brönstrup, M.; Menche, D. SAR Studies of the Leupyrrins: Design and Total Synthesis of Highly Potent Simplified Leupylogs. Chem. A Eur. J. 2020, 26, 15074–15078. [Google Scholar] [CrossRef] [PubMed]
- Chukwujekwu, J.C.; Lategan, C.A.; Smith, P.J.; Van Heerden, F.R.; Van Staden, J. Antiplasmodial and cytotoxic activity of isolated sesquiterpene lactones from the acetone leaf extract of Vernonia colorata. S. Afr. J. Bot. 2009, 75, 176–179. [Google Scholar] [CrossRef]
- Chen, H.; Wu, G.; Gao, S.; Guo, R.; Zhao, Z.; Yuan, H.; Liu, S.; Wu, J.; Lu, X.; Yuan, X.; et al. Discovery of Potent Small-Molecule Inhibitors of Ubiquitin-Conjugating Enzyme UbcH5c from α-Santonin Derivatives. J. Med. Chem. 2017, 60, 6828–6852. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Zhang, W. Tetrahydronaphtho[1,2-b]furan-2(3H)-One Derivatives and Their Preparation, Pharmaceutical Compositions and Use in the Treatment of Rheumatoid Arthritis. WO/2019/011285, 17 January 2019. [Google Scholar]
- Chen, L.Z.; Wu, J.; Li, K.; Wu, Q.Q.; Chen, R.; Liu, X.H.; Ruan, B.F. Novel phthalide derivatives: Synthesis and anti-inflammatory activity in vitro and in vivo. Eur. J. Med. Chem. 2020, 206, 112722–112737. [Google Scholar] [CrossRef]
- Ruan, B.; Li, Y. Resveratrol-Phthalide Hybrid Compound for Anti-Inflammatory Research and Its Preparation Method. CN110105316, 9 August 2019. [Google Scholar]
- Tran, Q.T.N.; Wong, W.S.F.; Chai, C.L.L. The identification of naturally occurring labdane diterpenoid calcaratarin D as a potential anti-inflammatory agent. Eur. J. Med. Chem. 2019, 174, 33–44. [Google Scholar] [CrossRef]
- Siedle, B.; García-Piñeres, A.J.; Murillo, R.; Schulte-Mönting, J.; Castro, V.; Rüngeler, P.; Klaas, C.A.; Da Costa, F.B.; Kisiel, W.; Merfort, I. Quantitative structure-activity relationship of sesquiterpene lactones as inhibitors of the transcription factor NF-κB. J. Med. Chem. 2004, 47, 6042–6054. [Google Scholar] [CrossRef] [PubMed]
- Nicolaou, K.C.; Sanchini, S.; Sarlah, D.; Lu, G.; Wu, T.R.; Nomura, D.K.; Cravatt, B.F.; Cubitt, B.; De La Torre, J.C.; Hessell, A.J.; et al. Design, synthesis, and biological evaluation of a biyouyanagin compound library. Proc. Natl. Acad. Sci. USA 2011, 108, 6715–6720. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.N.; Huang, X.Y.; Feng, Z.M.; Jiang, J.S.; Zhang, P.C. New Butyrolactone Type Lignans from Arctii Fructus and Their Anti-inflammatory Activities. J. Agric. Food Chem. 2015, 63, 7958–7966. [Google Scholar] [CrossRef] [PubMed]
- Singh, P.; Mittal, A.; Bhardwaj, A.; Kaur, S.; Kumar, S. 1-Toluene-sulfonyl-3-[(3′-hydroxy-5′-substituted)-γ-butyrolactone]-indoles: Synthesis, COX-2 inhibition and anti-cancer activities. Bioorg. Med. Chem. Lett. 2008, 18, 85–89. [Google Scholar] [CrossRef]
- Brethon, A.; Chantalat, L.; Christin, O.; Clary, L.; Fournier, J.F.; Gastreich, M.; Harris, C.S.; Isabet, T.; Pascau, J.; Thoreau, E.; et al. New Caspase-1 inhibitor by scaffold hopping into bio-inspired 3D-fragment space. Bioorg. Med. Chem. Lett. 2017, 27, 5373–5377. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K.; Itazaki, H.; Yoshida, T. Cinatrins, a novel family of phospholipase a2 inhibitors: II. Biological activities. J. Antibiot. 1992, 45, 50–55. [Google Scholar] [CrossRef] [PubMed]
- Pillay, P.; Vleggaar, R.; Maharaj, V.J.; Smith, P.J.; Lategan, C.A. Isolation and identification of antiplasmodial sesquiterpene lactones from Oncosiphon piluliferum. J. Ethnopharmacol. 2007, 112, 71–76. [Google Scholar] [CrossRef] [PubMed]
- Buskuhl, H.; De Oliviera, F.L.; Blind, L.Z.; De Freitas, R.A.; Barison, A.; Campos, F.R.; Corilo, Y.E.; Eberlin, M.N.; Caramori, G.F.; Biavatti, M.W. Sesquiterpene lactones from Vernonia scorpioides and their in vitro cytotoxicity. Phytochemistry 2010, 71, 1539–1544. [Google Scholar] [CrossRef]
- Taylor, P.G.; Dupuy Loo, O.A.; Bonilla, J.A.; Murillo, R. Anticancer activities of two sesquiterpene lactones, millerenolide and thieleanin isolated from Viguiera sylvatica and Decachaeta thieleana. Fitoterapia 2008, 79, 428–432. [Google Scholar] [CrossRef]
- Brisdelli, F.; Perilli, M.; Sellitri, D.; Piovano, M.; Garbarino, J.A.; Nicoletti, M.; Bozzi, A.; Amicosante, G.; Celenza, G. Cytotoxic activity and antioxidant capacity of purified lichen metabolites: An in vitro study. Phyther. Res. 2012, 27, 431–437. [Google Scholar] [CrossRef]
- Roy, P.K.; Roy, S.; Ueda, K. New cytotoxic cembranolides from an Okinawan soft coral, Lobophytum sp. Fitoterapia 2019, 136, 104162–104168. [Google Scholar] [CrossRef]
- Salaski, E.J.; Krishnamurthy, G.; Ding, W.D.; Yu, K.; Insaf, S.S.; Eid, C.; Shim, J.; Levin, J.I.; Tabei, K.; Toral-Barza, L.; et al. Pyranonaphthoquinone lactones: A new class of AKT selective kinase inhibitors alkylate a regulatory loop cysteine. J. Med. Chem. 2009, 52, 2181–2184. [Google Scholar] [CrossRef]
- Takano, S.; Hasuda, K.; Ito, A.; Koide, Y.; Ishii, F.; Haneda, I.; Chihara, S.; Koyama, Y. A new antibiotic, Medermycin. J. Antibiot. 1976, 29, 765–768. [Google Scholar] [CrossRef] [PubMed]
- Bergy, M.E. Kalafungin, a new broad spectrum antibiotic, isolation and characterization. J. Antibiot. 1968, 21, 454–457. [Google Scholar] [CrossRef]
- Iwai, Y.; Kōra, A.; Takahashi, Y.; Hayashi, T.; Awaya, J.; Masuma, R.; Ōiwa, R.; Ōmura, S. Production of deoxyfrenolicin and a new antibiotic, frenolicin B by Streptomyces Roseofulvus strain AM-3867. J. Antibiot. 1978, 31, 959–965. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.-H.; Rice, G.K.; Hall, I.H.; Amarnath, V. Antitumor agents. 86. Synthesis and cytotoxicity of α-methylene-γ-lactone-bearing purines. J. Med. Chem. 1987, 30, 586–588. [Google Scholar] [CrossRef] [PubMed]
- Huth, J.R.; Park, C.; Petros, A.M.; Kunzer, A.R.; Wendt, M.D.; Wang, X.; Lynch, C.L.; Mack, J.C.; Swift, K.M.; Judge, R.A.; et al. Discovery and Design of Novel HSP90 Inhibitors Using Multiple Fragment-based Design Strategies. Chem. Biol. Drug Des. 2007, 70, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Vivino, F.B.; Al-Hashimi, I.; Khan, Z.; LeVeque, F.; Salisbury, P.L.; Tran-Johnson, T.K.; Muscoplat, C.C.; Trivedi, M.; Goldlust, B.; Gallagher, S.C. Pilocarpine Tablets for the Treatment of Dry Mouth and Dry Eye Symptoms in Patients with Sjogren Syndrome. Arch. Intern. Med. 1999, 159, 174–181. [Google Scholar] [CrossRef] [PubMed]
- Kagawa, C.M.; Cella, J.A.; Van Arman, C.G. Action of New Steroids in Blocking Effects of Aldosterone and Deoxycorticosterone on Salt. Science 1957, 126, 1015–1016. [Google Scholar] [CrossRef] [PubMed]
- Struthers, A.; Krum, H.; Williams, G.H. A comparison of the aldosterone-blocking agents eplerenone and spironolactone. Clin. Cardiol. 2008, 31, 153–158. [Google Scholar] [CrossRef] [PubMed]
- Krattenmacher, R. Drospirenone: Pharmacology and pharmacokinetics of a unique progestogen. Contraception 2000, 62, 29–38. [Google Scholar] [CrossRef]
- Xu, H.; Lv, M.; Tian, X. A Review on Hemisynthesis, Biosynthesis, Biological Activities, Mode of Action, and Structure-Activity Relationship of Podophyllotoxins: 2003–2007. Curr. Med. Chem. 2009, 16, 327–349. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Bogni, A.; Schuetz, E.G.; Ratain, M.; Eileen Dolan, M.; McLeod, H.; Gong, L.; Thorn, C.; Relling, M.V.; Klein, T.E.; et al. Etoposide pathway. Pharm. Genom. 2009, 19, 552–553. [Google Scholar] [CrossRef] [PubMed]
- Clark, P.I.; Slevin, M.L. The Clinical Pharmacology of Etoposide and Teniposide. Clin. Pharm. 1987, 12, 223–252. [Google Scholar] [CrossRef] [PubMed]
- Tantry, U.S.; Liu, F.; Chen, G.; Gurbel, P.A. Vorapaxar in the secondary prevention of atherothrombosis. Expert. Rev. Cardiovasc. Ther. 2015, 13, 1293–1305. [Google Scholar] [CrossRef]
- Isbell, H.S. Reactions of Carbohydrates. Methods Carbohydr. Chem. 1963, 2, 13–15. [Google Scholar]
- Rauter, A.P.; Vogel, P.; Queneau, Y. (Eds.) Carbohydrates in Sustainable Development II; Springer: Berlin/Heidelberg, Germany, 2010; ISBN 978-3-642-15160-6. [Google Scholar] [CrossRef]
- Kwoh, D.; Pocalyko, D.J.; Carchi, A.J.; Harirchian, B.; Hargiss, L.O.; Wong, T.C. Regioselective synthesis and characterization of 6-O-alkanoylgluconolactones. Carbohydr. Res. 1995, 274, 111–121. [Google Scholar] [CrossRef]
- Garésio, F.; Kardos, N.; Bonnevie, C.; Petit, S.; Luche, J.-L. D-Gluconolactone as a precursor to new environmentally benign tensioactive agents. Green. Chem. 2000, 2, 33–36. [Google Scholar] [CrossRef]
- Lalot, J.; Manier, G.; Stasik, I.; Demailly, G.; Beaupère, D. Efficient syntheses of 1-S-alkyl-1-thio-L-ribitols, D-lyxitols and L-xylitols from D-pentono-1,4-lactones. Carbohydr. Res. 2001, 335, 55–61. [Google Scholar] [CrossRef]
- Chaveriat, L.; Stasik, I.; Demailly, G.; Beaupère, D. Direct syntheses of S-alkylthio-D-galactono-, D-mannono-1,4 lactones, S-alkylthio-L-galactitols and D-mannitols displaying amphiphilic and mesophasic properties. Carbohydr. Res. 2004, 339, 1817–1821. [Google Scholar] [CrossRef]
- Lalot, J.; Stasik, I.; Demailly, G.; Beaupère, D.; Godé, P. Synthesis and amphiphilic properties of S-alkylthiopentonolactones and their pentitol derivatives. J. Colloid. Interface Sci. 2004, 273, 604–610. [Google Scholar] [CrossRef]
- Lalot, J.; Stasik, I.; Demailly, G.; Beaupère, D.; Godé, P. Mesomorphism of s-alkylthiopentono-lactones and their itol derivatives. J. Therm. Anal. Calorim. 2003, 74, 77–83. [Google Scholar] [CrossRef]
- Schmitzer, A.; Franceschi, I.; Perez, E.; Rico-Lattes, I.; Lattes, A.; Thion, L.; Erard, M.; Vidal, C. Reactivity at the interface of chiral amphiphilic dendrimers. High asymmetric reduction by NaBH4 of various prochiral ketones. J. Am. Chem. Soc. 2001, 123, 5956–5961. [Google Scholar] [CrossRef]
- Csuk, R.; Kühn, M.; Ströhl, D. Easy access to C-glycosides from aldonolactones by a Claisen-type chain-extension reaction. Tetrahedron 1997, 53, 1311–1322. [Google Scholar] [CrossRef]
- Schweizer, F.; Inazu, T. Chain extension of sugar δ-lactones with the enolate of tert-butyl bromoacetate and elaboration into functionalized C-ketosides, C-glycosides, and C-glucosyl glycines. Org. Lett. 2001, 3, 4115–4118. [Google Scholar] [CrossRef] [PubMed]
- Taillefumier, C.; Lakhrissi, Y.M.; Lakhrissi, M.; Chapleur, Y. Efficient conditions for the synthesis of C-glycosylidene derivatives: A direct and stereoselective route to C-glycosyl compounds. Tetrahedron Asymmetry 2002, 13, 1707–1711. [Google Scholar] [CrossRef]
- Bourdon, B.; Corbet, M.; Fontaine, P.; Goekjian, P.G.; Gueyrard, D. Synthesis of enol ethers from lactones using modified Julia olefination reagents: Application to the preparation of tri- and tetrasubstituted exoglycals. Tetrahedron Lett. 2008, 49, 747–749. [Google Scholar] [CrossRef]
- Bililign, T.; Griffith, B.R.; Thorson, J.S. Structure, activity, synthesis and biosynthesis of aryl-C-glycosides. Nat. Prod. Rep. 2005, 22, 742–760. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.Y.W.; He, M. Recent advances in aryl C-glycoside synthesis. Curr. Top. Med. Chem. 2005, 5, 1333–1350. [Google Scholar] [CrossRef] [PubMed]
- Rauter, A.P.; Lopes, R.G.; Martins, A. C-Glycosylflavonoids: Identification, bioactivity and synthesis. Nat. Prod. Commun. 2007, 2, 1175–1196. [Google Scholar] [CrossRef]
- Song, X.; Hollingswortha, R.I. Facile syntheses of 1-deoxynojirimycin (DNJ) and 1-deoxymannojirimycin (DMJ). Tetrahedron Lett. 2007, 48, 3115–3118. [Google Scholar] [CrossRef]
- Gireaud, L.; Chaveriat, L.; Stasik, I.; Wadouachi, A.; Beaupère, D. Synthesis of 6-amino-6-deoxy-D-gulono-1,6-lactam and L-gulono-1,6-lactam derived from corresponding 5,6-O-sulfinyl hexono-1,4-lactones. Tetrahedron 2006, 62, 7455–7458. [Google Scholar] [CrossRef]
- Jiao, Y.; Fang, Z.J.; Jiang, Y.H.; Zheng, B.H.; Cheng, J. Practical and concise synthesis of 2-oxo-3,4,5,6-tetraethoxyazepane from D-glucono-1,5-lactone. Chin. Chem. Lett. 2008, 19, 795–796. [Google Scholar] [CrossRef]
- Falentin, C.; Beaupère, D.; Demailly, G.; Stasik, I. Efficient synthesis of new N-alkyl-D-ribono-1,5-lactams from D-ribono-1,4-lactone. Tetrahedron Lett. 2009, 50, 5364–5366. [Google Scholar] [CrossRef]
- Varela, O.P.; Zunszain, A. First synthesis of aldopentono-l,4-thiolactones. J. Org. Chem. 1993, 58, 7860–7864. [Google Scholar] [CrossRef]
- Lalot, J.; Stasik, I.; Demailly, G.; Beaupère, D. Efficient synthesis of 5-thio-D-arabinopyranose and 5-thio-D xylopyranose from the corresponding D-pentono-1,4-lactones. Carbohydr. Res. 2003, 338, 2241–2245. [Google Scholar] [CrossRef] [PubMed]
- Lalot, J.; Stasik, I.; Demailly, G.; Beaupère, D. An improved synthesis of 5-thio-D-ribose from D-ribono-1,4-lactone. Carbohydr. Res. 2002, 337, 1411–1416. [Google Scholar] [CrossRef] [PubMed]
- Rosenblum, S.B.; Bihovsky, R. Synthesis of the papulacandin C-arylglucosyl spiroketal nucleus. J. Am. Chem. Soc. 1990, 112, 2746–2748. [Google Scholar] [CrossRef]
- Czernecki, S.; Perlat, M.-C. C-Glycosides. 9. Stereospecific synthesis of C-glycosidic spiroketal of the papulacandins. J. Org. Chem. 1991, 56, 6289–6292. [Google Scholar] [CrossRef]
- McDonald, F.E.; Zhu, H.Y.H.; Holmquist, C.R. Rhodium-Catalyzed Alkyne Cyclotrimerization Strategies for C-Arylglycoside Synthesis. J. Am. Chem. Soc. 1995, 117, 6605–6606. [Google Scholar] [CrossRef]
- Yamamoto, Y.; Hashimoto, T.; Hattori, K.; Kikuchi, M.; Nishiyama, H. Synthesis of Spirocyclic C-Arylribosides via Cyclotrimerization. Org. Lett. 2006, 8, 3565–3568. [Google Scholar] [CrossRef] [PubMed]
- Shing, T.K.M.; Cheng, H.M. Short syntheses of Gabosine I and Gabosine G from δ-D-gluconolactone. J. Org. Chem. 2007, 72, 6610–6613. [Google Scholar] [CrossRef] [PubMed]
- Jeffrey, G.A.; Rosenstein, R.D.; Vlasse, M. The crystal structure of D-galactono-γ-lactone. Acta Crystallogr. 1967, 22, 725–733. [Google Scholar] [CrossRef] [PubMed]
- Shalaby, M.A.; Fronczek, F.A.; Vargas, D.; Younathan, E.S. Conformations and structure studies of sugar lactones. Part 111. The composition and conformation of d-mannurono-γ lactone in solution, and the structural analysis of its β anomer in the solid state. Carbohydr. Res. 1994, 265, 197–206. [Google Scholar] [CrossRef] [PubMed]
- Shalaby, M.A.; Fronczek, F.R.; Younathan, E.S. Conformations and structure studies of sugar lactones in the solid state. Part I. The molecular structure of l-rhamnono- and l-mannono-1,4-lactones. Carbohydr. Res. 1994, 264, 181–190. [Google Scholar] [CrossRef]
- Han, S.Y.; Joullié, M.M.; Faking, V.V.; Petasis, N.A. Spectroscopic, Crystallographic and Computational Studies of the Formation and Isomerization of Cyclic Acetals and Ketals of Pentonolactones. Tetrahedron Asymmetry 1994, 5, 2535–2562. [Google Scholar] [CrossRef]
- Shallard-Brown, H.A.; Harding, C.C.; Watkin, D.J.; Soengas, R.; Skytte, U.P.; Fleet, G.W.J. 2,3:5,6-Di-O-isopropylidene-2-C-hydroxy-methyl-D-talono-1,4lactone. Acta Crystallogr. Sect. E 2004, 60, o2163–o2164. [Google Scholar] [CrossRef]
- Punzo, F.; Watkin, D.J.; Hotchkiss, D.; Fleet, G.W.J. 2-C-Methyl-D-lyxono-1,4-lactone. Acta Cryst. Sect. E. 2006, 62, o98–o100. [Google Scholar] [CrossRef]
- Bream, R.; Watkin, D.; Soengas, R.; Eastwick-Field, V.; Fleet, G.W.J. 2,5-Di-O-acetyl-3-C-methyl-D-lyxono-1,4-lactone. Acta Cryst. Sect. E 2006, 62, o977–o979. [Google Scholar] [CrossRef]
- Jenkinson, S.F.; Rule, S.D.; Booth, K.V.; Fleet, G.W.J.; Watkin, D.J.; Petursson, S. 2-O-Benzhydryl-3,4-(S)-O-benzylidene-D-xylono-1,4-lactone. Acta Cryst. Section E 2008, 64, o1012. [Google Scholar] [CrossRef]
- Sá, M.M.; Silveira, G.P.; Caro, M.S.B.; Ellena, J. Synthesis of Novel O-Acylated-D-ribono-1,5-lactones and Structural Assignment Supported by Conventional NOESY-NMR and X-ray Analysis. J. Braz. Chem. Soc. 2008, 19, 18–23. [Google Scholar] [CrossRef]
- Bortoluzzi, A.J.; Sebrão, D.; Sá, M.M.; Nascimento, M.G. 5-O-Acetyl-D-ribono-1,4-lactone. Acta Cryst. Sect. E 2011, 67, o2778-o2778. [Google Scholar] [CrossRef] [PubMed]
- Bortoluzzi, A.J.; Silveira, G.P.; Sá, M.M. Crystal structure of 5-O-benzoyl-2,3-O-isopropylidene-D-ribono-1,4-lactone. Acta Cryst. Sect. E 2017, 73, 407–409. [Google Scholar] [CrossRef] [PubMed]
- Kinoshita, Y.; Ruble, J.R.; Jeffrey, G.A. The crystal structure of d-ribono-1,4-lactone at −150°. Carbohydr. Res. 1981, 92, 1–7. [Google Scholar] [CrossRef]
- Alén, R.; Valkonen, J. The Crystal and Molecular Structures of Five-Carbon and Six-Carbon Isosaccharino-1,4-lactones from Alkaline Pulping. Acta Chem. Scand. 1995, 49, 536–539. [Google Scholar] [CrossRef]
- Fun, H.-K.; Sivakumar, K. 2,3-Dideoxy-D-erythro-hex-2-enono-1,5-lactone and 1,4-Lactone Obtained from Tri-O-acetyl-d-glucal. Acta Crystallogr. Sect. C Cryst. Struct. Commun. 1995, C51, 1330–1333. [Google Scholar] [CrossRef]
- Suda, S.; Tateno, A.; Nakane, D.; Akitsu, T. Hirshfeld Surface Analysis for Investigation of Intermolecular Interaction of Molecular Crystals. Int. J. Org. Chem. 2023, 13, 57–85. [Google Scholar] [CrossRef]
- Krichen, F.; Walha, S.; Abdelmouleh, M. Hirshfeld surface analysis of the intermolecular interaction networks in cellulose Iα and Iβ. Carbohydr. Res. 2022, 518, 108600. [Google Scholar] [CrossRef]
- Skorupa, E.; Dmochowska, B.; Pellowska-Januszek, L.; Wojnowski, W.; Chojnacki, J.; Wiśniewski, A. Synthesis and structure of selected quaternary N-(1,4-anhydro-5-deoxy-2,3-O-isopropylidene-D,L-ribitol-5-yl)ammonium salts. Carbohydr. Res. 2004, 339, 2355–2362. [Google Scholar] [CrossRef]
- Dmochowska, B.; Ślusarz, R.; Chojnacki, J.; Samaszko-Fiertek, J.; Madaj, J. The quaternization reaction of 5-O-sulfonates of methyl 2,3-O-isopropylidene-β-D-ribofuranoside with selected heterocyclic and aliphatic amines. Molecules 2020, 25, 2161. [Google Scholar] [CrossRef] [PubMed]
- Batra, H.; Moriarty, R.M.; Penmasta, R.; Sharma, V.; Stanciuc, G.; Staszewski, J.P.; Tuladhar, S.M.; Walsh, D.A. A Concise, Efficient and Production-Scale Synthesis of a Protected L-Lyxonolactone Derivative: An Important Aldonolactone Core. Org. Process Res. Dev. 2006, 10, 484–486. [Google Scholar] [CrossRef]
- Mistry, B.D. A Handbook of Spectroscopic Data CHEMISTRY (UV, JR, PMR, JJCNMR and Mass Spectroscopy); Oxford Book Company: Jaipur, India, 2009; ISBN/ISSN 978-81-89473-86-0. [Google Scholar]
- Thompson, A.; Wolfrom, M.L. Isolation of Aldonic Acid Lactones through their Hydrazides. J. Am. Chem. Soc. 1946, 68, 1509–1510. [Google Scholar] [CrossRef]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Cryst. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT-Integrated Space-Group and Crystal-Structure Determination. Acta Cryst. Sect. A 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal Structure Refinement with SHELXL. Acta Cryst. Sect. C 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Spek, A.L. PLATON SQUEEZE: A tool for the calculation of the disordered solvent contribution to the calculated structure factors. Acta Cryst. Sect. C Struct. Chem. 2015, 71, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Spackman, P.R.; Turner, M.J.; McKinnon, J.J.; Wolff, S.K.; Grimwood, D.J.; Jayatilaka, D.; Spackman, M.A. CrystalExplorer: A program for Hirshfeld surface analysis, visualization and quantitative analysis of molecular crystals. J. Appl. Cryst. 2021, 54, 1006–1011. [Google Scholar] [CrossRef]


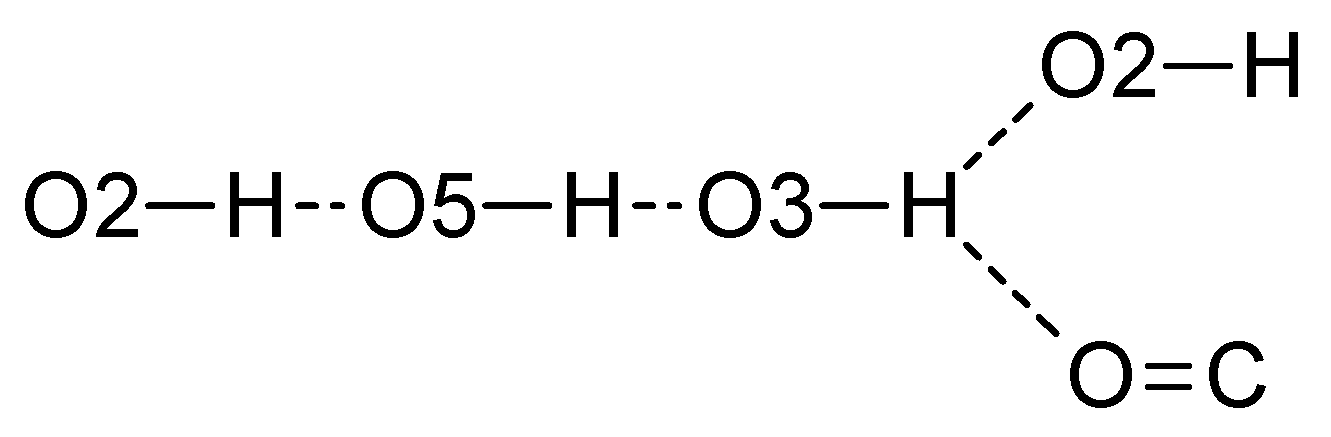

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| (2a) | (4) | |
|---|---|---|
| Crystal data | ||
| CCDC | 2387009 | 2387010 |
| Chemical formula | C5H8O5 | C15H18O7S |
| Mr | 148.11 | 342.35 |
| Crystal system, space group | Orthorhombic, P212121 | Orthorhombic, P212121 |
| Temperature (K) | 120 | 120 |
| a, b, c (Å) | 4.8905 (2), 10.7479 (6), 11.3738 (6) | 5.9288 (2), 15.1369 (5), 17.5364 (8) |
| V (Å3) | 597.84 (5) | 1573.78 (10) |
| Z | 4 | 4 |
| Radiation type | Mo Ka | Mo Ka |
| m (mm−1) | 0.15 | 0.24 |
| Crystal size (mm) | 0.43 × 0.05 × 0.04 | 0.45 × 0.04 × 0.03 |
| Data collection | ||
| Diffractometer | STOE IPDS 2T | STOE IPDS 2T |
| Absorption correction | none | Multi-scan STOE LANA |
| Tmin, Tmax | – | 0.159, 0.993 |
| No. of measured, independent and observed [I > 2s(I)] reflections | 7596, 1617, 1520 | 7534, 2969, 2584 |
| Rint | 0.032 | 0.080 |
| (sin q/l)max (Å−1) | 0.686 | 0.608 |
| Refinement | ||
| R[F2 > 2s(F2)], wR(F2), S | 0.034, 0.094, 1.04 | 0.073, 0.201, 1.08 |
| No. of reflections | 1617 | 2969 |
| No. of parameters | 101 | 211 |
| H-atom treatment | H atoms treated by a mixture of independent and constrained refinement | H-atom parameters constrained |
| Dρmax, Dρmin (e Å−3) | 0.36, −0.17 | 0.37, −0.49 |
| Absolute structure | Flack x determined using 588 quotients [(I+) − (I−)]/[(I+) + (I−)] (Parsons, Flack and Wagner, Acta Cryst. B69 (2013) 249–259). | Classical Flack method preferred over Parsons because s.u. lower. |
| Absolute structure parameter | 0.5 (10) | 0.3 (2) |
| Structure 2a | ||||
|---|---|---|---|---|
| D—H···A | D—H | H···A | D···A | D—H···A |
| O3—H3···O3 i | 0.83 (3) | 1.87 (3) | 2.6756 (10) | 162 (3) |
| O4—H4···O5 ii | 0.77 (3) | 2.07 (3) | 2.8155 (18) | 165 (3) |
| O5—H5···O2 iii | 0.91 (3) | 2.39 (3) | 3.1358 (18) | 139 (2) |
| O5—H5···O4 | 0.91 (3) | 2.18 (3) | 2.8202 (18) | 127 (2) |
| C2—H2···O2 iv | 1.00 | 2.29 | 3.269 (2) | 164 |
| C4—H4A···O1 v | 1.00 | 2.61 | 3.297 (2) | 126 |
| C5—H5A···O2 v | 0.99 | 2.55 | 3.535 (2) | 172 |
| Compound | Q(2)/Å | f(2)/° | d/° | Closest Conformation |
|---|---|---|---|---|
| 2a | 0.3953(17) | 278.4(2) | 15.5(2) | twisted on C2-C3 |
| 4 | 0.396(6) | 284.5(8) | 29.8(10) | envelope on C3 |
| Compound | H2 | H3 | H4 | H5 H5′ | C1 | C2 | C3 | C4 | C5 |
|---|---|---|---|---|---|---|---|---|---|
| 2a | 4.67 | 4.50 | 4.56 | 3.85 | 178.3 | 70.5 | 69.5 | 81.6 | 59.7 |
| 4 | 5.4 | 4.66 | 4.19 | 4.10 | 168.7 | 74.9 | 66.8 | 70.0 | 57.8 |
| Structure 4 | ||||
|---|---|---|---|---|
| D—H···A | D—H | H···A | D···A | D—H···A |
| C3—H3···O7 i | 1.00 | 2.40 | 3.290 (8) | 149 |
| C4—H4···O2 i | 1.00 | 2.50 | 3.236 (8) | 130 |
| C4—H4···O6 ii | 1.00 | 2.49 | 3.207 (7) | 128 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sosnowska, A.; Chojnacki, J.; Samaszko-Fiertek, J.; Madaj, J.; Dmochowska, B. Crystal Structures of d-Lyxono-1,4-lactone and Its O-Tosyl Derivative. Molecules 2025, 30, 287. https://doi.org/10.3390/molecules30020287
Sosnowska A, Chojnacki J, Samaszko-Fiertek J, Madaj J, Dmochowska B. Crystal Structures of d-Lyxono-1,4-lactone and Its O-Tosyl Derivative. Molecules. 2025; 30(2):287. https://doi.org/10.3390/molecules30020287
Chicago/Turabian StyleSosnowska, Anna, Jarosław Chojnacki, Justyna Samaszko-Fiertek, Janusz Madaj, and Barbara Dmochowska. 2025. "Crystal Structures of d-Lyxono-1,4-lactone and Its O-Tosyl Derivative" Molecules 30, no. 2: 287. https://doi.org/10.3390/molecules30020287
APA StyleSosnowska, A., Chojnacki, J., Samaszko-Fiertek, J., Madaj, J., & Dmochowska, B. (2025). Crystal Structures of d-Lyxono-1,4-lactone and Its O-Tosyl Derivative. Molecules, 30(2), 287. https://doi.org/10.3390/molecules30020287