
Human Pharmacokinetic Profiling and Comparative Analysis of Mangiferin and Its Monosodium Derivative from Mangifera indica Extracts Using UHPLC-MS/MS with 1H NMR and MALDI-TOF Confirmation

 , ,
, ,  , ,
, ,  and
and
Abstract
:1. Introduction
2. Results
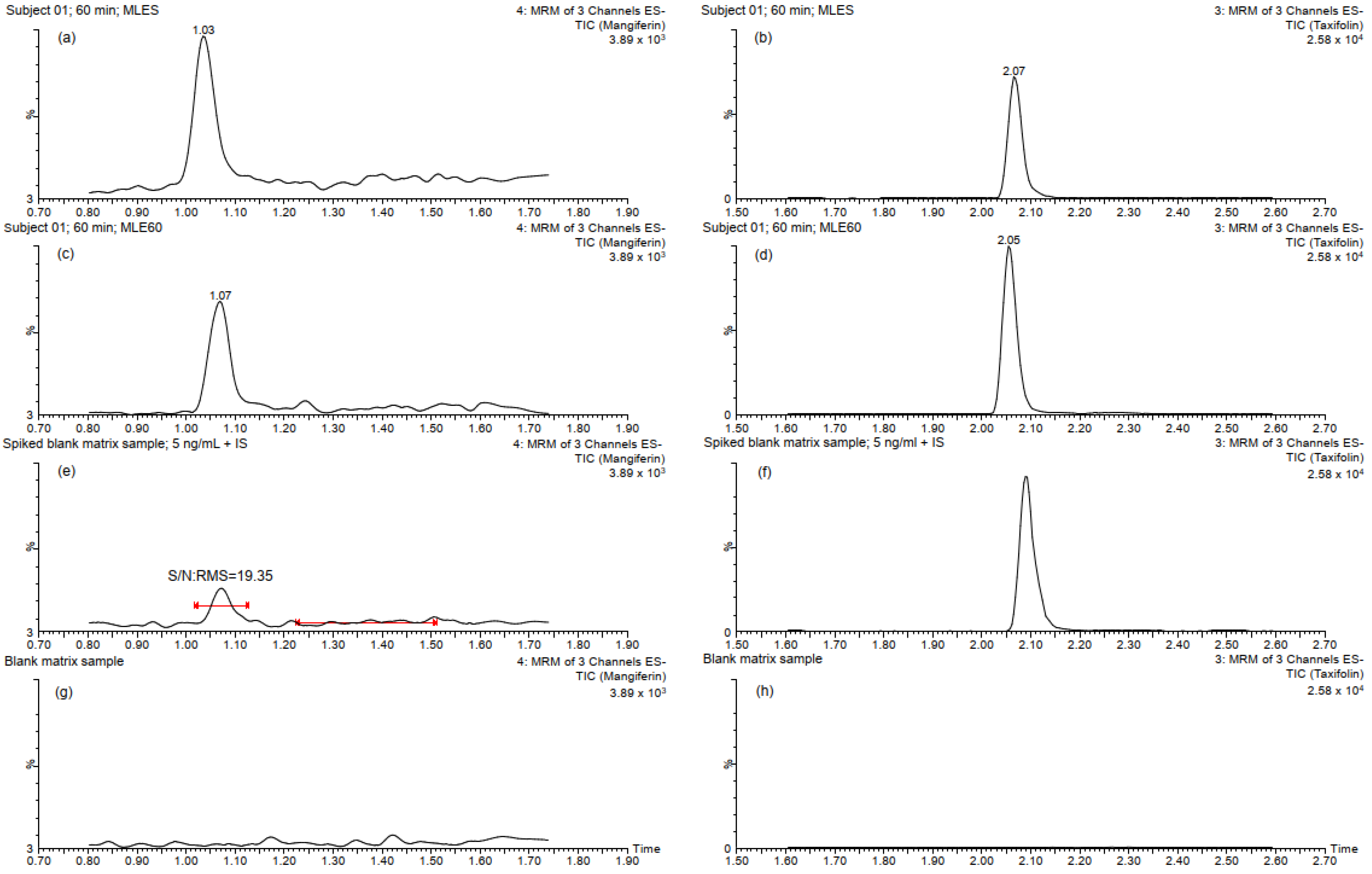
2.1. Sample Preparation and UHPLC-MS/MS Analysis
2.2. Method Validation
2.2.1. Specificity, Linearity and Sensitivity
2.2.2. Accuracy and Precision
2.2.3. Matrix Effect, Recovery, and Stability
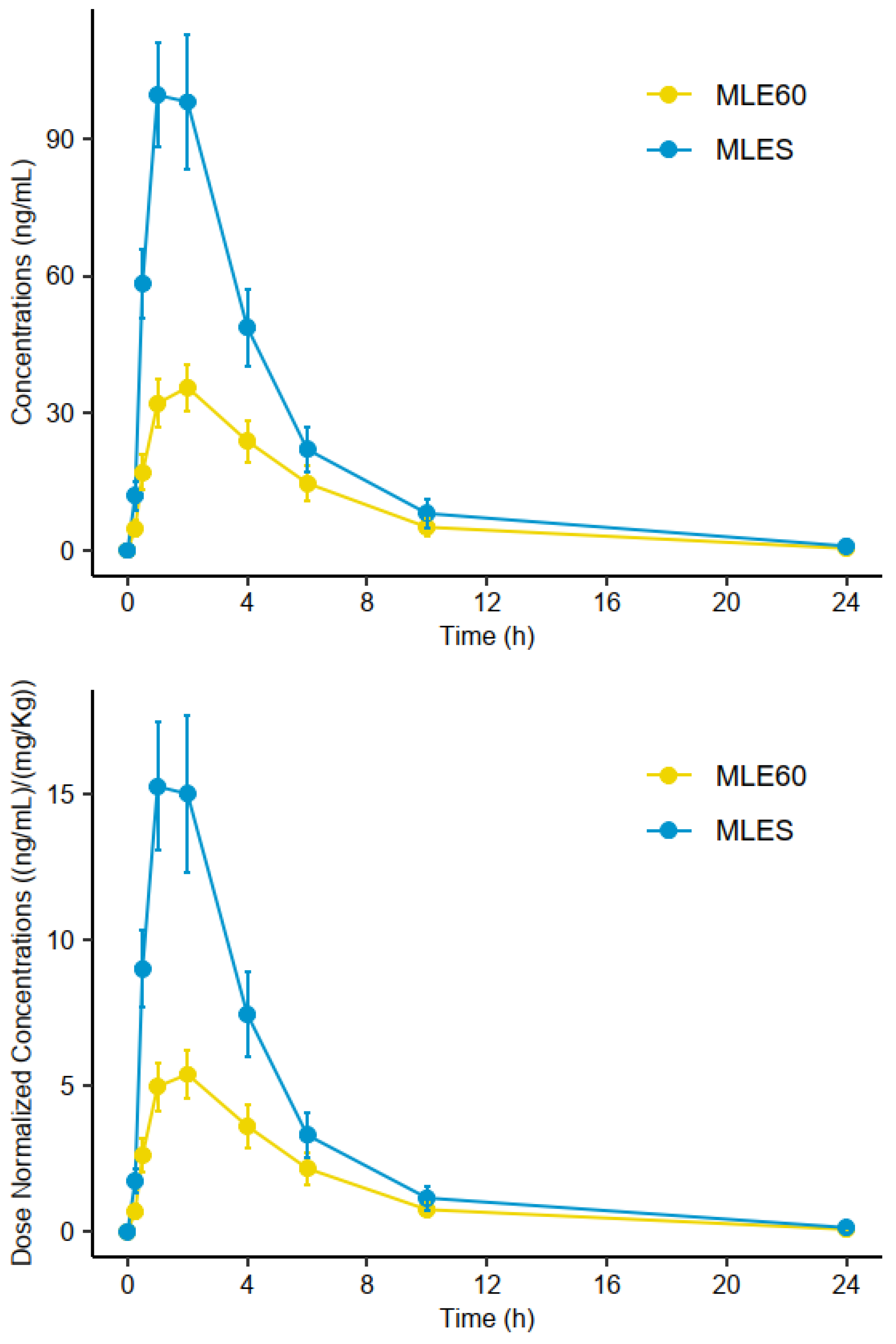
2.3. Analysis of Plasma Samples
2.3.1. Pharmacokinetic Parameters
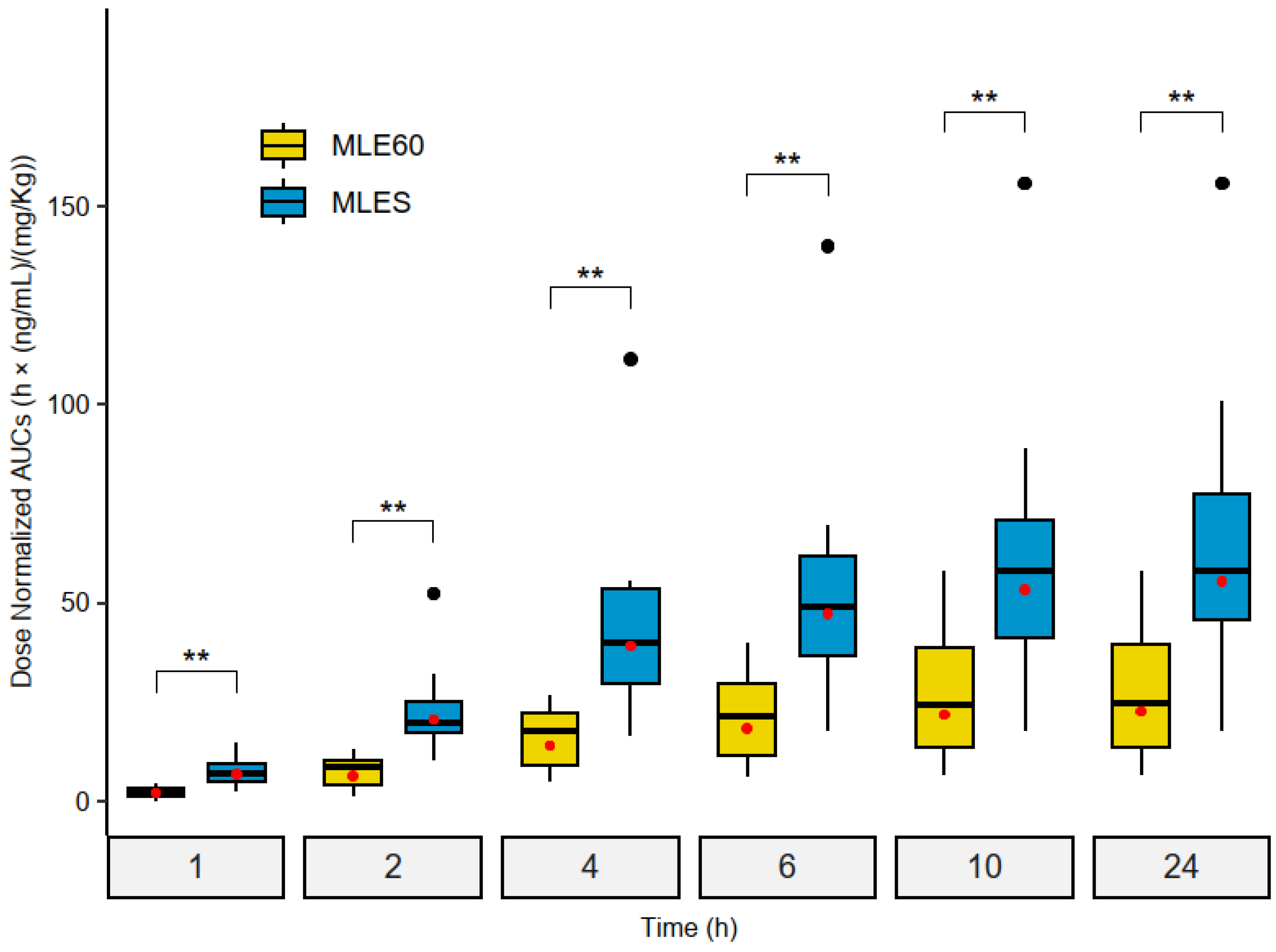
2.3.2. Relative Bioavailability (Frel) Assessment
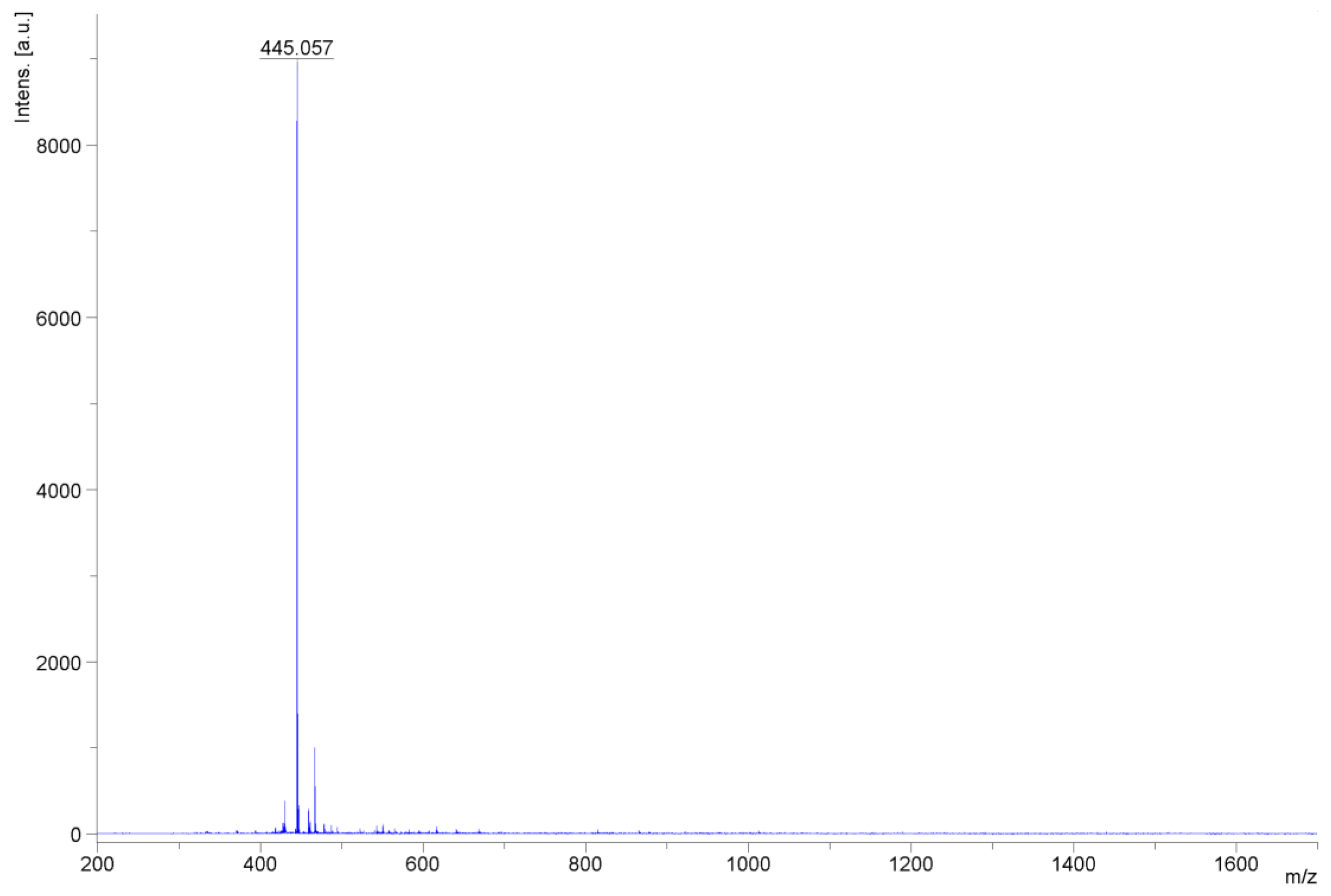
2.4. Characterization of Mangiferin Monosodium Salt
3. Discussion
4. Materials and Methods
4.1. Chemicals and Reagents
4.2. Plant Material
4.3. Mangifera Indica Leaf Extracts Preparations and Content of Mangiferin
4.4. Preparation of Mangiferin Stock Solution and Quality Control Samples
4.5. UHPLC-MS/MS Instrument and Conditions
4.6. Sample Preparation
4.6.1. Instrumentation
4.6.2. Plasma Samples
4.7. Method Validation
4.7.1. Specificity, Linearity and Sensitivity
4.7.2. Accuracy and Precision
4.7.3. Matrix Effect, Recovery and Stability
4.8. Pharmacokinetic Protocol
- Over 18 years old and with a normal body mass index (BMI) (<30), non-smokers;
- The subjects were determined to be healthy based on their medical history: normal liver and renal functions, without food allergies;
- No drug intake was allowed 2 weeks prior to or during this study;
- The subjects were asked not to consume meals containing mango fruit or mango leaf tea for three days before this study;
- Females were required to have regular menstrual cycles without the use of oral contraceptives.
4.9. Analysis of Pharmacokinetic Data
- C: concentration of mangiferin in the plasma samples in ng/mL;
- D: dose of mangiferin (479.8 mg for MLE60, 480.4 mg for MLES);
- BW: body weight in kg.
4.10. NMR and MALDI-TOF Confirmation Analysis
Samples Preparation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ross, I.A. Mangifera Indica. In Medicinal Plants of the World; Humana Press: Totowa, NJ, USA, 2003; pp. 315–328. [Google Scholar]
- Bally, I.S.E. Mangifera Indica (Mango). In Species Profile for Pacific Island Agroforestry; Elevich, C.R., Ed.; Permanent Agriculture Resources (PAR): Holualoa, HI, USA, 2006. [Google Scholar]
- Mukherjee, S.K. The Mango-Its Botany, Cultivation, Uses and Future Improvement, Especially as Observed in India. Econ. Bot. 1953, 7, 130–162. [Google Scholar] [CrossRef]
- Ediriweera, M.K.; Tennekoon, K.H.; Samarakoon, S.R. A Review on Ethnopharmacological Applications, Pharmacological Activities, and Bioactive Compounds of Mangifera Indica (Mango). Evid.-Based Complement. Altern. Med. 2017, 2017, 6949835. [Google Scholar] [CrossRef]
- Pan, J.; Yi, X.; Wang, Y.; Chen, G.; He, X. Benzophenones from Mango Leaves Exhibit α-Glucosidase and NO Inhibitory Activities. J. Agric. Food Chem. 2016, 64, 7475–7480. [Google Scholar] [CrossRef] [PubMed]
- Jhaumeer Laulloo, S.; Bhowon, M.G.; Soyfoo, S.; Chua, L.S. Nutritional and Biological Evaluation of Leaves of Mangifera Indica from Mauritius. J. Chem. 2018, 2018, 1–9. [Google Scholar] [CrossRef]
- Pan, J.; Yi, X.; Zhang, S.; Cheng, J.; Wang, Y.; Liu, C.; He, X. Bioactive Phenolics from Mango Leaves (Mangifera indica L.). Ind. Crops Prod. 2018, 111, 400–406. [Google Scholar] [CrossRef]
- Barreto, J.C.; Trevisan, M.T.S.; Hull, W.E.; Erben, G.; De Brito, E.S.; Pfundstein, B.; Würtele, G.; Spiegelhalder, B.; Owen, R.W. Characterization and Quantitation of Polyphenolic Compounds in Bark, Kernel, Leaves, and Peel of Mango (Mangifera indica L.). J. Agric. Food Chem. 2008, 56, 5599–5610. [Google Scholar] [CrossRef]
- Ge, D.D.; Zhang, Y.; Liu, E.W.; Wang, T.; Hu, L.M. Chemical Constituents of Mangifera Indica Leaves (I). Chin. Tradit. Herbal. Drugs 2011, 42, 428–431. [Google Scholar]
- Jyotshna; Khare, P.; Shanker, K. Mangiferin: A Review of Sources and Interventions for Biological Activities. BioFactors 2016, 42, 504–514. [Google Scholar] [CrossRef]
- Telang, M.; Dhulap, S.; Mandhare, A.; Hirwani, R. Therapeutic and Cosmetic Applications of Mangiferin: A Patent Review. Expert. Opin. Ther. Pat. 2013, 23, 1561–1580. [Google Scholar] [CrossRef] [PubMed]
- Wauthoz, N.; Balde, A. Ethnopharmacology of Mangifera Indica L. Bark and Pharmacological Studies of Its Main C-Glucosylxanthone, Mangiferin. Int. J. Biomed. Pharm. Sci. 2007, 1, 112–119. [Google Scholar] [CrossRef]
- Maenpuen, S.; Mee-udorn, P.; Pinthong, C.; Athipornchai, A.; Phiwkaow, K.; Watchasit, S.; Pimviriyakul, P.; Rungrotmongkol, T.; Tinikul, R.; Leartsakulpanich, U.; et al. Mangiferin Is a New Potential Antimalarial and Anticancer Drug for Targeting Serine Hydroxymethyltransferase. Arch. Biochem. Biophys. 2023, 745, 109712. [Google Scholar] [CrossRef]
- Jimenez del Rio, M.; Wiebe, J.C.; Lopez-Rios, L.; Vega-Morales, T.; Perez-Machin, R.; Sanchez-Rodriguez, A.; Mateos, C.; Gericke, N.P. Compositions for Enhancing Brain Activity. U.S. Patent US10537604B2, 21 January 2020. [Google Scholar]
- Wightman, E.L.; Jackson, P.A.; Forster, J.; Khan, J.; Wiebe, J.C.; Gericke, N.; Kennedy, D.O. Acute Effects of a Polyphenol-Rich Leaf Extract of Mangifera indica L. (Zynamite) on Cognitive Function in Healthy Adults: A Double-Blind, Placebo-Controlled Crossover Study. Nutrients 2020, 12, 2194. [Google Scholar] [CrossRef] [PubMed]
- Gelabert-Rebato, M.; Wiebe, J.C.; Martin-Rincon, M.; Gericke, N.; Perez-Valera, M.; Curtelin, D.; Galvan-Alvarez, V.; Lopez-Rios, L.; Morales-Alamo, D.; Calbet, J.A.L. Mangifera Indica L. Leaf Extract in Combination with Luteolin or Quercetin Enhances VO2peak and Peak Power Output, and Preserves Skeletal Muscle Function during Ischemia-Reperfusion in Humans. Front. Physiol. 2018, 9, 740. [Google Scholar] [CrossRef] [PubMed]
- Gelabert-Rebato, M.; Martin-Rincon, M.; Galvan-Alvarez, V.; Gallego-Selles, A.; Martinez-Canton, M.; Vega-Morales, T.; Wiebe, J.C.; Del Castillo, C.F.; Castilla-Hernandez, E.; Diaz-Tiberio, O.; et al. A Single Dose of the Mango Leaf Extract Zynamite® in Combination with Quercetin Enhances Peak Power Output during Repeated Sprint Exercise in Men and Women. Nutrients 2019, 11, 2592. [Google Scholar] [CrossRef] [PubMed]
- Gelabert-Rebato, M.; Wiebe, J.C.; Martin-Rincon, M.; Galvan-Alvarez, V.; Curtelin, D.; Perez-Valera, M.; Habib, J.J.; Pérez-López, A.; Vega, T.; Morales-Alamo, D.; et al. Enhancement of Exercise Performance by 48 Hours, and 15-Day Supplementation with Mangiferin and Luteolin in Men. Nutrients 2019, 11, 344. [Google Scholar] [CrossRef] [PubMed]
- Hou, S.; Wang, F.; Li, Y.; Li, Y.; Wang, M.; Sun, D.; Sun, C. Pharmacokinetic Study of Mangiferin in Human Plasma after Oral Administration. Food Chem. 2012, 132, 289–294. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Xu, F.; Zeng, X.; Yang, L.; Deng, Y.; Wu, Z.; Feng, Y.; Li, X. Application of a Liquid Chromatography/Tandem Mass Spectrometry Method to Pharmacokinetic Study of Mangiferin in Rats. J. Chromatogr. B 2010, 878, 3345–3350. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Deng, J.; Wang, Q.; Li, X.; WEI, H. Improvement in the Solubility of Mangiferin by HP-β-CD Inclusion. Chin. Tradit. Pat. Med. 2008, 30, 1123–1126. [Google Scholar]
- Tian, X.; Gao, Y.; Xu, Z.; Lian, S.; Ma, Y.; Guo, X.; Hu, P.; Li, Z.; Huang, C. Pharmacokinetics of Mangiferin and Its Metabolite—Norathyriol, Part 1: Systemic Evaluation of Hepatic First-Pass Effect In Vitro and In Vivo. BioFactors 2016, 42, 533–544. [Google Scholar] [CrossRef] [PubMed]
- Kesarwani, K.; Gupta, R. Bioavailability Enhancers of Herbal Origin: An Overview. Asian Pac. J. Trop. Biomed. 2013, 3, 253–266. [Google Scholar] [CrossRef]
- Tian, X.; Xu, Z.; Li, Z.; Ma, Y.; Lian, S.; Guo, X.; Hu, P.; Gao, Y.; Huang, C. Pharmacokinetics of Mangiferin and Its Metabolite—Norathyriol, Part 2: Influence of UGT, CYP450, P-Gp, and Enterobacteria and the Potential Interaction in Rhizoma Anemarrhenae Decoction with Timosaponin B2 as the Major Contributor. BioFactors 2016, 42, 545–555. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, F.d.R.; Valentim, I.B.; Ramones, E.L.C.; Trevisan, M.T.S.; Olea-Azar, C.; Perez-Cruz, F.; de Abreu, F.C.; Goulart, M.O.F. Antioxidant Activity of the Mangiferin Inclusion Complex with β-Cyclodextrin. LWT-Food Sci. Technol. 2013, 51, 129–134. [Google Scholar] [CrossRef]
- de Souza, J.R.R.; Feitosa, J.P.A.; Ricardo, N.M.P.S.; Trevisan, M.T.S.; de Paula, H.C.B.; Ulrich, C.M.; Owen, R.W. Spray-Drying Encapsulation of Mangiferin Using Natural Polymers. Food Hydrocoll. 2013, 33, 10–18. [Google Scholar] [CrossRef]
- Khurana, R.K.; Bansal, A.K.; Beg, S.; Burrow, A.J.; Katare, O.P.; Singh, K.K.; Singh, B. Enhancing Biopharmaceutical Attributes of Phospholipid Complex-Loaded Nanostructured Lipidic Carriers of Mangiferin: Systematic Development, Characterization and Evaluation. Int. J. Pharm. 2017, 518, 289–306. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.; Chen, H.; Sun, L.; Tong, L.; Zhang, T. Improving Permeability and Oral Absorption of Mangiferin by Phospholipid Complexation. Fitoterapia 2014, 93, 54–61. [Google Scholar] [CrossRef]
- Bhattacharyya, S.; Ahmmed, S.M.; Saha, B.P.; Mukherjee, P.K. Soya Phospholipid Complex of Mangiferin Enhances Its Hepatoprotectivity by Improving Its Bioavailability and Pharmacokinetics. J. Sci. Food Agric. 2014, 94, 1380–1388. [Google Scholar] [CrossRef] [PubMed]
- Gonzales, G.B.; Smagghe, G.; Grootaert, C.; Zotti, M.; Raes, K.; Camp, J. Van Flavonoid Interactions during Digestion, Absorption, Distribution and Metabolism: A Sequential Structure-Activity/Property Relationship-Based Approach in the Study of Bioavailability and Bioactivity. Drug Metab. Rev. 2015, 47, 175–190. [Google Scholar] [CrossRef]
- Guo, H.; Chen, M.; Li, M.; Hu, M.; Chen, B.; Zhou, C. Pharmacokinetic Comparisons of Mangiferin and Mangiferin Monosodium Salt in Rat Plasma by UPLC-MS/MS. J. Chem. 2019, 2019, 1–12. [Google Scholar] [CrossRef]
- Lin, H.; Teng, H.; Wu, W.; Li, Y.; Lv, G.; Huang, X.; Yan, W.; Lin, Z. Pharmacokinetic and Metabolomic Analyses of Mangiferin Calcium Salt in Rat Models of Type 2 Diabetes and Non-Alcoholic Fatty Liver Disease. BMC Pharmacol. Toxicol. 2020, 21, 59. [Google Scholar] [CrossRef]
- Guo, X.; Lankmayr, E. Phospholipid-Based Matrix Effects in LC-MS Bioanalysis. Bioanalysis 2011, 3, 349–352. [Google Scholar] [CrossRef]
- Panuwet, P.; Jr, R.E.H.; Souza, P.E.D.; Chen, X.; Radford, A.; Cohen, J.R.; Marder, M.E.; Kartavenka, K.; Barry, P.; Barr, D.B. Biological Matrix Effects in Quantitative Tandem Mass Spectrometry-Based Analytical Methods: Advancing Biomonitoring. Crit. Rev. Anal. Chem. 2016, 46, 93–105. [Google Scholar] [CrossRef] [PubMed]
- Le Bourvellec, C.; Renard, C.M.G.C. Interactions between Polyphenols and Macromolecules: Quantification Methods and Mechanisms. Crit. Rev. Food Sci. Nutr. 2012, 52, 213–248. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Lan, J.; Guan, M.; Sheng, F.; Zhang, H. Spectroscopic Investigation of Interaction between Mangiferin and Bovine Serum Albumin. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2009, 73, 936–941. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Deng, J.; Qian, Y.; Hou, X.T.; Zhu, Z.; Zhao, M.; Shang, E.; Qian, D.; Zeng, H.; Pang, H.; et al. Simultaneous Determination of Kaempferol, Quercetin, Mangiferin, Gallic Acid, p-Hydroxybenzoic Acid and Chlorpheniramine Maleate in Rat Plasma after Oral Administration of Mang-Guo-Zhi-Ke Tablets by UHPLC-MS/MS and Its Application to Pharmacokinetics. Biomed. Chromatogr. 2018, 32, 1–9. [Google Scholar] [CrossRef]
- Hara, H.; Ise, Y.; Morimoto, N.; Shimazawa, M.; Ichihashi, K.; Ohyama, M.; Iinuma, M. Laxative Effect of Agarwood Leaves and Its Mechanism. Biosci. Biotechnol. Biochem. 2008, 72, 335–345. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.; Liang, D.; Wang, Q.; Meng, X.; Li, Z. Total Synthesis of Mangiferin, Homomangiferin, and Neomangiferin. Org. Biomol. Chem. 2016, 14, 8821–8831. [Google Scholar] [CrossRef] [PubMed]
- Mendoza-Sarmiento, G.; Rojas-Hernández, A.; Galano, A.; Gutiérrez, A. A Combined Experimental-Theoretical Study of the Acid-Base Behavior of Mangiferin: Implications for Its Antioxidant Activity. RSC Adv. 2016, 6, 51171–51182. [Google Scholar] [CrossRef]
- Gómez-Zaleta, B.; Ramírez-Silva, M.T.; Gutiérrez, A.; González-Vergara, E.; Güizado-Rodríguez, M.; Rojas-Hernández, A. UV/Vis, 1H, and 13C NMR Spectroscopic Studies to Determine Mangiferin PKa Values. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2006, 64, 1002–1009. [Google Scholar] [CrossRef]
- Thilakarathna, S.H.; Rupasinghe, H.P.V. Flavonoid Bioavailability and Attempts for Bioavailability Enhancement. Nutrients 2013, 5, 3367–3387. [Google Scholar] [CrossRef] [PubMed]
- Peterson, B.; Weyers, M.; Steenekamp, J.H.; Steyn, J.D.; Gouws, C.; Hamman, J.H. Drug Bioavailability Enhancing Agents of Natural Origin (Bioenhancers) That Modulate Drug Membrane Permeation and Pre-Systemic Metabolism. Pharmaceutics 2019, 11, 33. [Google Scholar] [CrossRef]
- Sanugul, K.; Akao, T.; Li, Y.; Kakiuchi, N.; Nakamura, N.; Hattori, M. Isolation of a Human Intestinal Bacterium That Transforms Mangiferin to Norathyriol and Inducibility of the Enzyme That Cleaves a C-Glucosyl Bond. Biol. Pharm. Bull. 2005, 28, 1672–1678. [Google Scholar] [CrossRef]
- Denney, W.S.; Duvvuri, S.; Buckeridge, C. Simple, Automatic Noncompartmental Analysis: The PKNCA R Package. J. Pharmacokinet. Pharmacodyn. 2015, 42, 11–107. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2021; Available online: https://www.R-project.org/ (accessed on 10 October 2021).
- Beal, S.L. Ways to Fit a PK Model with Some Data below the Quantification Limit. J. Pharmacokinet. Pharmacodyn. 2001, 28, 481–504. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Nominal Concentration (ng/mL) | Accuracy | Precision | ||
|---|---|---|---|---|---|
| Calculated Concentration (ng/mL) ± SD | Accuracy (%) | Intra-Day (RSD, %) | Inter-Day (RSD, %) | ||
| Mangiferin | 5 (LLOQ) | 4.79 ± 0.40 | 95.74 | 8.28 | - |
| 50 | 51.11 ± 6.09 | 102.22 | 11.91 | 9.47 | |
| 500 | 517.83 ± 32.06 | 103.57 | 6.19 | 5.85 | |
| 1000 | 979.73 ± 24.34 | 97.93 | 2.48 | 2.05 | |
| Compound | Nominal Concentration (ng/mL) | Recovery (%) | Matrix Effect (%) | Stability (%, Mean ± RSD) | ||||
|---|---|---|---|---|---|---|---|---|
| Average | RSD | Average | RSD | Room Temp. | Autosampler | Freeze–Thaw | ||
| Mangiferin | 50 | 74.41 | 12.42 | 96.51 | 11.44 | 103.10 ± 3.66 | 109.92 ± 2.77 | 108.60 ± 3.87 |
| 500 | 74.90 | 4.18 | 109.99 | 3.60 | 101.27 ± 9.08 | 102.02 ± 8.81 | 101.68 ± 0.91 | |
| 1.000 | 81.11 | 4.66 | 106.48 | 6.12 | 101.75 ± 8.11 | 101.63 ± 8.50 | 102.64 ± 1.05 | |
| Parameter 1 | Units | MLE60 | MLES |
|---|---|---|---|
| AUC0–1h | h x ((ng/mL)/(mg/kg)) | 2.16 [81.5] | 6.88 [51.4] |
| AUC0–2h | h x ((ng/mL)/(mg/kg)) | 6.35 [75.7] | 20.6 [46.8] |
| AUC0–4h | h x ((ng/mL)/(mg/kg)) | 14.1 [64.5] | 39.2 [53.3] |
| AUC0–6h | h x ((ng/mL)/(mg/kg)) | 18.4 [72.7] | 47.3 [57.7] |
| AUC0–10h | h x ((ng/mL)/(mg/kg)) | 21.9 [83.8] | 53.3 [63.1] |
| AUC0–24h | h x ((ng/mL)/(mg/kg)) | 22.7 [87.8] | 55.5 [64.5] |
| AUC0–∞ | h x ((ng/mL)/(mg/kg)) | 24.9 [97.8] | 59.7 [68.4] |
| Cmax | ng/mL | 38.8 [41.1] | 103 [41.6] |
| Tmax | h | 2.00 [1.00, 4.00] | 2.00 [1.00, 2.00] |
| T1/2 | h | 3.19 [1.68] | 2.98 [2.04] |
| Vz/F | L/kg | 157.0 [46.4] | 58 [72.1] |
| Cl/F | (L/h)/kg | 40.1 [97.8] | 16.7 [68.4] |
| 0–1 h | 0–2 h | 0–4 h | 0–6 h | 0–10 h | 0–24 h | |
|---|---|---|---|---|---|---|
| MLES vs. MLE60 | 3.19 ** | 3.24 ** | 2.78 ** | 2.57 ** | 2.43 ** | 2.44 ** |
| Compound | Precursor Ion (m/z) | Product Ion [Collision Energy (eV)] | Cone Voltage (V) | Ion Mode | |
|---|---|---|---|---|---|
| Quantification Ion (m/z) | Qualification Ions (m/z) | ||||
| Mangiferin | 421.0 | 330.0 [24] | 300.9 [22]; 271.7 [36] | 48 | ESI− |
| Taxifolin (IS) | 303.0 | 285.0 [10] | 176.9 [10]; 124.9 [20] | 42 | ESI− |
| Subject Number | Male = M, Female = F | Body Weight (kg) | Experiment Day (P1 or P2) | Treatment | Mangiferin Dose (mg) | Normalized Dose (mg/kg BW) |
|---|---|---|---|---|---|---|
| 1 | M | 80.7 | P1 | MLE60 | 479.80 | 5.95 |
| 80.6 | P2 | MLES | 480.38 | 5.96 | ||
| 2 | F | 61.5 | P1 | MLE60 | 479.80 | 7.80 |
| 60.7 | P2 | MLES | 480.38 | 7.91 | ||
| 3 | F | 61.4 | P1 | MLE60 | 479.80 | 7.81 |
| 61.2 | P2 | MLES | 480.38 | 7.85 | ||
| 4 | M | 86.4 | P1 | MLES | 480.38 | 5.56 |
| 84.7 | P2 | MLE60 | 479.80 | 5.67 | ||
| 5 | M | 83.6 | P1 | MLES | 480.38 | 5.75 |
| 82.7 | P2 | MLE60 | 479.80 | 5.80 | ||
| 6 | F | 62.9 | P1 | MLES | 480.38 | 7.64 |
| 62.6 | P2 | MLE60 | 479.80 | 7.67 | ||
| 7 | F | 63.7 | P1 | MLES | 480.38 | 7.54 |
| 63.6 | P2 | MLE60 | 479.80 | 7.54 | ||
| 8 | F | 48.7 | P1 | MLE60 | 479.80 | 9.85 |
| 48.3 | P2 | MLES | 480.38 | 9.95 | ||
| 9 | M | 88.2 | P1 | MLE60 | 479.80 | 5.44 |
| 88.6 | P2 | MLES | 480.38 | 5.42 | ||
| 10 | F | B72 | P1 | MLE60 | 479.80 | 6.66 |
| 72.8 | P2 | MLES | 480.38 | 6.60 | ||
| 11 | M | 78.9 | P1 | MLES | 480.38 | 6.09 |
| 79 | P2 | MLE60 | 479.80 | 6.07 | ||
| 12 | M | 83.3 | P1 | MLES | 480.38 | 5.77 |
| 84.9 | P2 | MLE60 | 479.80 | 5.65 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fuentes-Rios, D.; Sanchez-Rodriguez, A.; Lopez-Rios, L.; Garcia-Gonzalez, E.; Martinez-Canton, M.; Galvan-Alvarez, V.; Gallego-Selles, A.; Martin-Rincon, M.; Calbet, J.A.L.; Vega-Morales, T. Human Pharmacokinetic Profiling and Comparative Analysis of Mangiferin and Its Monosodium Derivative from Mangifera indica Extracts Using UHPLC-MS/MS with 1H NMR and MALDI-TOF Confirmation. Molecules 2025, 30, 461. https://doi.org/10.3390/molecules30030461
Fuentes-Rios D, Sanchez-Rodriguez A, Lopez-Rios L, Garcia-Gonzalez E, Martinez-Canton M, Galvan-Alvarez V, Gallego-Selles A, Martin-Rincon M, Calbet JAL, Vega-Morales T. Human Pharmacokinetic Profiling and Comparative Analysis of Mangiferin and Its Monosodium Derivative from Mangifera indica Extracts Using UHPLC-MS/MS with 1H NMR and MALDI-TOF Confirmation. Molecules. 2025; 30(3):461. https://doi.org/10.3390/molecules30030461
Chicago/Turabian StyleFuentes-Rios, David, Alvaro Sanchez-Rodriguez, Laura Lopez-Rios, Eduardo Garcia-Gonzalez, Miriam Martinez-Canton, Victor Galvan-Alvarez, Angel Gallego-Selles, Marcos Martin-Rincon, Jose A. L. Calbet, and Tanausu Vega-Morales. 2025. "Human Pharmacokinetic Profiling and Comparative Analysis of Mangiferin and Its Monosodium Derivative from Mangifera indica Extracts Using UHPLC-MS/MS with 1H NMR and MALDI-TOF Confirmation" Molecules 30, no. 3: 461. https://doi.org/10.3390/molecules30030461
APA StyleFuentes-Rios, D., Sanchez-Rodriguez, A., Lopez-Rios, L., Garcia-Gonzalez, E., Martinez-Canton, M., Galvan-Alvarez, V., Gallego-Selles, A., Martin-Rincon, M., Calbet, J. A. L., & Vega-Morales, T. (2025). Human Pharmacokinetic Profiling and Comparative Analysis of Mangiferin and Its Monosodium Derivative from Mangifera indica Extracts Using UHPLC-MS/MS with 1H NMR and MALDI-TOF Confirmation. Molecules, 30(3), 461. https://doi.org/10.3390/molecules30030461