
Hemoglobin Variants as Targets for Stabilizing Drugs


Abstract
:1. Introduction
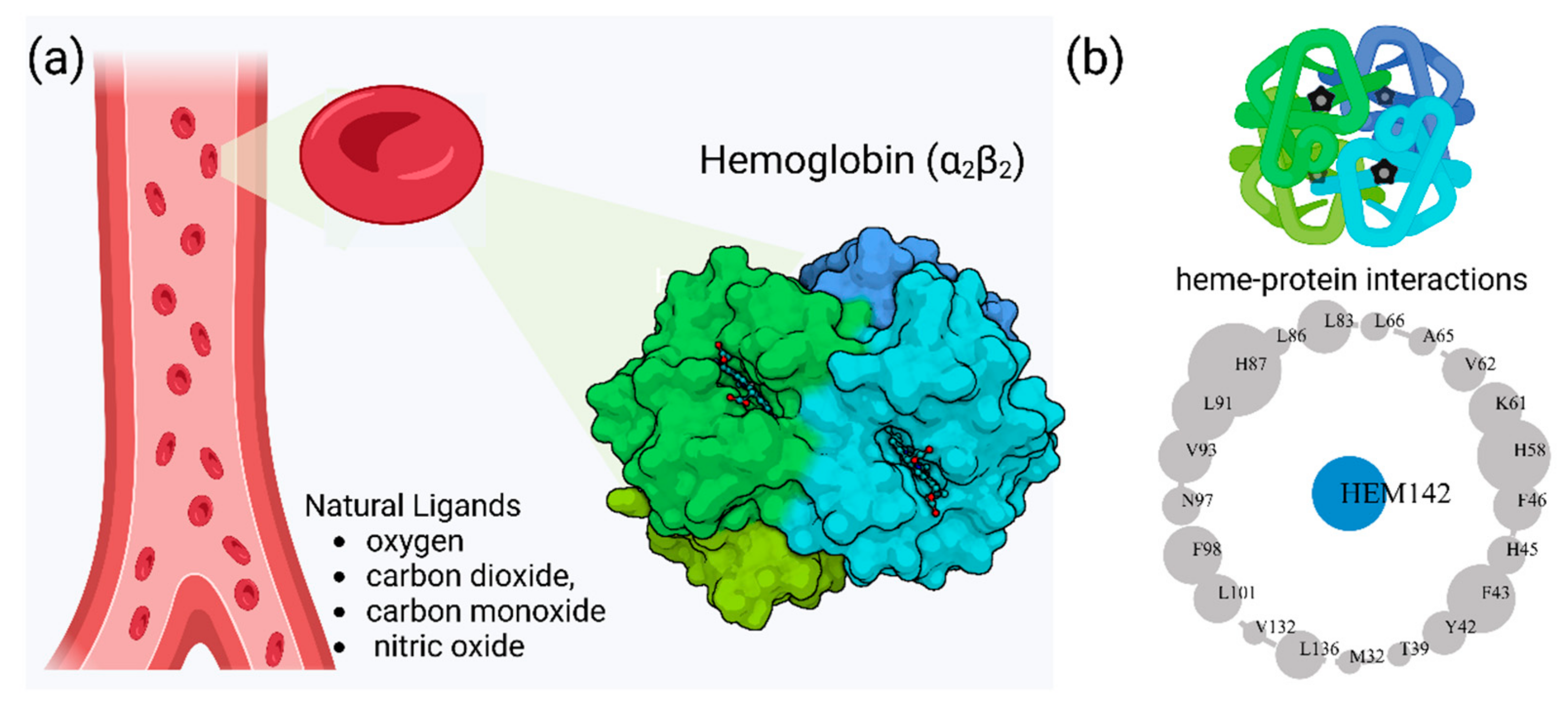
2. Hemoglobin Function and Clinical Variants
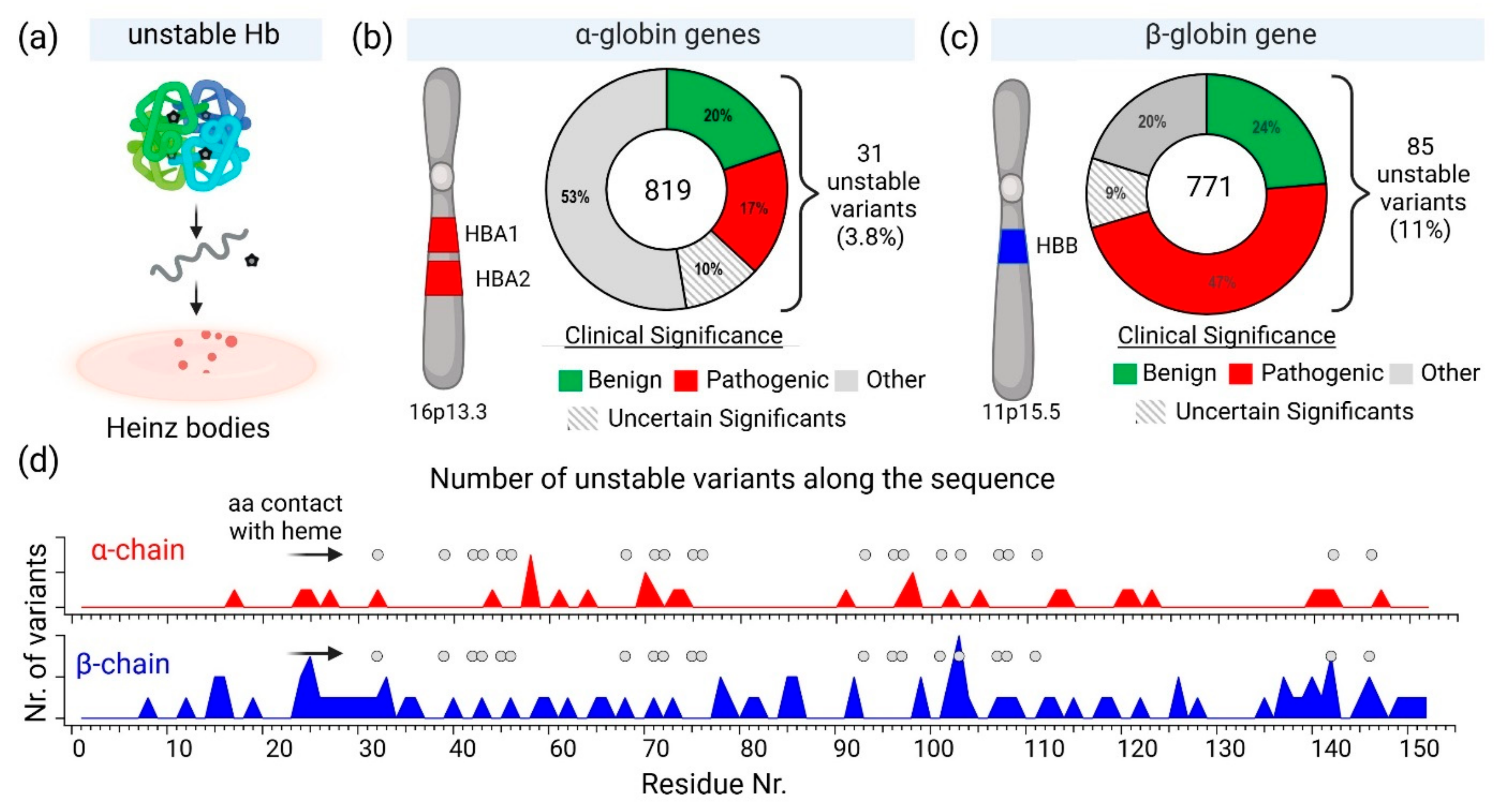
2.1. Diversity of Genetic Variants of Hemoglobin

{kind=link}
{kind=link}
{kind=link}
| Hb Variant | Mutation | Geo. Distribution | Clinical Features | Diag. Method | Notes |
|---|---|---|---|---|---|
| HbC | β-globin gene mutation (Glu → Lys at position 6) [52] | Common in West Africa | Mild chronic hemolysis, splenomegaly, jaundice in homozygotes; compound heterozygosity with HbS can worsen severity [52,53] | Hemoglobin electrophoresis, visualization of HbC crystals [54,55] | Affects RBC rigidity and may influence acquired immunity to malaria [56] |
| HbD-Punjab | β-globin gene mutation at codon 121 (Glu → Gln) [57] | Relatively low (~0.06% in one study) [57]; found in Indian subcontinent and other regions | Generally asymptomatic in heterozygotes; homozygotes may have mild hemolytic anemia and splenomegaly; severity increases when co-inherited with SCD or thalassemias [31,57,58,59,60,61,62] | Hemoglobin electrophoresis, molecular studies [60] | Treatment may include transfusions, hydroxyurea; HbSD-Punjab can mimic sickle cell disease [59] |
| HbE | β-globin gene mutation (Glu → Lys at position 26) [63] | Common in Southeast Asia; frequencies up to 0.433 in some Lao populations [64] | Variable severity from asymptomatic to severe anemia when co-inherited with β-thalassemia; altered redox properties and susceptibility to oxidative damage [65,66,67,68] | Hemoglobin electrophoresis, molecular analysis | Origin linked to malarial selection; minimal allosteric changes but altered redox properties [63,65] |
| Hb Bart’s (γ4) | α-globin gene deletions leading to excess γ chains (γ4 tetramers) [69] | High rates in regions with α-thalassemia (e.g., Saudi Arabia, Thailand) [70,71] | Indicates α-thalassemia severity; elevated levels correspond to number of α-globin gene deletions; high oxygen affinity but no cooperativity [69,72,73,74] | Quantification in cord blood; spectroscopic studies [74,75] | In some cases, elevated Hb Bart’s may be due to developmental asynchrony rather than true α-thalassemia [76] |
| Hb Chesapeake | α92 Arg → Leu mutation [77,78] | Reported in certain families; not widely prevalent | High oxygen affinity variant causing mild erythrocytosis; reduces tissue oxygen delivery, triggering increased RBC production [77,78,79,80] | Hemoglobin electrophoresis, oxygen dissociation studies, molecular analysis | Similar to other high-affinity variants (e.g., Hb Kempsey); fetal form also has increased O2 affinity [77,78] |
| Hb Lepore | Fusion of δ and β globin genes due to unequal crossover [81,82] | Prevalent in Southern Italy, found globally due to migration [83] | β-thalassemia minor phenotype in carriers; severe anemia in homozygotes or with co-inherited β-thalassemia [83,84,85,86] | Hematological, biochemical, and molecular analyses [82] | Variants include Lepore-Boston, Lepore-Hollandia, and Lepore-Washington-Boston; new variant Hb Lepore-Hong Kong identified [87] |
| Hb Constant Spring (CS) | Mutation in α2-globin termination codon, elongating α-chain [88] | Common in Southeast Asia; gene frequencies vary from 0.008 in Thailand to 0.143 in Vietnam [89,90] | Can lead to thalassemia intermedia when combined with α-thalassemia; rare homozygous cases cause fetal anemia and hydrops [91,92] | Selective enzymatic amplification of α2-globin DNA; hemoglobin electrophoresis [93] | Interferes with glycated hemoglobin measurements; important for genetic counseling [94] |
| Hb O-Arab | β121 Glu → Lys mutation [95,96] | Found in populations from the Middle East, North Africa, African Americans, West Africans [95,97,98] | Generally mild in homozygous form, but compound heterozygosity (e.g., Hb S/O Arab) can cause severe disease [95,99] | Hemoglobin electrophoresis with specialized techniques [95] | Management may involve transfusions and splenectomy [100] |
| Hb Seal Rock | Extended α-chain variant [101] | Rare, limited reports | Associated with mild Hb H disease and α-thalassemia-2 trait [101] | Hemoglobin electrophoresis, molecular studies | Impact depends on mutation location within the gene [102] |
| Hb Indianapolis | Rare, unstable β-globin variant [103] | Reported in a Brazilian patient [103] | Moderate hemolytic anemia and renal damage [103] | Routine DNA sequencing of globin genes [104] | Highlights the growing diversity of novel β-chain variants |
| Unstable Variants (e.g., Hb Madrid, Hb Showa-Yakushiji, Hb Santander, Hb Yokohama, Hb Seattle, Hb Miami, Hb Hershey, Hb Abington) | Various mutations in β-globin affecting amino acid positions [105,106,107,108,109,110,111] | Reported in disparate geographic locations (Spain, Republic of Korea, India, Japan, etc.) [105,106,107,108,109,110] | Mild to moderate hemolytic anemia; severity often increases with co-inherited thalassemia mutations [105,106,107,108,109,110,111] | Hemoglobin electrophoresis, DNA sequencing, RBC morphology analysis | Demonstrate clinical severity range and genetic complexity of unstable hemoglobin variants [104,105,106,107,108,109,110,111] |
2.2. Hemoglobin Instability and Heinz Bodies
2.3. Pharmacological Approaches to Hemoglobin Stabilization and Oxidative Stress in Hemolytic Disorders
2.4. Small-Molecule Binders to Hemoglobin
3. Search for Potentially New Hb Stabilisers: Hemoglobin Binding Proteins
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| Hb | Hemoglobin |
| SNO | S-nitrosohemoglobin |
| BPG | Bisphosphoglycerate |
| HbF | Fetal Hemoglobin |
| HbA | Adult Hemoglobin |
| HbC | Hemoglobin C |
| HbD | Hemoglobin D |
| HbE | Hemoglobin E |
| Hb CS | Hb Constant Spring |
| ACMG | American College of Medical Genetics and Genomics |
| SCD | Sickle Cell Disease |
| RBC | Red Blood Cell |
| ROS | Reactive Oxygen Species |
| AHSP | α-Hemoglobin Stabilizing Protein |
| G6PD | Glucose-6-phosphate Dehydrogenase |
| NAC | N-acetylcysteine |
| ALAS-1 | Aminolevulinic Acid Synthase |
| S1P | Sphingosine 1-phosphate |
| LPS | Lipopolysaccharide |
References
- Perutz, M.F. Stereochemical mechanism of oxygen transport by haemoglobin. Proc. R. Soc. Lond. Ser. B Biol. Sci. 1980, 208, 135–162. [Google Scholar]
- Bellelli, A. Hemoglobin and Cooperativity: Experiments and Theories. Curr. Protein Pept. Sci. 2010, 11, 2–36. [Google Scholar] [CrossRef] [PubMed]
- Riggs, A.F. The Bohr Effect. Annu. Rev. Physiol. 1988, 50, 181–204. [Google Scholar] [CrossRef] [PubMed]
- Tyuma, I.; Shimizu, K. Different response to organic phosphates of human fetal and adult hemoglobins. Arch. Biochem. Biophys. 1969, 129, 404–405. [Google Scholar] [CrossRef]
- Safo, M.K.; Ahmed, M.H.; Ghatge, M.S.; Boyiri, T. Hemoglobin–ligand binding: Understanding Hb function and allostery on atomic level. Biochim. Biophys. Acta (BBA) Proteins Proteom. 2011, 1814, 797–809. [Google Scholar] [CrossRef]
- Ahmed, M.H.; Ghatge, M.S.; Safo, M.K. Hemoglobin: Structure, Function and Allostery. Subcell. Biochem. 2020, 94, 345–382. [Google Scholar]
- Randad, R.S.; Mahran, M.A.; Mehanna, A.S.; Abraham, D.J. Allosteric modifiers of hemoglobin. 1. Design, synthesis, testing, and structure-allosteric activity relationship of novel hemoglobin oxygen affinity decreasing agents. J. Med. Chem. 1991, 34, 752–757. [Google Scholar] [CrossRef]
- Ronda, L.; Bruno, S.; Abbruzzetti, S.; Viappiani, C.; Bettati, S. Ligand reactivity and allosteric regulation of hemoglobin-based oxygen carriers. Biochim. Biophys. Acta (BBA) Proteins Proteom. 2008, 1784, 1365–1377. [Google Scholar] [CrossRef]
- Ciaccio, C.; Coletta, A.; De Sanctis, G.; Marini, S.; Coletta, M. Cooperativity and allostery in haemoglobin function. IUBMB Life 2008, 60, 112–123. [Google Scholar] [CrossRef]
- Maillett, D.H.; Simplaceanu, V.; Shen, T.-J.; Ho, N.T.; Olson, J.S.; Ho, C. Interfacial and Distal-Heme Pocket Mutations Exhibit Additive Effects on the Structure and Function of Hemoglobin. Biochemistry 2008, 47, 10551–10563. [Google Scholar] [CrossRef]
- Brunori, M.; Miele, A.E. Modulation of Allosteric Control and Evolution of Hemoglobin. Biomolecules 2023, 13, 572. [Google Scholar] [CrossRef] [PubMed]
- Storz, J.F. Hemoglobin structure and allosteric mechanism. In Hemoglobin; Oxford University Press: Oxford, UK, 2018; pp. 58–93. [Google Scholar]
- Ferguson, J.K.W.; Roughton, F.J.W. The chemical relationships and physiological importance of carbamino compounds of CO2 with hæmoglobin. J. Physiol. 1934, 83, 87–102. [Google Scholar] [CrossRef] [PubMed]
- Chan, N.-L.; Kavanaugh, J.S.; Rogers, P.H.; Arnone, A. Crystallographic Analysis of the Interaction of Nitric Oxide with Quaternary-T Human Hemoglobin. Biochemistry 2003, 43, 118–132. [Google Scholar] [CrossRef] [PubMed]
- Allen, B.W.; Stamler, J.S.; Piantadosi, C.A. Hemoglobin, nitric oxide and molecular mechanisms of hypoxic vasodilation. Trends Mol. Med. 2009, 15, 452–460. [Google Scholar] [CrossRef] [PubMed]
- Hamasaki, N.; Rose, Z.B. The Binding of Phosphorylated Red Cell Metabolites to Human Hemoglobin A. J. Biol. Chem. 1974, 249, 7896–7901. [Google Scholar] [CrossRef]
- Garby, L.; De Verdier, C.-H. Affinity of Human Hemoglobin a to 2,3—Diphosphoglycerate. Effect of Hemoglobin Concentration and of pH. Scand. J. Clin. Lab. Investig. 1971, 27, 345–350. [Google Scholar] [CrossRef]
- Beek, G.G.M.; Bruin, S.H. The pH Dependence of the Binding of d-Glycerate 2,3-Bisphosphate to Deoxyhemoglobin and Oxyhemoglobin. Determination of the Number of Binding Sites in Oxyhemoglobin. Eur. J. Biochem. 1979, 100, 497–502. [Google Scholar] [CrossRef]
- Isaacks, R.E. Can Metabolites Contribute in Regulating Blood Oxygen Affinity? In Advances in Experimental Medicine and Biology; Springer: New York, NY, USA, 1988; pp. 137–143. [Google Scholar]
- Mulquiney, P.J.; Kuchel, P.W. Model of 2,3-bisphosphoglycerate metabolism in the human erythrocyte based on detailed enzyme kinetic equations: Equations and parameter refinement. Biochem. J. 1999, 342, 581–596. [Google Scholar] [CrossRef]
- Kayikci, M.; Venkatakrishnan, A.J.; Scott-Brown, J.; Ravarani, C.N.J.; Flock, T.; Babu, M.M. Visualization and analysis of non-covalent contacts using the Protein Contacts Atlas. Nat. Struct. Mol. Biol. 2018, 25, 185–194. [Google Scholar] [CrossRef]
- López, F.J.B.; Centurion, I.F.; Díaz, F.J.P.; Pacheco, C.M.; Cortés, B.I.; Mendoza, B.M.T.; Flores-Jimenez, J.A.; Torre, L.D.C.R.-D.L. Structural Effect of Gγ and Aγ Globin Chains in Fetal Hemoglobin Tetramer. Blood 2023, 142 (Suppl. S1), 2291. [Google Scholar] [CrossRef]
- Sharma, S.K.; Lechner, R.B. Hematologic and coagulation disorders. In Obstetric Anesthesia: Principles and Practice, 2nd ed.; Chestnut, D.H., Ed.; Mosby Inc.: St. Louis, MO, USA, 1999; ISBN 0-3230-0383-4. [Google Scholar]
- Manca, L.; Masala, B. Disorders of the synthesis of human fetal hemoglobin. IUBMB Life 2008, 60, 94–111. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Rockwood, A.L.; Agarwal, A.M.; Anderson, L.C.; Weisbrod, C.R.; Hendrickson, C.L.; Marshall, A.G. Diagnosis of Hemoglobinopathy and β-Thalassemia by 21 Tesla Fourier Transform Ion Cyclotron Resonance Mass Spectrometry and Tandem Mass Spectrometry of Hemoglobin from Blood. Clin. Chem. 2019, 65, 986–994. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.-S.; Kim, H.S. Diagnosis of hemoglobinopathy and β-thalassemia by 21-Tesla Fourier transform ion cyclotron resonance mass spectrometry. Ann. Transl. Med. 2019, 7 (Suppl. S6), S239. [Google Scholar] [CrossRef] [PubMed]
- Carrocini, G.C.d.S.; Zamaro, P.J.A.; Bonini-Domingos, C.R. What influences Hb fetal production in adulthood? Rev. Bras. Hematol. Hemoter. 2011, 33, 231–236. [Google Scholar] [CrossRef]
- Hempe, J.M.; Craver, R.D. Laboratory Diagnosis of Structural Hemoglobinopathies and Thalassemias by Capillary Isoelectric Focusing. In Clinical Applications of Capillary Electrophoresis; Humana Press: Totowa, NJ, USA, 1999; pp. 81–98. [Google Scholar]
- Little, R.R.; Roberts, W.L. A Review of Variant Hemoglobins Interfering with Hemoglobin A1c Measurement. J. Diabetes Sci. Technol. 2009, 3, 446–451. [Google Scholar] [CrossRef]
- Fontana, L.; Alahouzou, Z.; Miccio, A.; Antoniou, P. Epigenetic Regulation of β-Globin Genes and the Potential to Treat Hemoglobinopathies through Epigenome Editing. Genes 2023, 14, 577. [Google Scholar] [CrossRef]
- Modell, B. Global epidemiology of haemoglobin disorders and derived service indicators. Bull. World Health Organ. 2008, 86, 480–487. [Google Scholar] [CrossRef]
- Turbpaiboon, C.; Wilairat, P. Alpha-hemoglobin stabilizing protein: Molecular function and clinical correlation. Front. Biosci. 2010, 15, 1–11. [Google Scholar] [CrossRef]
- Cardiero, G.; Musollino, G.; Prezioso, R.; Lacerra, G. mRNA Analysis of Frameshift Mutations with Stop Codon in the Last Exon: The Case of Hemoglobins Campania [α1 cod95 (−C)] and Sciacca [α1 cod109 (−C)]. Biomedicines 2021, 9, 1390. [Google Scholar] [CrossRef]
- Yang, P.; Chou, S.-J.; Li, J.; Hui, W.; Liu, W.; Sun, N.; Zhang, R.Y.; Zhu, Y.; Tsai, M.-L.; Lai, H.I.; et al. Supramolecular nanosubstrate–mediated delivery system enables CRISPR-Cas9 knockin of hemoglobin beta gene for hemoglobinopathies. Sci. Adv. 2020, 6, eabb7107. [Google Scholar] [CrossRef]
- Nualkaew, T.; Sii-Felice, K.; Giorgi, M.; McColl, B.; Gouzil, J.; Glaser, A.; Voon, H.P.; Tee, H.Y.; Grigoriadis, G.; Svasti, S.; et al. Coordinated β-globin expression and α2-globin reduction in a multiplex lentiviral gene therapy vector for β-thalassemia. Mol. Ther. 2021, 29, 2841–2853. [Google Scholar] [CrossRef] [PubMed]
- Kiem, H.-P.; I Arumugam, P.; Burtner, C.R.; Fox, C.F.; Beard, B.C.; Dexheimer, P.; E Adair, J.; Malik, P. Pigtailed macaques as a model to study long-term safety of lentivirus vector-mediated gene therapy for hemoglobinopathies. Mol. Ther. Methods Clin. Dev. 2014, 1, 14055. [Google Scholar] [CrossRef] [PubMed]
- Thom, C.S.; Dickson, C.F.; Gell, D.A.; Weiss, M.J. Hemoglobin Variants: Biochemical Properties and Clinical Correlates. Cold Spring Harb. Perspect. Med. 2013, 3, a011858. [Google Scholar] [CrossRef] [PubMed]
- Masson, E.; Zou, W.-B.; Génin, E.; Cooper, D.N.; Le Gac, G.; Fichou, Y.; Pu, N.; Rebours, V.; Férec, C.; Liao, Z.; et al. Expanding ACMG variant classification guidelines into a general framework. Hum. Genom. 2022, 16, 31. [Google Scholar] [CrossRef]
- Carss, K.; Goldstein, D.; Aggarwal, V.; Petrovski, S. Variant Interpretation and Genomic Medicine. In Handbook of Statistical Genomics; Balding, D., Moltke, I., Marioni, J., Eds.; John Wiley & Sons Ltd.: Hoboken, NJ, USA, 2019. [Google Scholar] [CrossRef]
- Chen, J.-M.; Masson, E.; Zou, W.-B.; Liao, Z.; Génin, E.; Cooper, D.N.; Férec, C. Validation of the ACMG/AMP guidelines-based seven-category variant classification system. medRxiv 2023. [Google Scholar] [CrossRef]
- Walsh, N.; Cooper, A.; Dockery, A.; O’Byrne, J.J. Variant reclassification and clinical implications. J. Med. Genet. 2024, 61, 207–211. [Google Scholar] [CrossRef]
- Sabath, D.E. Molecular Diagnosis of Thalassemias and Hemoglobinopathies. Am. J. Clin. Pathol. 2017, 148, 6–15. [Google Scholar] [CrossRef]
- Kountouris, P.; Stephanou, C.; Lederer, C.W.; Traeger-Synodinos, J.; Bento, C.; Harteveld, C.L.; Fylaktou, E.; Koopmann, T.T.; Halim-Fikri, H.; Michailidou, K.; et al. Adapting the ACMG/AMP variant classification framework: A perspective from the ClinGen Hemoglobinopathy Variant Curation Expert Panel. Hum. Mutat. 2021, 43, 1089–1096. [Google Scholar] [CrossRef]
- Turner, S.A.; Rao, S.K.; Morgan, R.H.; Vnencak-Jones, C.L.; Wiesner, G.L. The impact of variant classification on the clinical management of hereditary cancer syndromes. Anesth. Analg. 2019, 21, 426–430. [Google Scholar] [CrossRef]
- David, S.; Nora Syahirah, S.; Muhammad Nur Salam Bin, H.; Rajan, R. The Blood Blues: A Review on Methemoglobinemia. J. Pharmacol. Pharmacother. 2018, 9, 5. [Google Scholar]
- Boylston, M.; Beer, D. Methemoglobinemia: A case study. Crit. Care Nurse 2002, 22, 50–55. [Google Scholar] [CrossRef] [PubMed]
- Ashurst, J.; Wasson, M. Methemoglobinemia: A systematic review of the pathophysiology, detection, and treatment. Delw. Med. J. 2011, 83, 203–208. [Google Scholar] [PubMed]
- Nascimento, T.S.D.; Pereira, R.O.L.; de Mello, H.L.D.; Costa, J. Metemoglobinemia: Do diagnóstico ao tratamento. Braz. J. Anesthesiol. 2008, 58, 651–664. [Google Scholar] [CrossRef]
- Iolascon, A.; Bianchi, P.; Andolfo, I.; Russo, R.; Barcellini, W.; Fermo, E.; Toldi, G.; Ghirardello, S.; Rees, D.; Van Wijk, R.; et al. Recommendations for diagnosis and treatment of methemoglobinemia. Am. J. Hematol. 2021, 96, 1666–1678. [Google Scholar] [CrossRef] [PubMed]
- Ash-Bernal, R.; Wise, R.; Wright, S.M. Acquired Methemoglobinemia: A retrospective series of 138 cases at 2 teaching hospitals. Medicine 2004, 83, 265–273. [Google Scholar] [CrossRef]
- Friedman, N.; Scolnik, D.; McMurray, L.; Bryan, J. Acquired methemoglobinemia presenting to the pediatric emergency department: A clinical challenge. CJEM 2020, 22, 673–677. [Google Scholar] [CrossRef] [PubMed]
- Karna, B.; Jha, S.K.; Al Zaabi, E. Hemoglobin C Disease. 2023. Available online: https://www.ncbi.nlm.nih.gov/books/NBK559043/ (accessed on 12 August 2024).
- Charache, S.; Conley, C.L.; Waugh, D.F.; Ugoretz, R.J.; Spurrell, J.R. Pathogenesis of Hemolytic Anemia in Homozygous Hemoglobin C Disease. J. Clin. Investig. 1967, 46, 1795–1811. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, K.L.; Randolph, T.R. Development of a Microscopic Method to Identify Hemoglobin C Conditions for Use in Developing Countries. Am. Soc. Clin. Lab. Sci. 2019, 32, 61–66. [Google Scholar] [CrossRef]
- Ohiri, C.D.; Randolph, T.R. Development of an easy, inexpensive, and precise method to identify Hemoglobin C for use in underdeveloped countries. FASEB J. 2016, 30, 61–66. [Google Scholar] [CrossRef]
- Verra, F.; Simpore, J.; Warimwe, G.M.; Tetteh, K.K.; Howard, T.; Osier, F.H.A.; Bancone, G.; Avellino, P.; Blot, I.; Fegan, G.; et al. Haemoglobin C and S Role in Acquired Immunity against Plasmodium falciparum Malaria. PLoS ONE 2007, 2, e978. [Google Scholar] [CrossRef]
- Ghosh, A.; Basak, J.; Mukhopadhyay, A. Coexistence of rare variant HbD Punjab [α2β2121(Glu→Gln)] and alpha 3.7 kb deletion in a young boy of Hindu family in West Bengal, India. Cell. Mol. Biol. Lett. 2015, 20, 736–742. [Google Scholar] [CrossRef] [PubMed]
- Italia, K.; Upadhye, D.; Dabke, P.; Kangane, H.; Colaco, S.; Sawant, P.; Nadkarni, A.; Gorakshakar, A.; Jain, D.; Italia, Y.; et al. Clinical and hematological presentation among Indian patients with common hemoglobin variants. Clin. Chim. Acta 2014, 431, 46–51. [Google Scholar] [CrossRef] [PubMed]
- Oberoi, S.M.; Das, R.; Trehan, A.; Ahluwalia, J.; Bansal, D.M.; Malhotra, P.; Marwaha, R.K.M. HbSD-Punjab. J. Pediatr. Hematol. 2014, 36, e140–e144. [Google Scholar] [CrossRef] [PubMed]
- Pandey, S.; Mishra, R.M.; Pandey, S.; Shah, V.; Saxena, R. Molecular characterization of hemoglobin D Punjab traits and clinical-hematological profile of the patients. Sao Paulo Med. J. 2012, 130, 248–251. [Google Scholar] [CrossRef] [PubMed]
- A Petrenko, A.; Pivnik, A.V.; Kim, P.P.; Demidova, E.Y.; Surin, V.L.; O Abdullaev, A.; Sudarikov, A.B.; A Petrova, N.; A Maryina, S. Coinheritance of HbD-Punjab/β+-thalassemia (IVSI+5 G-C) in patient with Gilbert’s syndrome. Ter. Arkh. 2018, 90, 105–109. [Google Scholar] [CrossRef]
- Rieder, R. Globin chain synthesis in HbD (Punjab)-beta-thalassemia. Blood 1976, 47, 113–120. [Google Scholar] [CrossRef]
- Ohashi, J.; Naka, I.; Patarapotikul, J.; Hananantachai, H.; Brittenham, G.; Looareesuwan, S.; Clark, A.G.; Tokunaga, K. Extended Linkage Disequilibrium Surrounding the Hemoglobin E Variant Due to Malarial Selection. Am. J. Hum. Genet. 2004, 74, 1198–1208. [Google Scholar] [CrossRef]
- Flatz, G.; Sanguansermsri, T.; Sengchanh, S.; Horst, D.; Horst, J. The ‘Hot Spot’ of Hb E [β26(B8)Glu→Lys] in Southeast Asia: β-Globin Anomalies in the Lao Theung Population of Southern Laos. Hemoglobin 2004, 28, 197–204. [Google Scholar] [CrossRef]
- Roche, C.J.; Malashkevich, V.; Balazs, T.C.; Dantsker, D.; Chen, Q.; Moreira, J.; Almo, S.C.; Friedman, J.M.; Hirsch, R.E. Structural and Functional Studies Indicating Altered Redox Properties of Hemoglobin E. J. Biol. Chem. 2011, 286, 23452–23466. [Google Scholar] [CrossRef]
- Strader, M.B.; Kassa, T.; Meng, F.; Wood, F.B.; Hirsch, R.E.; Friedman, J.M.; Alayash, A.I. Oxidative instability of hemoglobin E (β26 Glu→Lys) is increased in the presence of free α subunits and reversed by α-hemoglobin stabilizing protein (AHSP): Relevance to HbE/β-thalassemia. Redox Biol. 2016, 8, 363–374. [Google Scholar] [CrossRef]
- Jamsai, D.; Zaibak, F.; Vadolas, J.; Voullaire, L.; Fowler, K.J.; Gazeas, S.; Peters, H.; Fucharoen, S.; Williamson, R.; Ioannou, P.A. A humanized BAC transgenic/knockout mouse model for HbE/β-thalassemia. Genomics 2006, 88, 309–315. [Google Scholar] [CrossRef] [PubMed]
- Naka, I.; Ohashi, J.; Nuchnoi, P.; Hananantachai, H.; Looareesuwan, S.; Tokunaga, K.; Patarapotikul, J. Lack of Association of the HbE Variant with Protection from Cerebral Malaria in Thailand. Biochem. Genet. 2008, 46, 708–711. [Google Scholar] [CrossRef] [PubMed]
- Kidd, R.D.; Baker, H.M.; Mathews, A.J.; Brittain, T.; Baker, E.N. Oligomerization and ligand binding in a homotetrameric hemoglobin: Two high-resolution crystal structures of hemoglobin Bart’s (γ4), a marker for α-thalassemia. Protein Sci. 2001, 10, 1739–1749. [Google Scholar] [CrossRef] [PubMed]
- Pootrakul, S.; Wasi, P.; Na-Nakorn, S. Haemoglobin Bart’s hydrops foetalis in Thailand. Ann. Hum. Genet. 1967, 30, 293–311. [Google Scholar] [CrossRef] [PubMed]
- Pembrey, M.E.; Weatherall, D.J.; Clegg, J.B.; Bunch, C.; Perrine, R.P. Haemoglobin Bart’s in Saudi Arabia. Br. J. Haematol. 1975, 29, 221–234. [Google Scholar] [CrossRef]
- Czerwinski, E.; Czerwinski, E.; Risk, M.; Risk, M.; Matustik, M.; Matustik, M. Crystallization and preliminary X-ray diffraction studies of methemoglobin Bart’s. J. Biol. Chem. 1981, 256, 13128–13129. [Google Scholar] [CrossRef]
- Papassotiriou, I.; Traeger-Synodinos, J.; Vlachou, C.; Karagiorga, M.; Metaxotou, A.; Kanavakis, E.; Stamoulakatou, A. Rapid and Accurate Quantitation of Hb Bart’s and Hb H Using Weak Cation Exchange High Performance Liquid Chromatography: Correlation with the α-Thalassemia Genotype. Hemoglobin 1999, 23, 203–211. [Google Scholar] [CrossRef]
- Rugless, M.J.; Fisher, C.A.; Stephens, A.D.; Amos, R.J.; Mohammed, T.; Old, J.M. Hb Bart’s in Cord Blood: An Accurate Indicator of α-Thalassemia. Hemoglobin 2006, 30, 57–62. [Google Scholar] [CrossRef]
- Sasazuki, T.; Isomoto, A.; Nakajima, H. Circular dichroism and absorption spectra of haemoglobin Bart’s. J. Mol. Biol. 1972, 65, 365–369. [Google Scholar] [CrossRef]
- Esan, G.J.F. Haemoglobin Bart’s in Newborn Nigerians. Br. J. Haematol. 1972, 22, 73–86. [Google Scholar] [CrossRef]
- Gibson, Q.H.; Nagel, R.L. Allosteric Transition and Ligand Binding in Hemoglobin Chesapeake. J. Biol. Chem. 1974, 249, 7255–7259. [Google Scholar] [CrossRef] [PubMed]
- Jones, C.M.; Charache, S.; Hathaway, P. The effect of hemoglobin F-Chesapeake (α292 Arg.→Leuγ2) on fetal oxygen affinity and erythropoiesis. Pediatr. Res. 1979, 13, 851–853. [Google Scholar] [CrossRef] [PubMed]
- Charache, S. A manifestation of abnormal hemoglobins of man: Altered oxygen affinity. Hemoglobin Chesapeake: From the clinic to the laboratory, and back again. Ann. N. Y. Acad. Sci. 1974, 241, 449–455. [Google Scholar] [CrossRef] [PubMed]
- Wajcman, H.; Galactéros, F. Hemoglobins with High Oxygen Affinity Leading to Erythrocytosis. New Variants and New Concepts. Hemoglobin 2005, 29, 91–106. [Google Scholar] [CrossRef] [PubMed]
- Harteveld, C.L.; Wijermans, P.W.; Arkesteijn, S.G.; Van Delft, P.; Kerkhoffs, J.-L.; Giordano, P.C. Hb Lepore-Leiden: A New δ/β Rearrangement Associated with a β-Thalassemia Minor Phenotype. Hemoglobin 2008, 32, 446–453. [Google Scholar] [CrossRef]
- Pirastru, M.; Manca, L.; Trova, S.; Mereu, P. Biochemical and Molecular Analysis of the Hb Lepore Boston Washington in a Syrian Homozygous Child. BioMed Res. Int. 2017, 2017, 1261972. [Google Scholar] [CrossRef]
- Mirabile, E.; Testa, R.; Consalvo, C.; Dickerhoff, R.; Schilirò, G. Association of Hb S/Hb lepore and δβ-thalassemia/Hb lepore in Sicilian patients: Review of the presence of Hb lepore in Sicily. Eur. J. Haematol. 1995, 55, 126–130. [Google Scholar] [CrossRef]
- Seward, D.P.; Ware, R.E.; Kinney, T.R. Hemoglobin sickle-lepore: Report of two siblings and review of the literature. Am. J. Hematol. 1993, 44, 192–195. [Google Scholar] [CrossRef]
- Efremov, G.D. Hemoglobins Lepore and Anti-Lepore. Hemoglobin 1978, 2, 197–233. [Google Scholar] [CrossRef]
- Chaibunruang, A.; Srivorakun, H.; Fucharoen, S.; Fucharoen, G.; Sae-Ung, N.; Sanchaisuriya, K. Interactions of hemoglobin Lepore (deltabeta hybrid hemoglobin) with v arious hemoglobinopathies: A molecular and hematological characteristi cs and differential diagnosis. Blood Cells Mol. Dis. 2010, 44, 140–145. [Google Scholar] [CrossRef]
- Jiang, F.; Tang, X.-W.; Li, J.; Zhou, J.-Y.; Zuo, L.-D.; Li, D.-Z. Hb Lepore-Hong Kong: First Report of a Novel δ/β-Globin Gene Fusion in a Chinese Family. Hemoglobin 2021, 45, 220–224. [Google Scholar] [CrossRef] [PubMed]
- Hunt, D.M.; Higgs, D.R.; Winichagoon, P.; Clegg, J.B.; Weatherall, D.J. Haemoglobin Constant Spring has an unstable α chain messenger RNA. Br. J. Haematol. 1982, 51, 405–413. [Google Scholar] [CrossRef] [PubMed]
- Thonglairoam, V.; Fucharoen, S.; Tanphaichitr, V.S.; Pung-Amritt, P.; Embury, S.H.; Winichagoon, P.; Wasi, P. Hemoglobin constant spring in bangkok: Molecular screening by selective enzymatic amplification of the α2-globin gene. Am. J. Hematol. 1991, 38, 277–280. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, V.H.; Sanchaisuriya, K.; Wongprachum, K.; Nguyen, M.D.; Phan, T.T.H.; Vo, V.T.; Sanchaisuriya, P.; Fucharoen, S.; Schelp, F.P. Hemoglobin Constant Spring is markedly high in women of an ethnic minority group in Vietnam: A community-based survey and hematologic features. Blood Cells Mol. Dis. 2014, 52, 161–165. [Google Scholar] [CrossRef]
- Jomoui, W.; Fucharoen, G.; Sanchaisuriya, K.; Nguyen, V.H.; Fucharoen, S. Hemoglobin Constant Spring among Southeast Asian Populations: Haplotypic Heterogeneities and Phylogenetic Analysis. PLoS ONE 2015, 10, e0145230. [Google Scholar] [CrossRef]
- Charoenkwan, P.; Sirichotiyakul, S.; Chanprapaph, P.; Tongprasert, F.; Taweephol, R.; Sae-Tung, R.; Sanguansermsri, T. Anemia and Hydrops in a Fetus with Homozygous Hemoglobin Constant Spring. J. Pediatr. Hematol. 2006, 28, 827–830. [Google Scholar] [CrossRef]
- Kropp, G.; Fucharoen, S.; Embury, S. Selective enzymatic amplification of alpha 2-globin DNA for detection of the hemoglobin Constant Spring mutation. Blood 1989, 73, 1987–1992. [Google Scholar] [CrossRef]
- Roberts, W.L. Hemoglobin Constant Spring Can Interfere with Glycated Hemoglobin Measurements by Boronate Affinity Chromatography. Clin. Chem. 2007, 53, 142–143. [Google Scholar] [CrossRef]
- Zimmerman, S.A.; O’Branski, E.E.; Rosse, W.F.; Ware, R.E. Hemoglobin S/O(Arab): Thirteen new cases and review of the lit-erature. Am. J. Hematol. 1999, 60, 279–284. [Google Scholar] [CrossRef]
- Dror, S. Clinical and hematological features of homozygous hemoglobin O-Arab [beta 121 Glu → Lys]. Pediatr. Blood Cancer 2012, 60, 506–507. [Google Scholar] [CrossRef]
- Sangaré, A.; Sanogo, I.; Meité, M.; Ambofo, Y.; Abesopie, V.; Ségbéna, A.; Tolo, A. Hemoglobin O Arab in Ivory Coast and western Africa. Med. Trop. (Mars) 1992, 52, 163–167. [Google Scholar] [PubMed]
- El-Hazmi, M.; Lehmann, H. Human Haemoglobins and Haemoglobinopathies in Arabia: Hb O Arab in Saudi Arabia. Acta Haematol. 1980, 63, 268–273. [Google Scholar] [CrossRef] [PubMed]
- Milner, P.F.; Miller, C.; Grey, R.; Seakins, M.; DeJong, W.W.; Went, L.N. Hemoglobin O Arab in Four Negro Families and Its Interaction with Hemoglobin S and Hemoglobin C. N. Engl. J. Med. 1970, 283, 1417–1425. [Google Scholar] [CrossRef] [PubMed]
- Hafsia, R.; Gouider, E.; Ben Moussa, S.; Ben Salah, N.; Elborji, W.; Hafsia, A. Hemoglobin O Arab: About 20 cases. Tunis. Med. 2007, 85, 637–640. [Google Scholar] [PubMed]
- Merritt, D.; Jones, R.T.; Head, C.; Thibodeau, S.N.; Fairbanks, V.F.; Steinberg, M.H.; Coleman, M.B.; Rodgers, G.P. HB Seal Rock [(α2)142 Term→glu, Codon 142 TAA→GAA]: An Extended α Chain Variant Associated with Anemia, Microcytosis, and α-Thalassemia-2 (-3.7 KB). Hemoglobin 1997, 21, 331–344. [Google Scholar] [CrossRef]
- Préhu, C.; Moradkhani, K.; Riou, J.; Bahuau, M.; Launay, P.; Martin, N.; Wajcman, H.; Goossens, M.; Galactéros, F. Chronic hemolytic anemia due to novel -globin chain variants: Critical location of the mutation within the gene sequence for a dominant effect. Haematologica 2009, 94, 1624–1625. [Google Scholar] [CrossRef]
- Fattori, A.; Kimura, E.; Albuquerque, D.; Oliveira, D.; Costa, F.; Sonati, M. Hb Indianapolis [β112 (G14) Cys→Arg] as the probable cause of moderate hemolytic anemia and renal damage in a Brazilian patient. Am. J. Hematol. 2007, 82, 672–675. [Google Scholar] [CrossRef]
- Henderson, S.J.; Timbs, A.T.; McCarthy, J.; Gallienne, A.E.; Proven, M.; Rugless, M.J.; Lopez, H.; Eglinton, J.; Dziedzic, D.; Beardsall, M.; et al. Ten Years of Routineα- andβ-Globin Gene Sequencing in UK Hemoglobinopathy Referrals Reveals 60 Novel Mutations. Hemoglobin 2016, 40, 75–84. [Google Scholar] [CrossRef]
- Outeirino, J.; Casey, R.; White, J.; Lehmann, H. Haemoglobin Madrid β115 (G17) Alanine→Proline: An Unstable Variant Associated with Haemolytic Anaemia. Acta Haematol. 1974, 52, 53–60. [Google Scholar] [CrossRef]
- Kim, J.Y.; Park, S.S.; Jung, H.L.; Keum, D.H.; Park, H.; Chang, Y.H.; Lee, Y.J.; Cho, H.I. Hb Madrid [β115(G17)Ala→Pro] in a Korean Family with Chronic Hemolytic Anemia. Hemoglobin 2000, 24, 133–138. [Google Scholar] [CrossRef]
- Edison, E.S.; Shaji, R.V.; Devi, S.G.; Kumar, S.S.; Srivastava, A.; Chandy, M. Hb Showa-Yakushiji [beta110(G12)Leu-->Pro] in four unrelated patients from west Bengal. Hemoglobin 2005, 29, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Villegas, A.; Ropero, P.; Nogales, A.; González, F.A.; Mateo, M.; Mazo, E.; Rodrigo, E.; Arias, M. Hb Santander [β34(B16)Val→Asp (GTC → GAC)]: A New Unstable Variant Found as a De Novo Mutation in a Spanish Patient. Hemoglobin 2003, 27, 31–35. [Google Scholar] [CrossRef] [PubMed]
- Nakatsuji, T.; Miwa, S.; Ohba, Y.; Hattori, Y.; Miyaji, T.; Hino, S.; Matsumoto, N. A New Unstable Hemoglobin, Hb Yokohama β31(B13)LEU → Pro, Causing Hemolytic Anemia. Hemoglobin 1981, 5, 667–678. [Google Scholar] [CrossRef] [PubMed]
- Huehns, E.R.; Hecht, F.; Yoshida, A.; Stamatoyannopoulos, G.; Hartman, J.; Motulsky, A.G. Hemoglobin-Seattle (α2Aβ276 Glu): An Unstable Hemoglobin Causing Chronic Hemolytic Anemia. Blood 1970, 36, 209–218. [Google Scholar] [CrossRef]
- Hoyer, J.D.; Baxter, J.K.; Moran, A.M.; Kubic, K.S.; Ehmann, W.C. Two Unstable β Chain Variants Associated with β-Thalassemia: Hb Miami [β116(G18)His→Pro], and Hb Hershey [β70(E14)Ala→Gly], and a Second Unstable Hb Variant at β70: Hb Abington [β70(E14)Ala→Pro]. Hemoglobin 2005, 29, 241–248. [Google Scholar] [CrossRef] [PubMed]
- Li, W. Biophysical Basis of Hb-S Polymerization in Red Blood Cell Sickling. bioRxiv 2019. [Google Scholar] [CrossRef]
- Karen, C. Sickle Cell Disease: A Genetic Disorder of Beta-Globin. Thalassemia and Other Hemolytic Anemias; IntechOpen: Rijeka, Croatia, 2018. [Google Scholar]
- Ellsworth, P.; Sparkenbaugh, E.M. Targeting the von Willebrand Factor–ADAMTS-13 axis in sickle cell disease. J. Thromb. Haemost. 2023, 21, 2–6. [Google Scholar] [CrossRef]
- Nader, E.; Romana, M.; Connes, P. The Red Blood Cell—Inflammation Vicious Circle in Sickle Cell Disease. Front. Immunol. 2020, 11, 454. [Google Scholar] [CrossRef]
- Ashley-Koch, A.; Yang, Q.; Olney, R.S. Sickle Hemoglobin (Hb S) Allele and Sickle Cell Disease: A HuGE Review. Am. J. Epidemiol. 2000, 151, 839–845. [Google Scholar] [CrossRef]
- Al-Fatlawi, A.C.Y. A Review on Sickle Cells Disease. Sci. J. Med. Res. 2019, 3, 146–148. [Google Scholar] [CrossRef]
- Poli, M.C.; Orange, J. CRISPR/Cas9β-globin Gene Targeting in Human Haematopoietic Stem Cells. Nature 2017, 140 (Suppl. S3), S226–S227. [Google Scholar]
- Demirci, S.; Gudmundsdottir, B.; Li, Q.; Haro-Mora, J.J.; Nassehi, T.; Drysdale, C.; Yapundich, M.; Gamer, J.; Seifuddin, F.; Tisdale, J.F.; et al. βT87Q-Globin Gene Therapy Reduces Sickle Hemoglobin Production, Allowing for Ex Vivo Anti-sickling Activity in Human Erythroid Cells. Mol. Ther. Methods Clin. Dev. 2020, 17, 912–921. [Google Scholar] [CrossRef] [PubMed]
- De Simone, G.; Quattrocchi, A.; Mancini, B.; di Masi, A.; Nervi, C.; Ascenzi, P. Thalassemias: From gene to therapy. Mol. Asp. Med. 2022, 84, 101028. [Google Scholar] [CrossRef]
- Angastiniotis, M.; Lobitz, S. Thalassemias: An Overview. Int. J. Neonatal Screen. 2019, 5, 16. [Google Scholar] [CrossRef]
- Weatherall, D.J. Fortnightly review: The thalassaemias. BMJ 1997, 314, 1675–1678. [Google Scholar] [CrossRef] [PubMed]
- Muncie, H.; James, C. Alpha and beta thalassemia. Am. Fam. Physician 2009, 80, 339–344. [Google Scholar]
- Vernimmen, D. Globins, from Genes to Physiology and Diseases. Blood Cells Mol. Dis. 2018, 70, 1. [Google Scholar] [CrossRef]
- Aksu, T.; Unal, S. Thalassemia. Trends Pediatr. 2021, 2, 1–7. [Google Scholar] [CrossRef]
- Meri, M.A.; Al-Hakeem, A.H.; Al-Abeadi, R.S. Overview on thalassemia: A review article. Med. Sci. J. Adv. Res. 2022, 3, 26–32. [Google Scholar] [CrossRef]
- Reiss, G.H.; Ranney, H.M.; Shaklai, N. Association of hemoglobin C with erythrocyte ghosts. J. Clin. Investig. 1982, 70, 946–952. [Google Scholar] [CrossRef]
- Singh, N.; Seth, T.; Tyagi, S. Review of Clinical and Hematological Profile of Hemoglobin D Cases in a Single Centre. J. Mar. Med. Soc. 2023, 25 (Suppl. S1), S74–S79. [Google Scholar] [CrossRef]
- Mukherjee, S.; Das, M.; Basu, K.; Sengupta, M.; Karmakar, S.; Jha, A.K.; Bandopadhyay, M. HbE Variants: An Experience from Tertiary Care Centre of Eastern India. Ann. Pathol. Lab. Med. 2020, 7, A570–A575. [Google Scholar] [CrossRef]
- Winterbourn, C.C. Oxidative denaturation in congenital hemolytic anemias: The unstable hemoglobins. Semin. Hematol. 1990, 27, 41–50. [Google Scholar] [PubMed]
- Taketa, F.; Huang, Y.; Libnoch, J.; Dessel, B. Hemoglobin wood β97(FG4) His → Leu: A new high-oxygen-affinity hemoglobin associated with familial erythrocytosis. Biochim. Biophys. Acta (BBA) Protein Struct. 1975, 400, 348–353. [Google Scholar] [CrossRef]
- Reed, C.S.; Hampson, R.; Gordon, S.; Jones, R.T.; Novy, M.J.; Brimhall, B.; Edwards, M.J.; Koler, R.D. Erythrocytosis Secondary to Increased Oxygen Affinity of a Mutant Hemoglobin, Hemoglobin Kempsey. Blood 1968, 31, 623–632. [Google Scholar] [CrossRef]
- Medri, C.; Méndez, A.; Hammerer-Lercher, A.; Rovó, A.; Angelillo-Scherrer, A. Unstable hemoglobin Montreal II uncovered in an adult with unexplained hemolysis exacerbated by a presumed viral infection: A case report. J. Med. Case Rep. 2022, 16, 145. [Google Scholar] [CrossRef]
- Fallon, J.A.; Smith, E.P.; Schoch, N.; Paruk, J.D.; Adams, E.A.; Evers, D.C.; Jodice, P.G.; Perkins, C.; Schulte, S.; Hopkins, W.A. Hematological indices of injury to lightly oiled birds from the Deepwater Horizon oil spill. Environ. Toxicol. Chem. 2018, 37, 451–461. [Google Scholar] [CrossRef]
- Harr, K.E.; Cunningham, F.L.; Pritsos, C.A.; Pritsos, K.L.; Muthumalage, T.; Dorr, B.S.; Horak, K.E.; Hanson-Dorr, K.C.; Dean, K.M.; Cacela, D.; et al. Weathered MC252 crude oil-induced anemia and abnormal erythroid morphology in double-crested cormorants (Phalacrocorax auritus) with light microscopic and ultrastructural description of Heinz bodies. Ecotoxicol. Environ. Saf. 2017, 146, 29–39. [Google Scholar] [CrossRef]
- Sözen, M.; Karaaslan, C.; Öner, R.; Gümrük, F.; Özdemir, M.; Altay, C.; Gürgey, A.; Öner, C. Severe hemolytic anemia associated with Hb Volga [β27(B9)Ala→Asp]: GCC→GAC at codon 27 in a Turkish family. Am. J. Hematol. 2004, 76, 378–382. [Google Scholar] [CrossRef]
- Wali, Y.; Al Zadjali, S.; Elshinawy, M.; Beshlawi, I.; Fawaz, N.; AlKindi, S.; Rawas, A.; Alsinani, S.; Daar, S.; Krishnamoorthy, R. Severity ranking of non-deletional alpha thalassemic alleles: Insights from an Omani family study. Eur. J. Haematol. 2011, 86, 507–511. [Google Scholar] [CrossRef]
- Gallagher, P.G. Diagnosis and management of rare congenital nonimmune hemolytic disease. Hematol. Am. Soc. Hematol. Educ. Program 2015, 2015, 392–399. [Google Scholar] [CrossRef] [PubMed]
- Fallon, J.A.; Hopkins, W.A.; Fox, L. A practical quantification method for Heinz bodies in birds applicable to rapid response field scenarios. Environ. Toxicol. Chem. 2013, 32, 401–405. [Google Scholar] [CrossRef] [PubMed]
- Drouilly, M.; Jourdan, L.; Gérard, D.; Russello, J.; Bobée, V.; Audouy, A.; Phulpin, A.; Perrin, J. Infantile pyknocytosis, a neonatal hemolytic anemia with Heinz bodies: A cohort study. Pediatr. Blood Cancer 2024, 71, e31078. [Google Scholar] [CrossRef]
- Furdak, P.; Bartosz, G.; Stefaniuk, I.; Cieniek, B.; Bieszczad-Bedrejczuk, E.; Soszyński, M.; Sadowska-Bartosz, I. Effect of Garlic Extract on the Erythrocyte as a Simple Model Cell. Int. J. Mol. Sci. 2024, 25, 5115. [Google Scholar] [CrossRef] [PubMed]
- Salgado, B.; Monteiro, L.; Rocha, N. Allium species poisoning in dogs and cats. J. Venom. Anim. Toxins Incl. Trop. Dis. 2011, 17, 4–11. [Google Scholar] [CrossRef]
- Ideguchi, H. Effects of abnormal Hb on red cell membranes. Rinsho Byori 1999, 47, 232–237. [Google Scholar]
- Sugawara, Y.; Hayashi, Y.; Shigemasa, Y.; Abe, Y.; Ohgushi, I.; Ueno, E.; Shimamoto, F. Molecular Biosensing Mechanisms in the Spleen for the Removal of Aged and Damaged Red Cells from the Blood Circulation. Sensors 2010, 10, 7099–7121. [Google Scholar] [CrossRef]
- Ronquist, G.; Theodorsson, E. Inherited, non-spherocytic haemolysis due to deficiency of glucose-6-phosphate dehydrogenase. Scand. J. Clin. Lab. Investig. 2007, 67, 105–111. [Google Scholar] [CrossRef]
- Jacob, H.S.; Winterhalter, K.H. The role of hemoglobin heme loss in Heinz body formation: Studies with a partially heme-deficient hemoglobin and with genetically unstable hemoglobins. J. Clin. Investig. 1970, 49, 2008–2016. [Google Scholar] [CrossRef]
- Jacob, H.; Winterhalter, K. Unstable Hemoglobins: The Role of Heme Loss in Heinz Body Formation. Proc. Natl. Acad. Sci. USA 1970, 65, 697–701. [Google Scholar] [CrossRef]
- Winterbourn, C.C.; Carrell, R.W. Studies of Hemoglobin Denaturation and Heinz Body Formation in the Unstable Hemoglobins. J. Clin. Investig. 1974, 54, 678–689. [Google Scholar] [CrossRef] [PubMed]
- Simmers, R.N.; Mulley, J.C.; Hyland, V.J.; Callen, D.F.; Sutherland, G.R. Mapping the human alpha globin gene complex to 16p13.2—Pter. J. Med. Genet. 1987, 24, 761–766. [Google Scholar] [CrossRef] [PubMed]
- Nicholls, R.D.; A Jonasson, J.; O McGee, J.; Patil, S.; Ionasescu, V.V.; Weatherall, D.J.; Higgs, D.R. High resolution gene mapping of the human alpha globin locus. J. Med. Genet. 1987, 24, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Daniels, R.J.; Peden, J.F.; Lloyd, C.; Horsley, S.W.; Clark, K.; Tufarelli, C.; Kearney, L.; Buckle, V.J.; Doggett, N.A.; Flint, J.; et al. Sequence, structure and pathology of the fully annotated terminal 2 Mb of the short arm of human chromosome 16. Hum. Mol. Genet. 2001, 10, 339–352. [Google Scholar] [CrossRef]
- Higgs, D.R.; Wainscoat, J.S.; Flint, J.; Hill, A.V.; Thein, S.L.; Nicholls, R.D.; Teal, H.; Ayyub, H.; E Peto, T.; Falusi, A.G. Analysis of the human alpha-globin gene cluster reveals a highly informative genetic locus. Proc. Natl. Acad. Sci. USA 1986, 83, 5165–5169. [Google Scholar] [CrossRef]
- Coelho, A.; Picanço, I.; Seuanes, F.; Seixas, M.T.; Faustino, P. Novel large deletions in the human α-globin gene cluster: Clarifying the HS-40 long-range regulatory role in the native chromosome environment. Blood Cells Mol. Dis. 2010, 45, 147–153. [Google Scholar] [CrossRef]
- Higgs, D.R.; Hill, A.V.S.; Nicholls, R.; Goodbourn, S.E.Y.; Ayyub, H.; Teal, H.; Clegg, J.B.; Weatherall, D.J. Molecular Rearrangements of the Human α-Globin Gene Cluster. Ann. N. Y. Acad. Sci. 1985, 445, 45–56. [Google Scholar] [CrossRef]
- Hatton, C.S.; Wilkie, A.O.; Drysdale, H.C.; Wood, W.G.; Vickers, M.A.; Sharpe, J.; Ayyub, H.; Pretorius, I.M.; Buckle, V.J.; Higgs, D.R. Alpha-thalassemia caused by a large (62 kb) deletion upstream of the human alpha globin gene cluster. Blood 1990, 76, 221–227. [Google Scholar] [CrossRef]
- Steinberg, M.H. A New Trans-Acting Modulator of Fetal Hemoglobin? Acta Haematol. 2018, 140, 112–113. [Google Scholar] [CrossRef]
- Cao, A.; Galanello, R. Beta-thalassemia. Anesth. Analg. 2010, 12, 61–76. [Google Scholar] [CrossRef]
- Moleirinho, A.; Seixas, S.; Lopes, A.M.; Bento, C.; Prata, M.J.; Amorim, A. Evolutionary Constraints in the β-Globin Cluster: The Signature of Purifying Selection at the δ-Globin (HBD) Locus and Its Role in Developmental Gene Regulation. Genome Biol. Evol. 2013, 5, 559–571. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.; Draper, P.; De Braekeleer, M. High-resolution chromosomal localization of the β-gene of the human β-globin gene complex by in situ hybridization. Cytogenet. Genome Res. 1985, 39, 269–274. [Google Scholar] [CrossRef] [PubMed]
- Gusella, J.; Varsanyi-Breiner, A.; Kao, F.T.; Jones, C.; Puck, T.T.; Keys, C.; Orkin, S.; Housman, D. Precise localization of human beta-globin gene complex on chromosome 11. Proc. Natl. Acad. Sci. USA 1979, 76, 5239–5242. [Google Scholar] [CrossRef] [PubMed]
- Deisseroth, A.; Nienhuis, A.; Lawrence, J.; Giles, R.; Turner, P.; Ruddle, F.H. Chromosomal localization of human β globin gene on human chromosome 11 in somatic cell hybrids. Proc. Natl. Acad. Sci. USA 1978, 75, 1456–1460. [Google Scholar] [CrossRef]
- E Bauer, D.; Orkin, S.H. Update on fetal hemoglobin gene regulation in hemoglobinopathies. Curr. Opin. Pediatr. 2011, 23, 1–8. [Google Scholar] [CrossRef]
- Manning, J.M.; Dumoulin, A.; Li, X.; Manning, L.R. Normal and Abnormal Protein Subunit Interactions in Hemoglobins. J. Biol. Chem. 1998, 273, 19359–19362. [Google Scholar] [CrossRef]
- Kidd, R.D.; Russell, J.E.; Watmough, N.J.; Baker, E.N.; Brittain, T. The Role of β Chains in the Control of the Hemoglobin Oxygen Binding Function: Chimeric Human/Mouse Proteins, Structure, and Function. Biochemistry 2001, 40, 15669–15675. [Google Scholar] [CrossRef]
- Yamaguchi, T.; Pang, J.; Reddy, K.S.; Surrey, S.; Adachi, K. Role of β112 Cys (G14) in Homo-(β4) and Hetero-(α2β2) Tetramer Hemoglobin Formation. J. Biol. Chem. 1998, 273, 14179–14185. [Google Scholar] [CrossRef]
- Borgstahl, G.E.; Rogers, P.H.; Arnone, A. The 1·8 Å Structure of Carbonmonoxy-β4 Hemoglobin: Analysis of a Homotetramer with the R Quaternary Structure of Liganded α2β2 Hemoglobin. J. Mol. Biol. 1994, 236, 817–830. [Google Scholar] [CrossRef]
- A Walder, J.; Chatterjee, R.; Steck, T.L.; Low, P.S.; Musso, G.F.; Kaiser, E.T.; Rogers, P.H.; Arnone, A. The interaction of hemoglobin with the cytoplasmic domain of band 3 of the human erythrocyte membrane. J. Biol. Chem. 1984, 259, 10238–10246. [Google Scholar] [CrossRef]
- Gell, D.; Kong, Y.; Eaton, S.A.; Weiss, M.J.; Mackay, J.P. Biophysical Characterization of the α-Globin Binding Protein α-Hemoglobin Stabilizing Protein. J. Biol. Chem. 2002, 277, 40602–40609. [Google Scholar] [CrossRef] [PubMed]
- Domingues-Hamdi, E.; Vasseur, C.; Fournier, J.-B.; Marden, M.C.; Wajcman, H.; Baudin-Creuza, V. Role of α-Globin H Helix in the Building of Tetrameric Human Hemoglobin: Interaction with α-Hemoglobin Stabilizing Protein (AHSP) and Heme Molecule. PLoS ONE 2014, 9, e111395. [Google Scholar] [CrossRef] [PubMed]
- Feng, L.; Gell, D.A.; Zhou, S.; Gu, L.; Kong, Y.; Li, J.; Hu, M.; Yan, N.; Lee, C.; Rich, A.M.; et al. Molecular Mechanism of AHSP-Mediated Stabilization of α-Hemoglobin. Cell 2004, 119, 629–640. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Kong, Y.; Dore, L.C.; Abdulmalik, O.; Katein, A.M.; Zhou, S.; Choi, J.K.; Gell, D.; Mackay, J.P.; Gow, A.J.; et al. An erythroid chaperone that facilitates folding of α-globin subunits for hemoglobin synthesis. J. Clin. Investig. 2007, 117, 1856–1865. [Google Scholar] [CrossRef]
- Mollan, T.L.; Yu, X.; Weiss, M.J.; Olson, J.S. The Role of Alpha-Hemoglobin Stabilizing Protein in Redox Chemistry, Denaturation, and Hemoglobin Assembly. Antioxid. Redox Signal. 2010, 12, 219–231. [Google Scholar] [CrossRef]
- Yu, X.; Mollan, T.L.; Butler, A.; Gow, A.J.; Olson, J.S.; Weiss, M.J. Analysis of human α globin gene mutations that impair binding to the α hemoglobin stabilizing protein. Blood 2009, 113, 5961–5969. [Google Scholar] [CrossRef]
- Voon, H.P.J.; Vadolas, J. Controlling-globin: A review of-globin expression and its impact on -thalassemia. Haematologica 2008, 93, 1868–1876. [Google Scholar] [CrossRef]
- Ho, H.-Y.; Cheng, M.-L.; Chiu, D.T.-Y. Glucose-6-phosphate dehydrogenase—From oxidative stress to cellular functions and degenerative diseases. Redox Rep. 2007, 12, 109–118. [Google Scholar] [CrossRef]
- Arese, P.; Gallo, V.; Pantaleo, A.; Turrini, F. Life and Death of Glucose-6-Phosphate Dehydrogenase (G6PD) Deficient Erythrocytes—Role of Redox Stress and Band 3 Modifications. Transfus. Med. Hemotherapy 2012, 39, 328–334. [Google Scholar] [CrossRef]
- Janney, S.; Joist, J.; Fitch, C. Excess release of ferriheme in G6PD-deficient erythrocytes: Possible cause of hemolysis and resistance to malaria. Blood 1986, 67, 331–333. [Google Scholar] [CrossRef]
- Scott, M.D.; Zuo, L.; Lubin, B.H.; Chiu, D.T. NADPH, not glutathione, status modulates oxidant sensitivity in normal and glucose-6-phosphate dehydrogenase-deficient erythrocytes. Blood 1991, 77, 2059–2064. [Google Scholar] [CrossRef]
- Nicol, C.J.; Zielenski, J.; Tsui, L.; Wells, P.G. An embryoprotective role for glucose-6-phosphate dehydrogenase in developmental oxidative stress and chemical teratogenesis. FASEB J. 2000, 14, 111–127. [Google Scholar] [CrossRef] [PubMed]
- Francis, R.O.; Jhang, J.S.; Pham, H.P.; Hod, E.A.; Zimring, J.C.; Spitalnik, S.L. Glucose-6-phosphate dehydrogenase deficiency in transfusion medicine: The unknown risks. Vox Sang. 2013, 105, 271–282. [Google Scholar] [CrossRef] [PubMed]
- Fibach, E.; Rachmilewitz, E. The Role of Oxidative Stress in Hemolytic Anemia. Curr. Mol. Med. 2008, 8, 609–619. [Google Scholar] [CrossRef] [PubMed]
- Haghpanah, S.; Hosseini-Bensenjan, M.; Zekavat, O.R.; Bordbar, M.; Karimi, M.; Ramzi, M.; Asmarian, N. The Effect of N-Acetyl Cysteine and Vitamin E on Oxidative Status and Hemoglobin Level in Transfusion-Dependent Thalassemia Patients: A Systematic Review and Meta-Analysis. Iran. J. Blood Cancer 2021, 15, 22–35. [Google Scholar] [CrossRef]
- Halima, W.M.A.B.; Hannemann, A.; Rees, D.; Brewin, J.; Gibson, J. The Effect of Antioxidants on the Properties of Red Blood Cells from P atients with Sickle Cell Anemia. Front. Physiol. 2019, 10, 976. [Google Scholar]
- Pallotta, V.; Gevi, F.; D’Alessandro, A.; Zolla, L. Storing red blood cells with vitamin C and N-acetylcysteine prevents oxidative stress-related lesions: A metabolomics overview. Blood Transfus. 2014, 12, 376–387. [Google Scholar]
- Delesderrier, E.; Curioni, C.; Omena, J.; Macedo, C.R.; Cople-Rodrigues, C.; Citelli, M. Antioxidant nutrients and hemolysis in sickle cell disease. Clin. Chim. Acta 2020, 510, 381–390. [Google Scholar] [CrossRef]
- Fibach, E.; Rachmilewitz, E.A. The role of antioxidants and iron chelators in the treatment of oxidative stress in thalassemia. Ann. N. Y. Acad. Sci. 2010, 1202, 10–16. [Google Scholar] [CrossRef]
- Bonkovsky, H.L.; Guo, J.T.; Hou, W.; Li, T.; Narang, T.; Thapar, M. Porphyrin and heme metabolism and the porphyrias. Compr. Physiol. 2013, 3, 365–401. [Google Scholar]
- Bissell, D.M.; Lai, J.C.; Meister, R.K.; Blanc, P.D. Role of Delta-aminolevulinic Acid in the Symptoms of Acute Porphyria. Am. J. Med. 2014, 128, 313–317. [Google Scholar] [CrossRef] [PubMed]
- Watson, C. Hematin and Porphyria. N. Engl. J. Med. 1975, 293, 605–607. [Google Scholar] [CrossRef] [PubMed]
- Tenhunen, R.; Mustajoki, P. Acute Porphyria: Treatment with Heme. Semin. Liver Dis. 1998, 18, 53–55. [Google Scholar] [CrossRef] [PubMed]
- Tian, Q.; Li, T.; Hou, W.; Zheng, J.; Schrum, L.W.; Bonkovsky, H.L. Lon Peptidase 1 (LONP1)-dependent Breakdown of Mitochondrial 5-Aminolevulinic Acid Synthase Protein by Heme in Human Liver Cells. J. Biol. Chem. 2011, 286, 26424–26430. [Google Scholar] [CrossRef]
- Hift, R.J.; Thunell, S.; Brun, A. Drugs in porphyria: From observation to a modern algorithm-based system for the prediction of porphyrogenicity. Pharmacol. Ther. 2011, 132, 158–169. [Google Scholar] [CrossRef]
- Gardner, L.; Smith, S.; Cox, T. Biosynthesis of delta-aminolevulinic acid and the regulation of heme formation by immature erythroid cells in man. J. Biol. Chem. 1991, 266, 22010–22018. [Google Scholar] [CrossRef]
- Steinberg, M.H. Hydroxyurea Treatment for Sickle Cell Disease. Sci. World J. 2002, 2, 1706–1728. [Google Scholar] [CrossRef]
- Steinberg, M.H. Therapies to increase fetal hemoglobin in sickle cell disease. Curr. Hematol. Rep. 2003, 2, 95–101. [Google Scholar]
- Fathallah, H.; Atweh, G.F. Induction of Fetal Hemoglobin in the Treatment of Sickle Cell Disease. Hematol. Am. Soc. Hematol. Educ. Program 2006, 2006, 58–62. [Google Scholar] [CrossRef]
- Cokic, V.P.; Smith, R.D.; Beleslin-Cokic, B.B.; Njoroge, J.M.; Miller, J.L.; Gladwin, M.T.; Schechter, A.N. Hydroxyurea induces fetal hemoglobin by the nitric oxide–dependent activation of soluble guanylyl cyclase. J. Clin. Investig. 2003, 111, 231–239. [Google Scholar] [CrossRef]
- Rodgers, G.P.; Dover, G.J.; Noguchi, C.T.; Schechter, A.N.; Nienhuis, A.W. Hematologic Responses of Patients with Sickle Cell Disease to Treatment with Hydroxyurea. N. Engl. J. Med. 1990, 322, 1037–1045. [Google Scholar] [CrossRef] [PubMed]
- Anderson, N. Hydroxyurea therapy: Improving the lives of patients with sickle cell disease. Pediatr. Nurs. 2006, 32, 541–543. [Google Scholar]
- Goldberg, M.A.; Brugnara, C.; Dover, G.J.; Schapira, L.; Charache, S.; Bunn, H.F. Treatment of Sickle Cell Anemia with Hydroxyurea and Erythropoietin. N. Engl. J. Med. 1990, 323, 366–372. [Google Scholar] [CrossRef] [PubMed]
- Dover, G.; Samuel, C. Hydroxyurea induction of fetal hemoglobin synthesis in sickle-cell disease. Semin. Oncol. 1992, 19, 5. [Google Scholar]
- Grace, R.F.; Glenthøj, A.; Barcellini, W.; Verhovsek, M.; Rothman, J.A.; Morado, M.; Layton, D.M.; Andres, O.; Galactéros, F.; van Beers, E.J.; et al. Long-Term Hemoglobin Response and Reduction in Transfusion Burden Are Maintained in Patients with Pyruvate Kinase Deficiency Treated with Mitapivat. Blood 2022, 140, 5313–5315. [Google Scholar] [CrossRef]
- Grace, R.F.; Rose, C.; Layton, D.M.; Galactéros, F.; Barcellini, W.; Morton, D.H.; van Beers, E.J.; Yaish, H.; Ravindranath, Y.; Kuo, K.H.; et al. Safety and Efficacy of Mitapivat in Pyruvate Kinase Deficiency. N. Engl. J. Med. 2019, 381, 933–944. [Google Scholar] [CrossRef]
- Kuo, K.; Layton, D.; Lal, A.; Al-Samkari, H.; Bhatia, J.; Tong, B.; Lynch, M.; Uhlig, K.; Vichinsky, E. Results from a phase 2 study of mitapivat in adults with non–transfusion-dependent alpha- or beta-thalassemia. Hematol. Transfus. Cell Ther. 2021, 43, S28. [Google Scholar] [CrossRef]
- Kuo, K.H.; Layton, D.M.; Lal, A.; Al-Samkari, H.; Kosinski, P.A.; Tong, B.; Estepp, J.H.; Uhlig, K.; Vichinsky, E.P. Mitapivat Improves Markers of Erythropoietic Activity in Long-Term Study of Adults with Alpha- or Beta-Non-Transfusion-Dependent Thalassemia. Blood 2022, 140 (Suppl. S1), 2479–2480. [Google Scholar] [CrossRef]
- Kuo, K.; Layton, D.; Lal, A.; Al-Samkari, H.; Bhatia, J.; Kosinski, P.; Tong, B.; Lynch, M.; Uhlig, K.; Vichinsky, E. S116: Long-term efficacy and safety of the oral pyruvate kinase activator mitapivat in adults with non—Transfusion-dependent alpha- or beta-thalassemia. HemaSphere 2022, 6, 8–9. [Google Scholar] [CrossRef]
- Xu, J.Z.; Conrey, A.; Frey, I.; Gwaabe, E.; A Menapace, L.; Tumburu, L.; Lundt, M.; Li, Q.; Glass, K.; Iyer, V.; et al. Mitapivat (AG-348) Demonstrates Safety, Tolerability, and Improvements in Anemia, Hemolysis, Oxygen Affinity, and Hemoglobin S Polymerization Kinetics in Adults with Sickle Cell Disease: A Phase 1 Dose Escalation Study. Blood 2021, 138 (Suppl. S1), 10. [Google Scholar] [CrossRef]
- Pilo, F.; Angelucci, E. Mitapivat for sickle cell disease and thalassemia. Drugs Today 2023, 59, 125–134. [Google Scholar] [CrossRef] [PubMed]
- Musallam, K.M.; Taher, A.T.; Cappellini, M.D. Right in time: Mitapivat for the treatment of anemia in α- and β-thalassemia. Cell Rep. Med. 2022, 3, 100790. [Google Scholar] [CrossRef] [PubMed]
- Olubiyi, O.O.; Olagunju, M.O.; Strodel, B. Rational Drug Design of Peptide-Based Therapies for Sickle Cell Disease. Molecules 2019, 24, 4551. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.T.; Ogu, U.O. Sickle cell disease in the new era: Advances in drug treatment. Transfus. Apher. Sci. 2022, 61, 103555. [Google Scholar] [CrossRef]
- Ali, M.A.; Ahmad, A.; Chaudry, H.; Aiman, W.; Aamir, S.; Anwar, M.Y.; Khan, A. Efficacy and safety of recently approved drugs for sickle cell disease: A review of clinical trials. Exp. Hematol. 2020, 92, 11–18.e1. [Google Scholar] [CrossRef]
- Ataga, K.I.; Desai, P.C. Advances in new drug therapies for the management of sickle cell disease. Expert Opin. Orphan Drugs 2018, 6, 329–343. [Google Scholar] [CrossRef]
- Torres, L.; Conran, N. Emerging pharmacotherapeutic approaches for the management of sickle cell disease. Expert Opin. Pharmacother. 2018, 20, 173–186. [Google Scholar] [CrossRef]
- Carden, M.A.; Little, J. Emerging disease-modifying therapies for sickle cell disease. Haematologica 2019, 104, 1710–1719. [Google Scholar] [CrossRef]
- Huang, B.; Ghatge, M.S.; Donkor, A.K.; Musayev, F.N.; Deshpande, T.M.; Al-Awadh, M.; Alhashimi, R.T.; Zhu, H.; Omar, A.M.; Telen, M.J.; et al. Design, Synthesis, and Investigation of Novel Nitric Oxide (NO)-Releasing Aromatic Aldehydes as Drug Candidates for the Treatment of Sickle Cell Disease. Molecules 2022, 27, 6835. [Google Scholar] [CrossRef]
- Nakagawa, A.; Lui, F.E.; Wassaf, D.; Yefidoff-Freedman, R.; Casalena, D.; Palmer, M.A.; Meadows, J.; Mozzarelli, A.; Ronda, L.; Abdulmalik, O.; et al. Identification of a Small Molecule that Increases Hemoglobin Oxygen Affinity and Reduces SS Erythrocyte Sickling. ACS Chem. Biol. 2014, 9, 2318–2325. [Google Scholar] [CrossRef]
- Kassa, T.; Strader, M.B.; Nakagawa, A.; Zapol, W.M.; Alayash, A.I. Targeting βCys93 in hemoglobin S with an antisickling agent possessing dual allosteric and antioxidant effects. Metallomics 2017, 9, 1260–1270. [Google Scholar] [CrossRef] [PubMed]
- Kassa, T.; Wood, F.; Strader, M.B.; Alayash, A.I. Antisickling Drugs Targeting βCys93 Reduce Iron Oxidation and Oxidative Changes in Sickle Cell Hemoglobin. Front. Physiol. 2019, 10, 931. [Google Scholar] [CrossRef] [PubMed]
- Garel, M.C.; Domenget, C.; Caburi-Martin, J.; Prehu, C.; Galacteros, F.; Beuzard, Y. Covalent binding of glutathione to hemoglobin. I. Inhibition of hemoglobin S polymerization. J. Biol. Chem. 1986, 261, 14704–14709. [Google Scholar] [CrossRef] [PubMed]
- Garel, M.; Domenget, C.; Galacteros, F.; Martincaburi, J.; Beuzard, Y. Inhibition of erythrocyte sickling by thiol reagents. Mol. Pharmacol. 1984, 26, 559–565. [Google Scholar]
- Abdulmalik, O.; Safo, M.K.; Chen, Q.; Yang, J.; Brugnara, C.; Ohene-Frempong, K.; Abraham, D.J.; Asakura, T. 5-hydroxymethyl-2-furfural modifies intracellular sickle haemoglobin and inhibits sickling of red blood cells†,‡. Br. J. Haematol. 2005, 128, 552–561. [Google Scholar] [CrossRef]
- Xu, G.G.; Pagare, P.P.; Ghatge, M.S.; Safo, R.P.; Gazi, A.; Chen, Q.; David, T.; Alabbas, A.B.; Musayev, F.N.; Venitz, J.; et al. Design, Synthesis, and Biological Evaluation of Ester and Ether Derivatives of Antisickling Agent 5-HMF for the Treatment of Sickle Cell Disease. Mol. Pharm. 2017, 14, 3499–3511. [Google Scholar] [CrossRef]
- Stern, W.; Mathews, D.; McKew, J.; Shen, X.; Kato, G.J. A Phase 1, First-in-Man, Dose-Response Study of Aes-103 (5-HMF), an Anti-Sickling, Allosteric Modifier of Hemoglobin Oxygen Affinity in Healthy Norman Volunteers. Blood 2012, 120, 3210. [Google Scholar] [CrossRef]
- Lucas, A.; Ao-Ieong, E.S.Y.; Williams, A.T.; Jani, V.P.; Muller, C.R.; Yalcin, O.; Cabrales, P. Increased Hemoglobin Oxygen Affinity With 5-Hydroxymethylfurfural Supports Cardiac Function During Severe Hypoxia. Front. Physiol. 2019, 10, 1350. [Google Scholar] [CrossRef]
- Hannemann, A.; Cytlak, U.M.; Rees, D.C.; Tewari, S.; Gibson, J.S. Effects of 5-hydroxymethyl-2-furfural on the volume and membrane permeability of red blood cells from patients with sickle cell disease. J. Physiol. 2014, 592, 4039–4049. [Google Scholar] [CrossRef]
- Alhashimi, R.T.; Ghatge, M.S.; Donkor, A.K.; Deshpande, T.M.; Anabaraonye, N.; Alramadhani, D.; Danso-Danquah, R.; Huang, B.; Zhang, Y.; Musayev, F.N.; et al. Design, Synthesis, and Antisickling Investigation of a Nitric Oxide-Releasing Prodrug of 5HMF for the Treatment of Sickle Cell Disease. Biomolecules 2022, 12, 696. [Google Scholar] [CrossRef]
- Safo, M.K.; Abdulmalik, O.; Danso-Danquah, R.; Burnett, J.C.; Nokuri, S.; Joshi, G.S.; Musayev, F.N.; Asakura, T.; Abraham, D.J. Structural Basis for the Potent Antisickling Effect of a Novel Class of Five-Membered Heterocyclic Aldehydic Compounds. J. Med. Chem. 2004, 47, 4665–4676. [Google Scholar] [CrossRef] [PubMed]
- Safo, M.K.; Kato, G.J. Therapeutic Strategies to Alter the Oxygen Affinity of Sickle Hemoglobin. Hematol. Clin. N. Am. 2014, 28, 217–231. [Google Scholar] [CrossRef] [PubMed]
- Bonaventura, C.; Fago, A.; Henkens, R.; Crumbliss, A.L. Critical Redox and Allosteric Aspects of Nitric Oxide Interactions with Hemoglobin. Antioxid. Redox Signal. 2004, 6, 979–991. [Google Scholar]
- Gladwin, M.T.; Ognibene, F.P.; Pannell, L.K.; Nichols, J.S.; Pease-Fye, M.E.; Shelhamer, J.H.; Schechter, A.N. Relative role of heme nitrosylation and β-cysteine 93 nitrosation in the transport and metabolism of nitric oxide by hemoglobin in the human circulation. Proc. Natl. Acad. Sci. USA 2000, 97, 9943–9948. [Google Scholar] [CrossRef] [PubMed]
- Sonveaux, P.; Lobysheva, I.I.; Feron, O.; McMahon, T.J. Transport and Peripheral Bioactivities of Nitrogen Oxides Carried by Red Blood Cell Hemoglobin: Role in Oxygen Delivery. Physiology 2007, 22, 97–112. [Google Scholar] [CrossRef]
- Stamler, J.S.; Jia, L.; Eu, J.P.; McMahon, T.J.; Demchenko, I.T.; Bonaventura, J.; Gernert, K.; Piantadosi, C.A. Blood Flow Regulation by S-Nitrosohemoglobin in the Physiological Oxygen Gradient. Science 1997, 276, 2034–2037. [Google Scholar] [CrossRef]
- Frehm, E.; Bonaventura, J.; Gow, A. Serial Review: Biomedical Implications for Hemoglobin Interactions with Nitric Oxide Serial Review Editors: Mark T. Gladwin and Rakesh Patel S-nitrosohemoglobin: An allosteric mediator of no group function in m ammalian vasculature. Free Radic. Biol. Med. 2004, 37, 11. [Google Scholar]
- Su, H.; Liu, X.; Du, J.; Deng, X.; Fan, Y. The role of hemoglobin in nitric oxide transport in vascular system. Med. Nov. Technol. Devices 2020, 5, 100034. [Google Scholar] [CrossRef]
- Lancaster, J.; Hutchings, A.; Kerby, J.D.; Patel, R.P. The hemoglobin-nitric oxide axis: Implications for transfusion therapeutics. Transfus. Altern. Transfus. Med. 2007, 9, 273–280. [Google Scholar] [CrossRef]
- Omar, A.M.; Abdulmalik, O.; Ghatge, M.S.; Muhammad, Y.A.; Paredes, S.D.; El-Araby, M.E.; Safo, M.K. An Investigation of Structure-Activity Relationships of Azolylacryloyl Derivatives Yielded Potent and Long-Acting Hemoglobin Modulators for Reversing Erythrocyte Sickling. Biomolecules 2020, 10, 1508. [Google Scholar] [CrossRef]
- Pagare, P.P.; Ghatge, M.S.; Musayev, F.N.; Deshpande, T.M.; Chen, Q.; Braxton, C.; Kim, S.; Venitz, J.; Zhang, Y.; Abdulmalik, O.; et al. Rational design of pyridyl derivatives of vanillin for the treatment of sickle cell disease. Bioorg. Med. Chem. 2018, 26, 2530–2538. [Google Scholar] [CrossRef] [PubMed]
- Pagare, P.P.; Rastegar, A.; Abdulmalik, O.; Omar, A.M.; Zhang, Y.; Fleischman, A.; Safo, M.K. Modulating hemoglobin allostery for treatment of sickle cell disease: Current progress and intellectual property. Expert Opin. Ther. Patents 2021, 32, 115–130. [Google Scholar] [CrossRef] [PubMed]
- Gopalsamy, A.; Aulabaugh, A.E.; Barakat, A.; Beaumont, K.C.; Cabral, S.; Canterbury, D.P.; Casimiro-Garcia, A.; Chang, J.S.; Chen, M.Z.; Choi, C.; et al. PF-07059013: A Noncovalent Modulator of Hemoglobin for Treatment of Sickle Cell Disease. J. Med. Chem. 2020, 64, 326–342. [Google Scholar] [CrossRef] [PubMed]
- Saunthararajah, Y. Targeting sickle cell disease root-cause pathophysiology with small molecules. Haematologica 2019, 104, 1720–1730. [Google Scholar] [CrossRef] [PubMed]
- Knee, K.M.; Jasuja, R.; Barakat, A.; Rao, D.; Wenzel, Z.; Sahasrabudhe, P.; Narula, J.; Jasti, J.; Chang, J.S.; Beaumont, K.; et al. A Novel Non-Covalent Modulator of Hemoglobin Improves Anemia and Reduces Sickling in a Mouse Model of Sickle Cell Disease. Blood 2019, 134 (Suppl. S1), 207. [Google Scholar] [CrossRef]
- Oder, E.; Safo, M.K.; Abdulmalik, O.; Kato, G.J. New developments in anti-sickling agents: Can drugs directly prevent the polymerization of sickle haemoglobin in vivo? Br. J. Haematol. 2016, 175, 24–30. [Google Scholar] [CrossRef]
- Metcalf, B.; Chuang, C.; Dufu, K.; Patel, M.P.; Silva-Garcia, A.; Johnson, C.; Lu, Q.; Partridge, J.R.; Patskovska, L.; Patskovsky, Y.; et al. Discovery of GBT440, an Orally Bioavailable R-State Stabilizer of Sickle Cell Hemoglobin. ACS Med. Chem. Lett. 2017, 8, 321–326. [Google Scholar] [CrossRef]
- Kapoor, S.; Little, J.A.; Pecker, L.H. Advances in the Treatment of Sickle Cell Disease. Mayo Clin. Proc. 2018, 93, 1810–1824. [Google Scholar] [CrossRef]
- Archer, N.; Galacteros, F.; Brugnara, C. 2015 Clinical trials update in sickle cell anemia. Am. J. Hematol. 2015, 90, 934–950. [Google Scholar] [CrossRef]
- Deshpande, T.M.; Pagare, P.P.; Ghatge, M.S.; Chen, Q.; Musayev, F.N.; Venitz, J.; Zhang, Y.; Abdulmalik, O.; Safo, M.K. Rational modification of vanillin derivatives to stereospecifically destabilize sickle hemoglobin polymer formation. Acta Crystallogr. Sect. D Struct. Biol. 2018, 74, 956–964. [Google Scholar] [CrossRef]
- Scipioni, M.; Kay, G.; Megson, I.L.; Lin, P.K.T. Synthesis of novel vanillin derivatives: Novel multi-targeted scaffold ligands against Alzheimer’s disease. MedChemComm 2019, 10, 764–777. [Google Scholar] [CrossRef] [PubMed]
- Beaudry, F.; Ross, A.; Lema, P.P.; Vachon, P. Pharmacokinetics of vanillin and its effects on mechanical hypersensitivity in a rat model of neuropathic pain. Phytother. Res. 2009, 24, 525–530. [Google Scholar] [CrossRef] [PubMed]
- Ashraf, Z.; Rafiq, M.; Seo, S.-Y.; Babar, M.M.; Zaidi, N.-U.S. Synthesis, kinetic mechanism and docking studies of vanillin derivatives as inhibitors of mushroom tyrosinase. Bioorg. Med. Chem. 2015, 23, 5870–5880. [Google Scholar] [CrossRef] [PubMed]
- Abraham, D.; Mehanna, A.; Wireko, F.; Whitney, J.; Thomas, R.; Orringer, E. Vanillin, a potential agent for the treatment of sickle cell anemia. Blood 1991, 77, 1334–1341. [Google Scholar] [CrossRef]
- Abdulmalik, O.; Pagare, P.P.; Huang, B.; Xu, G.G.; Ghatge, M.S.; Xu, X.; Chen, Q.; Anabaraonye, N.; Musayev, F.N.; Omar, A.M.; et al. VZHE-039, a novel antisickling agent that prevents erythrocyte sickling under both hypoxic and anoxic conditions. Sci. Rep. 2020, 10, 20277. [Google Scholar] [CrossRef]
- Pagare, P.P.; Ghatge, M.S.; Chen, Q.; Musayev, F.N.; Venitz, J.; Abdulmalik, O.; Zhang, Y.; Safo, M.K. Exploration of Structure-Activity Relationship of Aromatic Aldehydes Bearing Pyridinylmethoxy-Methyl Esters as Novel Antisickling Agents. J. Med. Chem. 2020, 63, 14724–14739. [Google Scholar] [CrossRef]
- Lee, C.; Maestre-Reyna, M.; Hsu, K.; Wang, H.; Liu, C.; Jeng, W.; Lin, L.; Wood, R.; Chou, C.; Yang, J.; et al. Crowning Proteins: Modulating the Protein Surface Properties using Crown Ethers. Angew. Chem. Int. Ed. Engl. 2014, 53, 13054–13058. [Google Scholar] [CrossRef]
- Leonard, M.; Dellacherie, E. Acylation of human hemoglobin with polyoxyethylene derivatives. Biochim. Biophys. Acta (BBA) Protein Struct. Mol. Enzymol. 1984, 791, 219–225. [Google Scholar] [CrossRef]
- Abraham, D.J.; Wireko, F.C.; Randad, R.S.; Poyart, C.; Kister, J.; Bohn, B.; Liard, J.F.; Kunert, M.P. Allosteric modifiers of hemoglobin: 2-[4-[[(3,5-disubstituted anilino)carbonyl]methyl]phenoxy]-2-methylpropionic acid derivatives that lower the oxygen affinity of hemoglobin in red cell suspensions, in whole blood, and in vivo in rats. Biochemistry 1992, 31, 9141–9149. [Google Scholar] [CrossRef]
- Sun, K.; D’alessandro, A.; Ahmed, M.H.; Zhang, Y.; Song, A.; Ko, T.-P.; Nemkov, T.; Reisz, J.A.; Wu, H.; Adebiyi, M.; et al. Structural and Functional Insight of Sphingosine 1-Phosphate-Mediated Pathogenic Metabolic Reprogramming in Sickle Cell Disease. Sci. Rep. 2017, 7, 15281. [Google Scholar] [CrossRef]
- Kaca, W.; Roth, R.; Levin, J. Hemoglobin, a newly recognized lipopolysaccharide (LPS)-binding protein that enhances LPS biological activity. J. Biol. Chem. 1994, 269, 25078–25084. [Google Scholar] [CrossRef] [PubMed]
- Bahl, N.; Du, R.; Winarsih, I.; Ho, B.; Tucker-Kellogg, L.; Tidor, B.; Ding, J.L. Delineation of Lipopolysaccharide (LPS)-binding Sites on Hemoglobin: From in silico predictions to biophysical characterization. J. Biol. Chem. 2011, 286, 37793–37803. [Google Scholar] [CrossRef] [PubMed]
- Lechuga, G.C.; Souza-Silva, F.; Sacramento, C.Q.; Trugilho, M.R.O.; Valente, R.H.; Napoleão-Pêgo, P.; Dias, S.S.G.; Fintelman-Rodrigues, N.; Temerozo, J.R.; Carels, N.; et al. SARS-CoV-2 Proteins Bind to Hemoglobin and Its Metabolites. Int. J. Mol. Sci. 2021, 22, 9035. [Google Scholar] [CrossRef] [PubMed]
- Fischer, S.; Nagel, R.L.; Bookchin, R.M.; Roth, E.F.; Tellez-Nagel, I. The binding of hemoglobin to membranes of normal and sickle erythrocytes. Biochim. Biophys. Acta (BBA) Biomembr. 1975, 375, 422–433. [Google Scholar] [CrossRef]
- Shaklai, N.; Yguerabide, J.; Ranney, H.M. Interaction of hemoglobin with red blood cell membranes as shown by a fluorescent chromophore. Biochemistry 1977, 16, 5585–5592. [Google Scholar] [CrossRef]
- Yenamandra, A.; Marjoncu, D. Voxelotor: A Hemoglobin S Polymerization Inhibitor for the Treatment of Sickle Cell Disease. J. Adv. Pract. Oncol. 2020, 11, 873–877. [Google Scholar] [CrossRef]
- Singh, J.; Maggo, S.; Sadananden, U.K. Voxelotor: Novel drug for sickle cell disease. Int. J. Basic Clin. Pharmacol. 2020, 9, 513–517. [Google Scholar] [CrossRef]
- Han, J.; Saraf, S.L.; Gordeuk, V.R. Systematic Review of Voxelotor: A First-in-Class Sickle Hemoglobin Polymerization Inhibitor for Management of Sickle Cell Disease. Pharmacother. J. Hum. Pharmacol. Drug Ther. 2020, 40, 525–534. [Google Scholar] [CrossRef]
- Howard, J.; Hemmaway, C.J.; Telfer, P.; Layton, D.M.; Porter, J.; Awogbade, M.; Mant, T.; Gretler, D.D.; Dufu, K.; Hutchaleelaha, A.; et al. A phase 1/2 ascending dose study and open-label extension study of voxelotor in patients with sickle cell disease. Blood 2019, 133, 1865–1875. [Google Scholar] [CrossRef]
- Hutchaleelaha, A.; Patel, M.; Washington, C.; Siu, V.; Allen, E.; Oksenberg, D.; Gretler, D.D.; Mant, T.; Lehrer-Graiwer, J. Pharmacokinetics and pharmacodynamics of voxelotor (GBT440) in healthy adults and patients with sickle cell disease. Br. J. Clin. Pharmacol. 2019, 85, 1290–1302. [Google Scholar] [CrossRef]
- Vissa, M.; Vichinsky, E. Voxelotor for the treatment of sickle cell disease. Expert Rev. Hematol. 2021, 14, 253–262. [Google Scholar] [CrossRef] [PubMed]
- Vasseur, C.; Baudin-Creuza, V. Role of alpha-hemoglobin molecular chaperone in the hemoglobin formation and clinical expression of some hemoglobinopathies. Transfus. Clin. Biol. 2015, 22, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Favero, M.E.; Costa, F.F. Alpha-Hemoglobin-Stabilizing Protein: An Erythroid Molecular Chaperone. Biochem. Res. Int. 2011, 2011, 373859. [Google Scholar] [CrossRef] [PubMed]
- Weiss, M.J.; Zhou, S.; Feng, L.; Gell, D.A.; Mackay, J.P.; Shi, Y.; Gow, A.J. Role of Alpha Hemoglobin-Stabilizing Protein in Normal Erythropoiesis and β-Thalassemia. Ann. N. Y. Acad. Sci. 2005, 1054, 103–117. [Google Scholar] [CrossRef]
- Khandros, E.; Mollan, T.L.; Yu, X.; Wang, X.; Yao, Y.; D’Souza, J.; Gell, D.A.; Olson, J.S.; Weiss, M.J. Insights into Hemoglobin Assembly through in Vivo Mutagenesis of α-Hemoglobin Stabilizing Protein. J. Biol. Chem. 2012, 287, 11325–11337. [Google Scholar] [CrossRef]
- Eggleson, K.K.; Duffin, K.L.; Goldberg, D.E. Identification and Characterization of Falcilysin, a Metallopeptidase Involved in Hemoglobin Catabolism within the Malaria Parasite Plasmodium falciparum. J. Biol. Chem. 1999, 274, 32411–32417. [Google Scholar] [CrossRef]
- Christina, E.M.; Goldberg, D. Plasmodium falciparum falcilysin: A metalloprotease with dual specificity. J. Biol. Chem. 2003, 278, 38022–38028. [Google Scholar]
- Ralph, S.A. Subcellular multitasking—Multiple destinations and roles for the Plasmodium falcilysin protease. Mol. Microbiol. 2007, 63, 309–313. [Google Scholar] [CrossRef]
- Ponpuak, M.; Klemba, M.; Park, M.; Gluzman, I.Y.; Lamppa, G.K.; Goldberg, D.E. A role for falcilysin in transit peptide degradation in the Plasmodium falciparum apicoplast. Mol. Microbiol. 2006, 63, 314–334. [Google Scholar] [CrossRef]
- Chance, J.; Hannah, F.; Hernandez, O.; Istvan, E.; Armann, A.; Maslov, N.; Ruby, A.; Teodulo, C.; Huyen, N.; Brian, V.; et al. Development of piperazine-based hydroxamic acid inhibitors against fal cilysin, an essential malarial protease. Bioorg. Med. Chem. Lett. 2018, 28, 1846–1848. [Google Scholar] [CrossRef]
- Shen, F.; Zheng, G.; Setegne, M.; Tenglin, K.; Izada, M.; Xie, H.; Zhai, L.; Orkin, S.H.; Dassama, L.M.K. A cell-permeant nano-body-based degrader that induces fetal hemoglobin. ACS Cent. Sci. 2022, 8, 1695–1703. [Google Scholar] [CrossRef] [PubMed]
- Maolu, Y.; Manizheh, I.; Karin, T.; Thibault, V.; Liting, Z.; Ge, Z.; Arthanari, H.; Laura, M.K.D.; Orkin, S. Evolution of nanobodies specific for BCL11A. Proc. Natl. Acad. Sci. USA 2022, 120, e2218959120. [Google Scholar]
- Gülgün, A.; Andaç, M.; Denizli, A.; Duman, M. Recognition of human hemoglobin with macromolecularly imprinted polyme ric nanoparticles using non-covalent interactions. J. Mol. Recognit. 2021, 34, e2935. [Google Scholar]
- Khakurel, K.P.; Žoldák, G.; Angelov, B.; Andreasson, J. On the feasibility of time-resolved X-ray powder diffraction of macromolecules using laser-driven ultrafast X-ray sources. J. Appl. Crystallogr. 2024, 57 Pt 4, 1205–1211. [Google Scholar] [CrossRef]
- Khakurel, K.P.; Nemergut, M.; Džupponová, V.; Kropielnicki, K.; Savko, M.; Žoldák, G.; Andreasson, J. Design and fabrication of 3D-printed in situ crystallization plates for probing microcrystals in an external electric field. J. Appl. Crystallogr. 2024, 57 Pt 4, 842–847. [Google Scholar] [CrossRef]
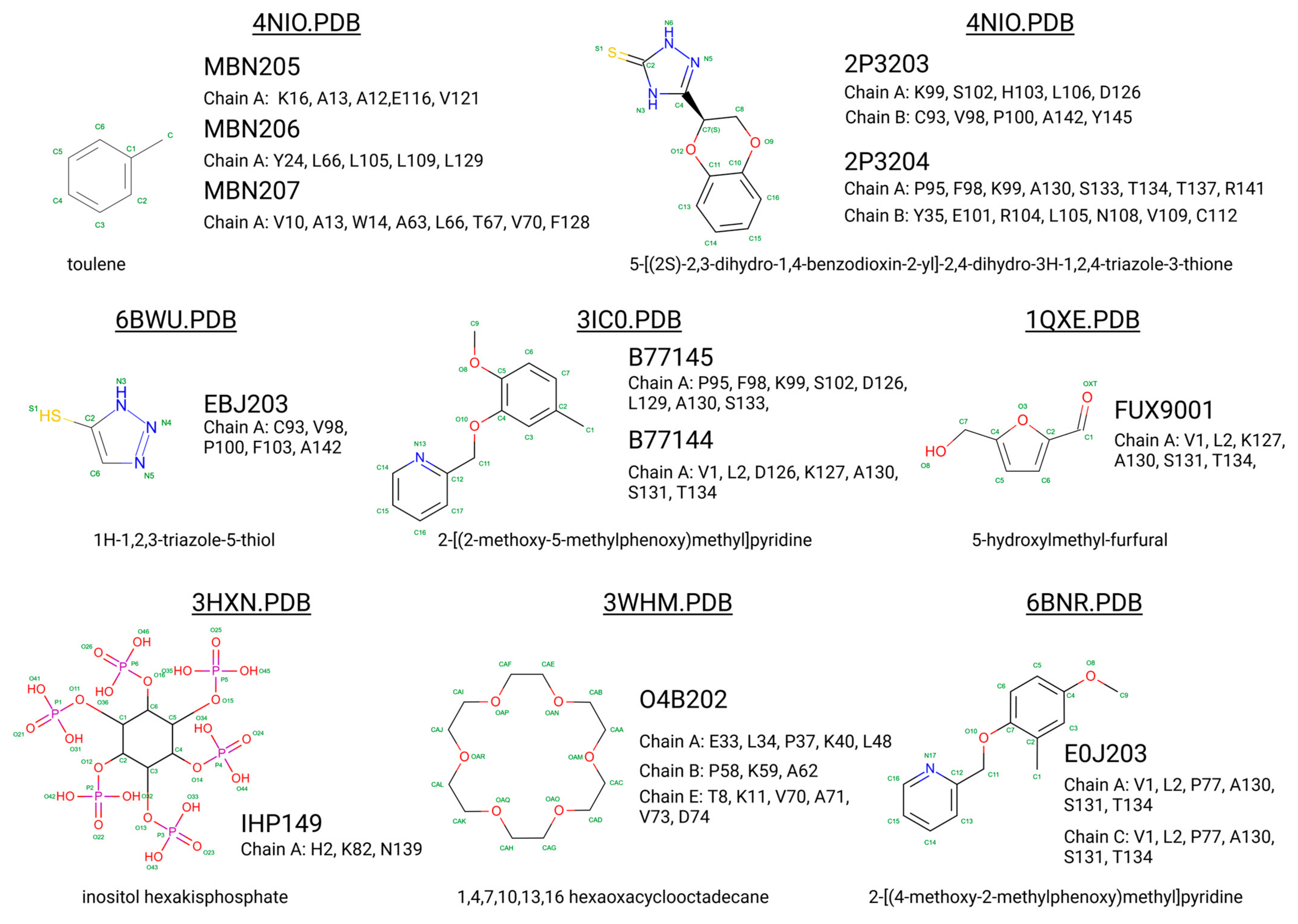
| Small Molecule/Binder | Appearance Time | Main Interaction with Hemoglobin | Therapeutic or Research Impact |
|---|---|---|---|
VZHE-039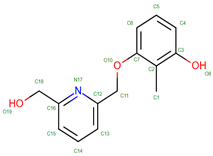 | 2020 | Enhances oxygen transport efficiency | Potential therapeutic agent to improve oxygen delivery |
Compound 23 (PF-07059013) | 2020 | Noncovalent binder that enhances hemoglobin stability | Explored for reducing sickling in sickle cell disease |
INN-310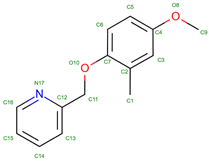 | 2017 | Vanillin derivative that influences hemoglobin stability | Explored for impacts on oxygen affinity and hemoglobin stability |
TD3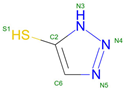 | 2017 | Affects oxygen binding dynamics | Potential for therapeutic use in modifying hemoglobin function |
GBT440 (Voxeltor) | 2015 | Increases oxygen affinity to prevent sickle hemoglobin polymerization | Used in treating sickle cell disease |
18-crown-6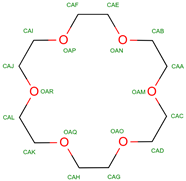 | 2013 | Alters oxygen-binding properties | Used in studies for potential modulation of hemoglobin function |
Toluene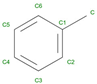 | 2013 | Solvent in structural studies | Used to understand hemoglobin structure |
TD1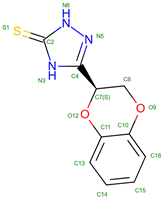 | 2013 | Alters hemoglobin’s structural stability | Studied for potential therapeutic impacts on hemoglobin function |
INN-298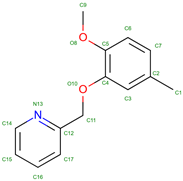 | 2005 | Modifies hemoglobin function | Investigated for its effects on hemoglobin and potential treatments |
L35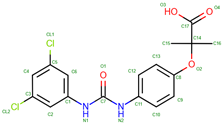 | 2005 | Modifies oxygen affinity and function | Investigated for its potential to treat hemoglobinopathies |
5-Hydroxymethylfurfural (5HMF) | 2003 | Increases oxygen affinity, reducing sickling | Explored for treatment of sickle cell disease |
Inositol Hexakisphosphate (IHP) | 1974 | Stabilizes deoxyhemoglobin, reduces oxygen affinity | Research tool for studying oxygen release mechanics |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Žoldáková, M.; Novotný, M.; Khakurel, K.P.; Žoldák, G. Hemoglobin Variants as Targets for Stabilizing Drugs. Molecules 2025, 30, 385. https://doi.org/10.3390/molecules30020385
Žoldáková M, Novotný M, Khakurel KP, Žoldák G. Hemoglobin Variants as Targets for Stabilizing Drugs. Molecules. 2025; 30(2):385. https://doi.org/10.3390/molecules30020385
Chicago/Turabian StyleŽoldáková, Miroslava, Michal Novotný, Krishna P. Khakurel, and Gabriel Žoldák. 2025. "Hemoglobin Variants as Targets for Stabilizing Drugs" Molecules 30, no. 2: 385. https://doi.org/10.3390/molecules30020385
APA StyleŽoldáková, M., Novotný, M., Khakurel, K. P., & Žoldák, G. (2025). Hemoglobin Variants as Targets for Stabilizing Drugs. Molecules, 30(2), 385. https://doi.org/10.3390/molecules30020385