
Comprehensive Profiling of Illicit Amphetamines Seized in Poland: Insights from Gas Chromatography–Mass Spectrometry and Chemometric Analysis



Abstract
:1. Introduction
2. Results and Discussion
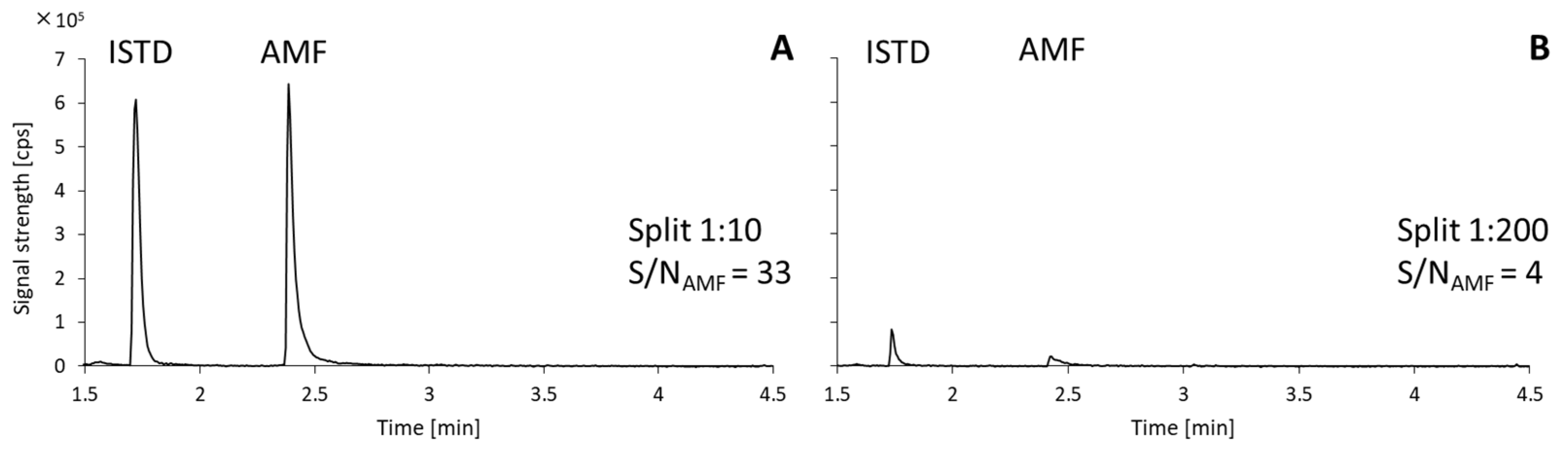
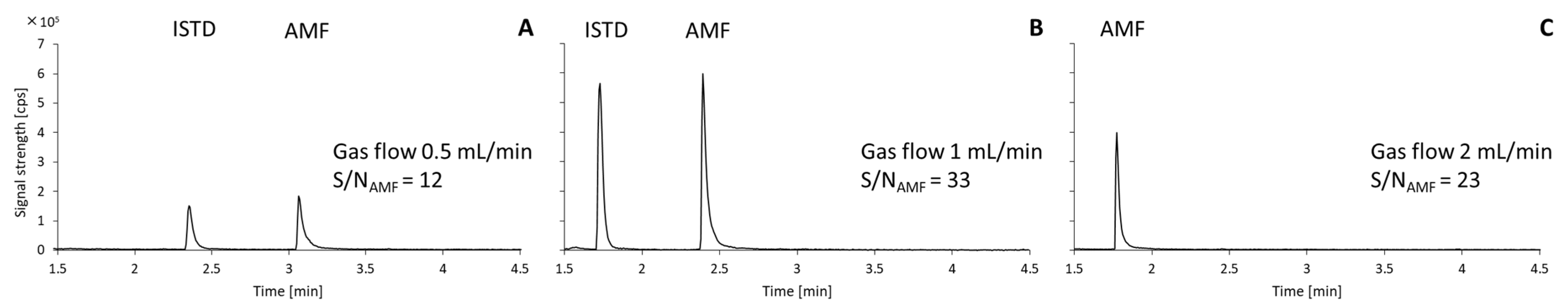
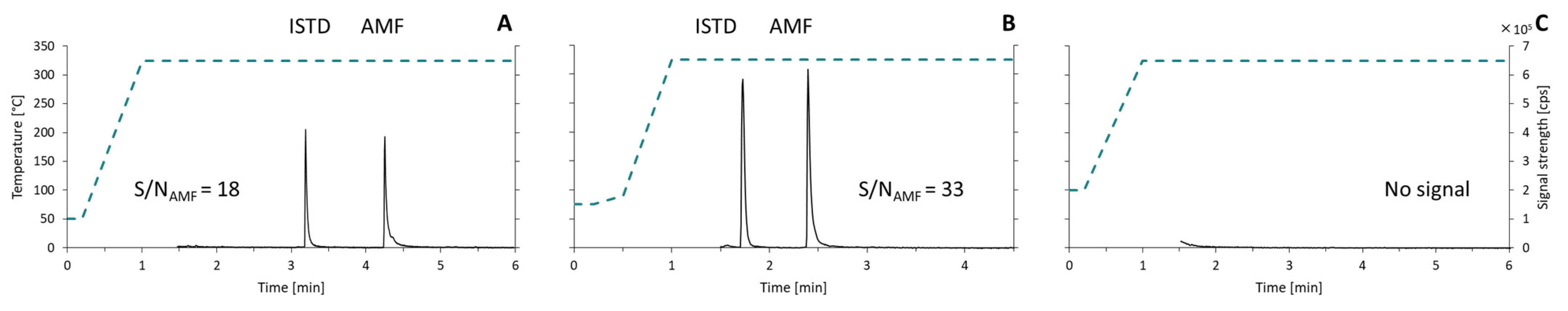
2.1. Optimization of Chromatographic Method
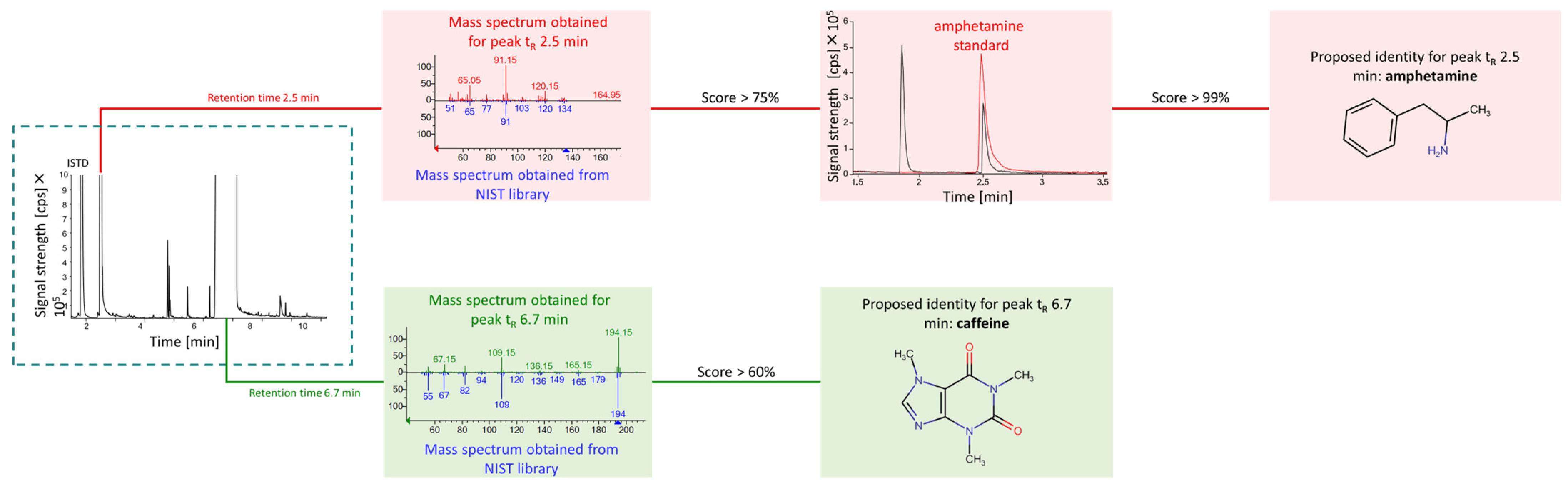
2.2. Confirmation of Identity for Amphetamine and Qualitative Analysis for Other Compounds
2.3. Determination of Amphetamine in Obtained Samples
- The number of intoxicating doses, based on a standard dose of 10 mg of pure amphetamine;
- The number of doses for “heavy users”, defined as 50 mg of pure amphetamine per dose;
- The mass of the smallest intoxicating dose;
- The number of commercial portions, assumed to be 1 g per portion;
- The market value, calculated at 10 EUR per gram.
2.4. Data Preparation for Impurity Profiling
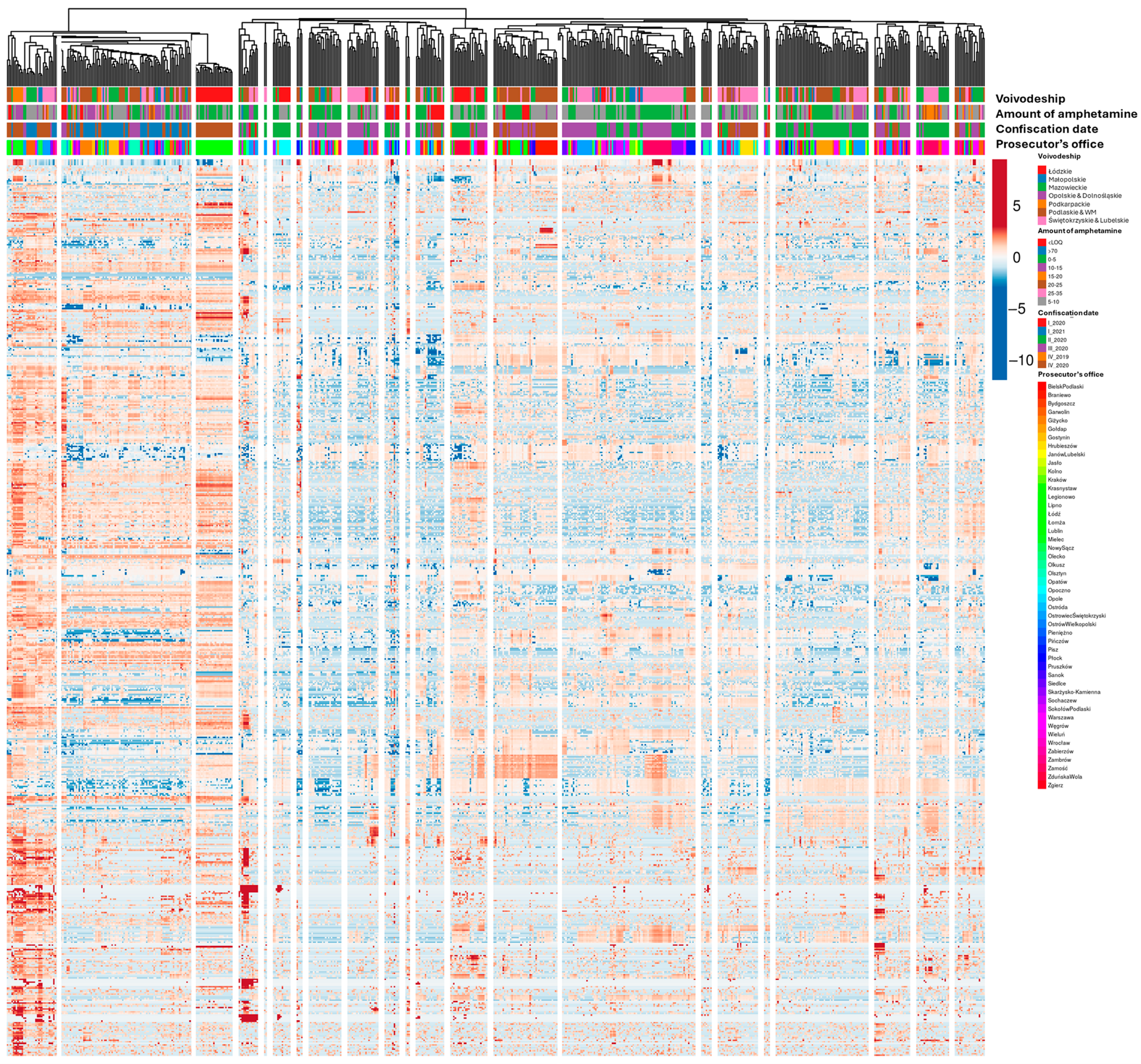
2.5. Impurity Profiling
3. Materials and Methods
3.1. Chemicals and Reagents
3.2. Registration of Evidence and Collection of Analytical Samples
3.3. Sample Preparation Procedure
3.4. Instrumentation
3.5. Calibration of Method for Determination of Amphetamine
3.6. Precision of Quantitative Method
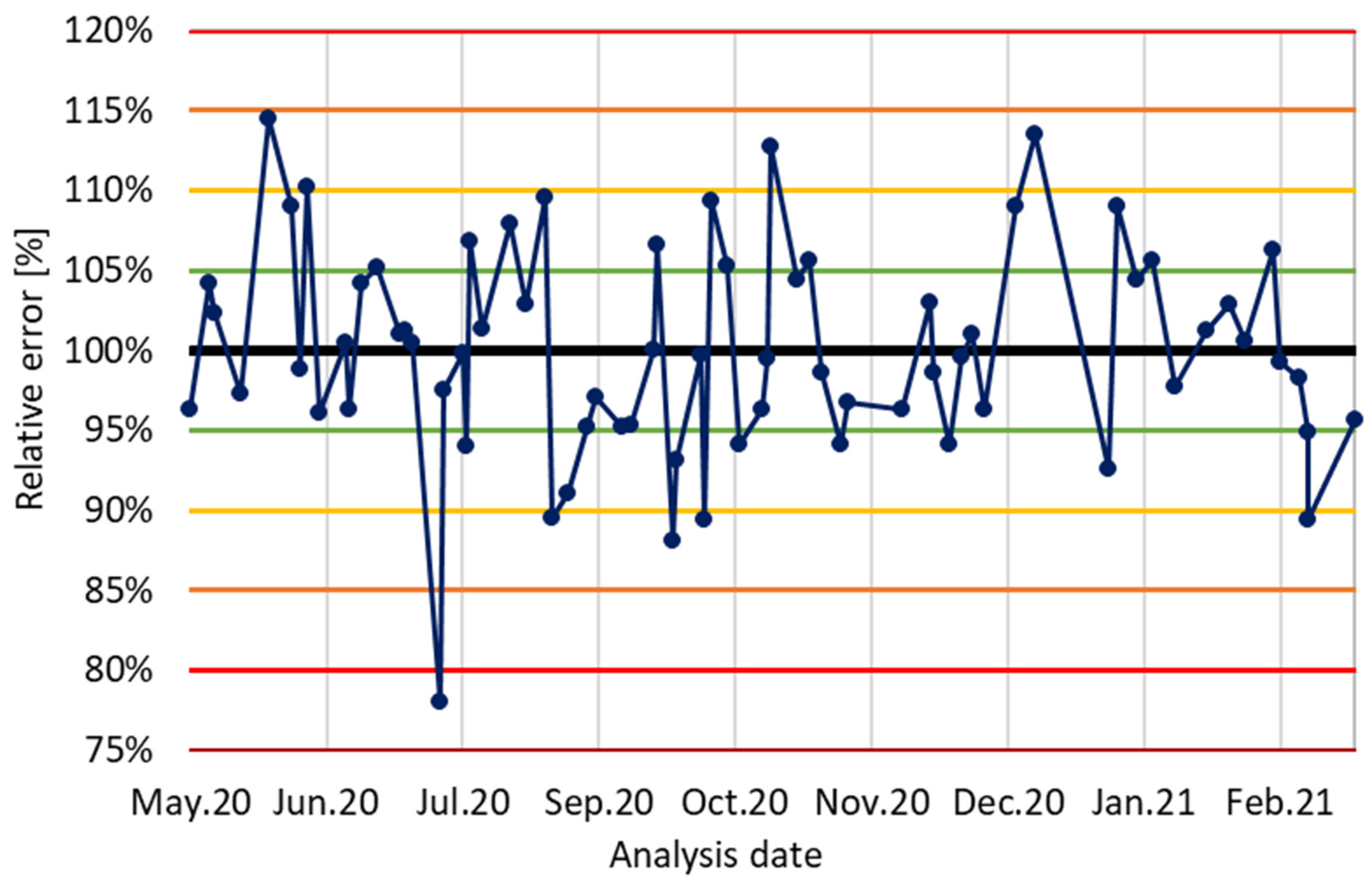
3.7. Long-Term Precision and Quality Assurance for GC-MS
3.8. Data Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Raviña, E. The Evolution of Drug Discovery: From Traditional Medicines to Modern Drugs; Wiley-VCH: Weinheim, Germany, 2011; ISBN 978-3-527-32669-3. [Google Scholar]
- Gootenberg, P. The Oxford Handbook of Global Drug History; Oxford Handbooks; Oxford University Press: New York, NY, USA, 2022; ISBN 978-0-19-084264-2. [Google Scholar]
- European Monitoring Centre for Drugs and Drug Addiction. European Drug Report 2024: Trends and Developments; Publications Office of the European Union: Luxembourg, 2024; ISBN 978-92-9497-975-9. [Google Scholar]
- European Monitoring Centre for Drugs and Drug Addiction. EU Drug Market: Amphetamine: In-Depth Analysis; Publications Office of the European Union: Luxembourg, 2023; ISBN 978-92-9497-851-6. [Google Scholar]
- Barberet, R.; Smith, C.J.; Zhang, S. Routledge Handbook of International Criminology; Routledge International Handbooks; Routledge: Abinqdon, UK, 2011; ISBN 978-1-135-19385-0. [Google Scholar]
- Blickman, T. The Ecstasy Industry. Exploring the Global Market. TNI Brief. Ser. 2004, 9, 1–32. [Google Scholar]
- Hauser, F.M.; Rößler, T.; Hulshof, J.W.; Weigel, D.; Zimmermann, R.; Pütz, M. Identification of Specific Markers for Amphetamine Synthesised from the Pre-precursor APAAN Following the Leuckart Route and Retrospective Search for APAAN Markers in Profiling Databases from Germany and the Netherlands. Drug Test. Anal. 2018, 10, 671–680. [Google Scholar] [CrossRef] [PubMed]
- Zábranský, T. Methamphetamine in the Czech Republic. J. Drug Issues 2007, 37, 155–180. [Google Scholar] [CrossRef]
- Green, M.K.; Ciesielski, A.L.; Wagner, J.R. Detection of One Pot Methamphetamine Laboratory Byproducts in Wastewater via Solid Phase Extraction and Liquid Chromatography-Tandem Mass Spectrometry. Forensic Chem. 2020, 19, 100253. [Google Scholar] [CrossRef]
- Trynda, A.; Duszyńska, A. Current Trends in Purity, Quantity and Prices of the Most Popular Drugs in Poland. Issues Forensic Sci. 2019, 306, 48–58. [Google Scholar] [CrossRef]
- Alabdalla, M.A. Chemical Characterization of Counterfeit Captagon Tablets Seized in Jordan. Forensic Sci. Int. 2005, 152, 185–188. [Google Scholar] [CrossRef]
- Pergolizzi, J., Jr.; LeQuang, J.A.K.; Vortsman, E.; Magnusson, P.; EL-Tallawy, S.N.; Wagner, M.; Salah, R.; Varrassi, G. The Emergence of the Old Drug Captagon as a New Illicit Drug: A Narrative Review. Cureus 2024, 16, e55053. [Google Scholar] [CrossRef] [PubMed]
- European Monitoring Centre for Drugs and Drug Addiction; Europol. EU Drug Markets Report: A Strategic Analysis; Publications Office of the European Union: Luxembourg, 2013; ISBN 978-92-9168-595-0. [Google Scholar]
- Bajda, K. Criminological and Forensic Aspects of Selected Forms of Contemporary Organized Crime in Poland. Przegląd Prawno-Ekon. 2019, 25, 9–24. [Google Scholar] [CrossRef]
- Hołyst, B. Technika Kryminalistyczna w Pierwszej Połowie XXI Wieku: Wybrane Problemy; Wydawnictwo Naukowe PWN: Warszawa, Poland, 2014; ISBN 978-83-01-18129-1. [Google Scholar]
- Krawczyk, W.S. Amfetamina i Jej Pochodne: Metody Nielegalnej Produkcji; Wydawnictwo Centralnego Laboratorium Kryminalistycznego Policji—Instytutu Badawczego: Warszawa, Poland, 2015; ISBN 978-83-63420-06-2. [Google Scholar]
- European Monitoring Centre for Drugs and Drug Addiction. Perspectives on Drugs: Synthetic Drug Production in Europe; Publications Office of the European Union: Luxembourg, 2015. [Google Scholar]
- Umar, Q.; Luo, M. A Brief Review: Advancement in the Synthesis of Amine through the Leuckart Reaction. Reactions 2023, 4, 117–147. [Google Scholar] [CrossRef]
- Stojanovska, N.; Fu, S.; Tahtouh, M.; Kelly, T.; Beavis, A.; Kirkbride, K.P. A Review of Impurity Profiling and Synthetic Route of Manufacture of Methylamphetamine, 3,4-Methylenedioxymethylamphetamine, Amphetamine, Dimethylamphetamine and p-Methoxyamphetamine. Forensic Sci. Int. 2013, 224, 8–26. [Google Scholar] [CrossRef] [PubMed]
- Chłopaś, A.; Bańka, K.; Buszewicz, G. Amphetamine in Illegally Produced Phenylethylamine—Intentional Action or Failed Synthesis of a Designer Drug. Curr. Issues Pharm. Med. Sci. 2015, 26, 16–20. [Google Scholar] [CrossRef]
- International Narcotics Control Board. Report on Precursors. Precursors and Chemicals Frequently Used in the Illicit Manufacture of Narcotic Drugs and Psychotropic Substances; United Nations Publications: Vienna, Austria, 2023; ISBN 978-92-1-003054-0. [Google Scholar]
- International Narcotics Control Board. Bulletin on Narcotics. Science in Drug Control: The Role of Laboratory and Scientific Expertise; United Nations Publications: Vienna, Austria, 2005; ISBN 978-92-1-148221-8. [Google Scholar]
- Hauser, F.M.; Pütz, M.; Rößler, T.; Hulshof, J.W. Identification of Specific Markers for Amphetamines Synthesized from Glycidic Acid Pre-precursors and Retrospective Search in German Profiling Database. Drug Test. Anal. 2020, 12, 41–52. [Google Scholar] [CrossRef] [PubMed]
- Power, J.D.; Kavanagh, P.; McLaughlin, G.; Barry, M.; Dowling, G.; Brandt, S.D. ‘APAAN in the Neck’—A Reflection on Some Novel Impurities Found in Seized Materials Containing Amphetamine in Ireland during Routine Forensic Analysis. Drug Test. Anal. 2017, 9, 966–976. [Google Scholar] [CrossRef] [PubMed]
- European Monitoring Centre for Drugs and Drug Addiction; Europol. EU Drug Markets Report 2016: In-Depth Analysis; Publications Office of the European Union: Luxembourg, 2016; ISBN 978-92-9168-842-5. [Google Scholar]
- Galarda, M.; Bachliński, R.; Central Forensic Laboratory of the Police. Chemistry Department Identification of 2-Acetyl-2-Phenylacetamide (APAA)—The Precursor for Production of Benzylmethylketone (BMK, P2P). Issues Forensic Sci. 2019, 304, 83–95. [Google Scholar] [CrossRef]
- Tsujikawa, K.; Okada, Y.; Segawa, H.; Kuwayama, K.; Yamamuro, T.; Kanamori, T.; Iwata, Y.T. Analysis of Potential Phenylacetone Precursors (Ethyl 3-oxo-2-phenylbutyrate, Methyl 3-oxo-4-phenylbutyrate, and Ethyl 3-oxo-4-phenylbutyrate) by Gas Chromatography/Mass Spectrometry and Their Conversion to Phenylacetone. Drug Test. Anal. 2022, 14, 439–449. [Google Scholar] [CrossRef] [PubMed]
- Emke, E.; Vughs, D.; Kolkman, A.; De Voogt, P. Wastewater-Based Epidemiology Generated Forensic Information: Amphetamine Synthesis Waste and Its Impact on a Small Sewage Treatment Plant. Forensic Sci. Int. 2018, 286, e1–e7. [Google Scholar] [CrossRef] [PubMed]
- DeRuiter, J.; Clark, C.R.; Noggle, F.T. Gas Chromatographic and Mass Spectral Analysis of Amphetamine Products Synthesized from 1-Phenyl-2-Nitropropene. J. Chromatogr. Sci. 1994, 32, 511–519. [Google Scholar] [CrossRef]
- General Assembly of the United Nations. Action Plan Against Illicit Manufacture, Trafficking and Abuse of Amphetamine-Type Stimulants and Their Precursors; United Nations Publications: Vienna, Austria, 1998. [Google Scholar]
- Strömberg, L. Comparative Gas Chromatographic Analysis of Narcotics: II Amphetamine Sulphate. J. Chromatogr. A 1975, 106, 335–342. [Google Scholar] [CrossRef]
- Strömberg, L.; Maehly, A.C. Comparative Gas Chromatographic Analysis of Narcotics: III Phenmetrazine Hydrochloride. J. Chromatogr. A 1975, 109, 67–72. [Google Scholar] [CrossRef]
- Strömberg, L.; Bergkvist, H.; Edirisinghe, E.A.M.K. Comparative Gas Chromatographic Analysis of Narcotics: IV Methamphetamine Hydrochloride. J. Chromatogr. A 1983, 258, 65–72. [Google Scholar] [CrossRef]
- Dayrit, F.M.; Dumlao, M.C. Impurity Profiling of Methamphetamine Hydrochloride Drugs Seized in the Philippines. Forensic Sci. Int. 2004, 144, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.X.; Zhang, D.M.; Han, X.G. Identification of Impurities and Statistical Classification of Methamphetamine Hydrochloride Drugs Seized in China. Forensic Sci. Int. 2008, 182, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Jonson, C.S.L. Amphetamine Profiling—Improvements of Data Processing. Forensic Sci. Int. 1994, 69, 45–54. [Google Scholar] [CrossRef]
- King, L.A.; Clarke, K.; Orpet, A.J. Amphetamine Profiling in the UK. Forensic Sci. Int. 1994, 69, 65–75. [Google Scholar] [CrossRef]
- Besacier, F.; Chaudron-Thozet, H.; Rousseau-Tsangaris, M.; Girard, J.; Lamotte, A. Comparative Chemical Analyses of Drug Samples: General Approach and Application to Heroin. Forensic Sci. Int. 1997, 85, 113–125. [Google Scholar] [CrossRef]
- Yüksel, B. Quantitative GC-FID Analysis of Heroin for Seized Drugs. Ann. Clin. Anal. Med. 2020, 11, 38–42. [Google Scholar] [CrossRef]
- Chan, K.-W.; Tan, G.-H.; Wong, R.C.S. Investigation of Illicit Heroin Seized in Malaysia: Physical Characteristics and Chemical Profiling. Aust. J. Forensic Sci. 2012, 44, 353–369. [Google Scholar] [CrossRef]
- Barnfield, C.; Burns, S.; Byrom, D.L.; Kemmenoe, A.V. The Routine Profiling of Forensic Heroin Samples. Forensic Sci. Int. 1988, 39, 107–117. [Google Scholar] [CrossRef]
- Puthaviriyakorn, V.; Siriviriyasomboon, N.; Phorachata, J.; Pan-ox, W.; Sasaki, T.; Tanaka, K. Identification of Impurities and Statistical Classification of Methamphetamine Tablets (Ya-Ba) Seized in Thailand. Forensic Sci. Int. 2002, 126, 105–113. [Google Scholar] [CrossRef]
- Palhol, F.; Boyer, S.; Naulet, N.; Chabrillat, M. Impurity Profiling of Seized MDMA Tablets by Capillary Gas Chromatography. Anal. Bioanal. Chem. 2002, 374, 274–281. [Google Scholar] [CrossRef] [PubMed]
- Moore, J.M.; Allen, A.C.; Cooper, D.A. Determination of Manufacturing Impurities in Heroin by Capillary Gas Chromatography with Electron Capture Detection after Derivatization with Heptafluorobutyric Anhydride. Anal. Chem. J. 1984, 56, 642–646. [Google Scholar] [CrossRef]
- Alhazmi, H.A.; Ahsan, W.; Al Bratty, M.; Khalid, A.; Sultana, S.; Najmi, A.; Makeen, H.A.; Attafi, I.M.; Abualsail, F.M.; Arishy, M.A.; et al. Chemo-Profiling of Illicit Amphetamine Tablets Seized from Jazan, Saudi Arabia, Using Gas Chromatography-Mass Spectrometry and Chemometric Techniques. J. Chem. 2021, 2021, 1517785. [Google Scholar] [CrossRef]
- Langone, D.; Painter, B.; Nash, C.; Hulshof, J.; Oldenhof, S.; Johnston, M.R.; Kirkbride, K.P. Impurity Profiling of Methamphetamine Synthesized from Methyl α-acetylphenylacetate. Drug Test. Anal. 2022, 14, 1310–1324. [Google Scholar] [CrossRef]
- Afshar Etemadi, N.A. Impurity Profiling of Street Methamphetamine Samples Seized in Kermanshah, Iran with Special Focus on Methamphetamine Impurities Health Hazards. J. Clin. Toxicol. 2015, 5, 1000258. [Google Scholar] [CrossRef]
- Mat Desa, W.N.S.; Ismail, D. Impurity Profiling of Amphetamine and Methamphetamine Using Gas Chromatography Mass Spectrometry (GCMS) Harmonised Methods. Sains Malays. 2017, 46, 149–156. [Google Scholar] [CrossRef]
- Qi, Y.; Evans, I.; McCluskey, A. New Impurity Profiles of Recent Australian Imported ‘Ice’: Methamphetamine Impurity Profiling and the Identification of (Pseudo)Ephedrine and Leuckart Specific Marker Compounds. Forensic Sci. Int. 2007, 169, 173–180. [Google Scholar] [CrossRef]
- Laposchan, S.; Kranenburg, R.F.; Van Asten, A.C. Impurities, Adulterants and Cutting Agents in Cocaine as Potential Candidates for Retrospective Mining of GC-MS Data. Sci. Justice 2022, 62, 60–75. [Google Scholar] [CrossRef] [PubMed]
- Allen, A.C.; Cooper, D.A.; Moore, J.M.; Gloger, M.; Neumann, H. Illicit Heroin Manufacturing By-Products: Capillary Gas Chromatographic Determination and Structural Elucidation of Narcotine- and Norlaudanosine-Related Compounds. Anal. Chem. J. 1984, 56, 2940–2947. [Google Scholar] [CrossRef] [PubMed]
- Morello, D.R.; Cooper, S.D.; Panicker, S.; Casale, J.F. Signature Profiling and Classification of Illicit Heroin by GC-MS Analysis of Acidic and Neutral Manufacturing Impurities. J. Forensic Sci. 2010, 55, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Myors, R.B.; Skopec, S.V.; Wells, R.J.; Crisp, P.T. Investigation of Heroin Profiling Using Trace Organic Impurities. Analyst 2001, 126, 679–689. [Google Scholar] [CrossRef]
- Morello, D.; Meyers, R. Qualitative and Quantitative Determination of Residual Solvents in Illicit Cocaine HCl and Heroin HCl. J. Forensic Sci. 1995, 40, 957–963. [Google Scholar] [CrossRef]
- Stride Nielsen, L.; Villesen, P.; Lindholst, C. Stability of Amphetamine Impurity Profiles during 12 Months of Storage. Forensic Sci. Int. 2018, 290, 129–136. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K.; Ohmori, T.; Inoue, T. Analysis of Impurities in Illicit Methamphetamine. Forensic Sci. Int. 1992, 56, 157–165. [Google Scholar] [CrossRef]
- Gimeno, P.; Besacier, F.; Chaudron-Thozet, H.; Girard, J.; Lamotte, A. A Contribution to the Chemical Profiling of 3,4-Methylenedioxymethamphetamine (MDMA) Tablets. Forensic Sci. Int. 2002, 127, 1–44. [Google Scholar] [CrossRef]
- Milliet, Q.; Weyermann, C.; Esseiva, P. The Profiling of MDMA Tablets: A Study of the Combination of Physical Characteristics and Organic Impurities as Sources of Information. Forensic Sci. Int. 2009, 187, 58–65. [Google Scholar] [CrossRef]
- Weyermann, C.; Marquis, R.; Delaporte, C.; Esseiva, P.; Lock, E.; Aalberg, L.; Bozenko, J.S.; Dieckmann, S.; Dujourdy, L.; Zrcek, F. Drug Intelligence Based on MDMA Tablets Data: I. Organic Impurities Profiling. Forensic Sci. Int. 2008, 177, 11–16. [Google Scholar] [CrossRef]
- Nielsen, L.S.; Villesen, P.; Lindholst, C. Stability of Cocaine Impurity Profiles during 12 Months of Storage. Forensic Sci. Int. 2016, 264, 56–62. [Google Scholar] [CrossRef]
- Taner, B.; Hüseyin, Ç. Statistical Assessment Using Chemical Profiling of Ecstasy Samples Seized in Turkey. J. Anal. Chem. 2018, 73, 1020–1028. [Google Scholar] [CrossRef]
- Verovšek, T.; Heath, D.; Heath, E. Enantiomeric Profiling of Amphetamines in Wastewater Using Chiral Derivatisation with Gas Chromatographic-Tandem Mass Spectrometric Detection. Sci. Total Environ. 2022, 835, 155594. [Google Scholar] [CrossRef]
- Kochana, J.; Tomaszewski, W.; Moszczyński, T.; Zakrzewska, A.; Parczewski, A. Application of Carbon Adsorbents for Extraction of MDMA Impurities in TLC Drug Profiling. J. Liq. Chromatogr. Relat. Technol. 2008, 31, 819–827. [Google Scholar] [CrossRef]
- Renton, R.J.; Cowie, J.S.; Oon, M.C.H. A Study of the Precursors, Intermediates and Reaction by-Products in the Synthesis of 3,4-Methylenedioxymethylamphetamine and Its Application to Forensic Drug Analysis. Forensic Sci. Int. 1993, 60, 189–202. [Google Scholar] [CrossRef]
- Hays, P.; Remaud, G.; Jamin, É.; Martin, Y.-L. Geographic Origin Determination of Heroin and Cocaine Using Site-Specific Isotopic Ratio Deuterium NMR. J. Forensic Sci. 2000, 45, 552–562. [Google Scholar] [CrossRef]
- Balayssac, S.; Retailleau, E.; Bertrand, G.; Escot, M.-P.; Martino, R.; Malet-Martino, M.; Gilard, V. Characterization of Heroin Samples by 1H NMR and 2D DOSY 1H NMR. Forensic Sci. Int. 2014, 234, 29–38. [Google Scholar] [CrossRef] [PubMed]
- Lambrechts, M.; Tönnesen, F.; Rasmussen, K.E. Profiling of Impurities in Illicit Amphetamine Samples by High-Performance Liquid Chromatography Using Column Switching. J. Chromatogr. A 1986, 369, 365–377. [Google Scholar] [CrossRef] [PubMed]
- Makino, Y.; Urano, Y.; Nagano, T. Impurity Profiling of Ephedrines in Methamphetamine by High-Performance Liquid Chromatography. J. Chromatogr. A 2002, 947, 151–154. [Google Scholar] [CrossRef]
- Huizer, H. Analytical Studies on Illicit Heroin II. Comparison of Samples. J. Forensic Sci. 1983, 28, 40–48. [Google Scholar] [CrossRef] [PubMed]
- Lurie, I.S.; Bailey, C.G.; Anex, D.S.; Bethea, M.J.; McKibben, T.D.; Casale, J.F. Profiling of Impurities in Illicit Methamphetamine by High-Performance Liquid Chromatography and Capillary Electrochromatography. J. Chromatogr. A 2000, 870, 53–68. [Google Scholar] [CrossRef] [PubMed]
- Lurie, I.S.; Carr, S.M. The Quantitation of Heroin and Selected Basic Impurities Via Reversed Phase HPLC. I. The Analysis of Unadulterated Heroin Samples. J. Liq. Chromatogr. 1986, 9, 2485–2509. [Google Scholar] [CrossRef]
- Collins, M.; Casale, E.; Hibbert, D.B.; Panicker, S.; Robertson, J.; Vujic, S. Chemical Profiling of Heroin Recovered from the North Korean Merchant Vessel Pong Su. J. Forensic Sci. 2006, 51, 597–602. [Google Scholar] [CrossRef] [PubMed]
- Lurie, I.S.; Driscoll, S.E.; Cathapermal, S.S.; Panicker, S. Determination of Heroin and Basic Impurities for Drug Profiling by Ultra-High-Pressure Liquid Chromatography. Forensic Sci. Int. 2013, 231, 300–305. [Google Scholar] [CrossRef] [PubMed]
- Huizer, H. Analytical Studies on Illicit Heroin I. The Occurrence of O 3-Monoacetylmorphine. J. Forensic Sci. 1983, 28, 32–39. [Google Scholar] [CrossRef]
- Dams, R.; Benijts, T.; Günther, W.; Lambert, W.; De Leenheer, A. Sonic Spray Ionization Technology: Performance Study and Application to a LC/MS Analysis on a Monolithic Silica Column for Heroin Impurity Profiling. Anal. Chem. J. 2002, 74, 3206–3212. [Google Scholar] [CrossRef]
- Li, L.; Brown, J.L.; Toske, S.G. Simultaneous Detection and Quantitation of Organic Impurities in Methamphetamine by Ultra-high-performance Liquid Chromatography–Tandem Mass Spectrometry, a Complementary Technique for Methamphetamine Profiling. Drug Test. Anal. 2018, 10, 1209–1219. [Google Scholar] [CrossRef]
- Jovanov, P.; Petrin-Miličević, M.; Radosavljević-Stevanović, N.; Vraneš, M.; Belić, S.; Sakač, M.; Nikolov, J.; Gadžurić, S. Rapid Determination of the Primary Alkaloids in Illicit Heroin by High-Performance Liquid Chromatography with Tandem Mass Spectrometry (HPLC–MS/MS). Anal. Lett. 2021, 54, 1224–1232. [Google Scholar] [CrossRef]
- Li, L.; Panicker, S.; Casale, E.M. UHPLC–MS/MS Quantitation of Porphyroxine in Opium and Application of Porphyroxine-acetylated Products as Signature Markers for Heroin. Drug Test. Anal. 2019, 11, 999–1008. [Google Scholar] [CrossRef] [PubMed]
- Debrus, B.; Broséus, J.; Guillarme, D.; Lebrun, P.; Hubert, P.; Veuthey, J.-L.; Esseiva, P.; Rudaz, S. Innovative Methodology to Transfer Conventional GC-MS Heroin Profiling to UHPLC-MS/MS. Anal. Bioanal. Chem. 2011, 399, 2719–2730. [Google Scholar] [CrossRef] [PubMed]
- Lurie, I.S.; Toske, S.G. Applicability of Ultra-Performance Liquid Chromatography-Tandem Mass Spectrometry for Heroin Profiling. J. Chromatogr. A 2008, 1188, 322–326. [Google Scholar] [CrossRef] [PubMed]
- Alhazmi, H.A.; Ahsan, W.; Al Bratty, M.; Javed, S.A.; El-Sharkawy, K.A.; Khalid, A.; Alsalem, H.M.; Hakami, A.M.; Attafi, M.A.; Oraiby, M.E. Analysis of Amphetamine and Methamphetamine Contents in Seized Tablets from Jazan, Saudi Arabia by Liquid Chromatography-Mass Spectroscopy (LC-MS/MS) and Chemometric Techniques. Saudi Pharm. J. 2020, 28, 703–709. [Google Scholar] [CrossRef]
- Liu, C.; Hua, Z.; Meng, X. Applicability of Ultra-high Performance Liquid Chromatography-quadrupole-time of Flight Mass Spectrometry for Cocaine Profiling. Drug Test. Anal. 2017, 9, 1152–1161. [Google Scholar] [CrossRef] [PubMed]
- Carby-Robinson, D.; Dalsgaard, P.W.; Mollerup, C.B.; Linnet, K.; Rasmussen, B.S. Cocaine Profiling Method Retrospectively Developed with Nontargeted Discovery of Markers Using Liquid Chromatography with Time-of-flight Mass Spectrometry Data. Drug Test. Anal. 2022, 14, 462–473. [Google Scholar] [CrossRef]
- Lurie, I.S.; Anex, D.S.; Fintschenko, Y.; Choi, W.-Y. Profiling of Impurities in Heroin by Capillary Electrochromatography and Laser-Induced Fluorescence Detection. J. Chromatogr. A 2001, 924, 421–427. [Google Scholar] [CrossRef]
- Macchia, M.; Manetto, G.; Mori, C.; Papi, C.; Di Pietro, N.; Salotti, V.; Bortolotti, F.; Tagliaro, F. Use of β-Cyclodextrin in the Capillary Zone Electrophoretic Separation of the Components of Clandestine Heroin Preparations. J. Chromatogr. A 2001, 924, 499–506. [Google Scholar] [CrossRef] [PubMed]
- Iwata, Y.T.; Inoue, H.; Kuwayama, K.; Kanamori, T.; Tsujikawa, K.; Miyaguchi, H.; Kishi, T. Forensic Application of Chiral Separation of Amphetamine-Type Stimulants to Impurity Analysis of Seized Methamphetamine by Capillary Electrophoresis. Forensic Sci. Int. 2006, 161, 92–96. [Google Scholar] [CrossRef]
- Lurie, I.S.; Chan, K.C.; Spratley, T.K.; Casale, J.F.; Issaq, H.J. Separation and Detection of Acidic and Neutral Impurities in Illicit Heroin via Capillary Electrophoresis. J. Chromatogr. B Biomed. Sci. Appl. 1995, 669, 3–13. [Google Scholar] [CrossRef]
- Lurie, I.; Hays, P.; Valentino, A. Analysis of Carbohydrates in Seized Heroin Using Capillary Electrophoresis. J. Forensic Sci. 2006, 51, 39–44. [Google Scholar] [CrossRef]
- Zhang, Z.; Yan, B.; Liu, K.; Liao, Y.; Liu, H. CE-MS Analysis of Heroin and Its Basic Impurities Using a Charged Polymer-protected Gold Nanoparticle-coated Capillary. Electrophoresis 2009, 30, 379–387. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Hua, Z.; Meng, X. Profiling of Illicit Cocaine Seized in China by ICP-MS Analysis of Inorganic Elements. Forensic Sci. Int. 2017, 276, 77–84. [Google Scholar] [CrossRef]
- Liu, C.; Hua, Z.; Bai, Y.; Liu, Y. Profiling and Classification of Illicit Heroin by ICP-MS Analysis of Inorganic Elements. Forensic Sci. Int. 2014, 239, 37–43. [Google Scholar] [CrossRef]
- NicDaéid, N.; Jayaram, S.; Kerr, W.J. Elemental Profiling Using ICPMS of Methylamphetamine Hydrochloride Prepared from Proprietary Medication Using the Moscow and Hypophosphorous Synthesis. Sci. Justice 2013, 53, 278–285. [Google Scholar] [CrossRef]
- Marumo, Y.; Inoue, T.; Seta, S. Analysis of Inorganic Impurities in Seized Methamphetamine Samples. Forensic Sci. Int. 1994, 69, 89–95. [Google Scholar] [CrossRef]
- Kuras, M.J.; Wachowicz, M.J. Cannabis Profiling Based on Its Elemental Composition-Is It Possible?: Cannibard profiling based on its elemental composition. J. Forensic Sci. 2011, 56, 1250–1255. [Google Scholar] [CrossRef] [PubMed]
- Bermejobarrera, P.; Moredapineiro, A.; Moredapineiro, J.; Bermejobarrera, A. Effectiveness of Palladium as a Chemical Modifier for Direct Silver and Manganese Determination in Cocaine and Heroin by Electrothermal Atomic Absorption Spectrometry. Talanta 1996, 43, 1783–1792. [Google Scholar] [CrossRef] [PubMed]
- Aalberg, L.; Andersson, K.; Bertler, C.; Borén, H.; Cole, M.D.; Dahlén, J.; Finnon, Y.; Huizer, H.; Jalava, K.; Kaa, E.; et al. Development of a harmonised method for the profiling of amphetamines: I. Synthesis of standards and compilation of analytical data. Forensic Sci. Int. 2005, 149, 219–229. [Google Scholar] [CrossRef]
- Aalberg, L.; Andersson, K.; Bertler, C.; Cole, M.D.; Finnon, Y.; Huizer, H.; Jalava, K.; Kaa, E.; Lock, E.; Lopes, A. Development of a harmonized method for the profiling of amphetamines. II. Stability of impurities in organic solvents. Forensic Sci. Int. 2005, 149, 231–241. [Google Scholar] [CrossRef] [PubMed]
- Andersson, K.; Lock, E.; Jalava, K.; Huizer, H.; Jonson, S.; Kaa, E.; Lopes, A.; Poortman-van Der Meer, A.; Sippola, E.; Dujourdy, L.; et al. Development of a Harmonised Method for the Profiling of Amphetamines VI. Forensic Sci. Int. 2007, 169, 86–99. [Google Scholar] [CrossRef]
- Andersson, K.; Jalava, K.; Lock, E.; Finnon, Y.; Huizer, H.; Kaa, E.; Lopes, A.; Poortman-van Der Meer, A.; Cole, M.D.; Dahlén, J. Development of a harmonised method for the profiling of amphetamines: III. Development of the gas chromatographic method. Forensic Sci. Int. 2007, 169, 50–63. [Google Scholar] [CrossRef]
- Andersson, K.; Jalava, K.; Lock, E.; Huizer, H.; Kaa, E.; Lopes, A.; Poortman-van Der Meer, A.; Cole, M.D.; Dahlén, J.; Sippola, E. Development of a harmonised method for the profiling of amphetamines: IV. Optimisation of sample preparation. Forensic Sci. Int. 2007, 169, 64–76. [Google Scholar] [CrossRef]
- Lock, E.; Aalberg, L.; Andersson, K.; Dahlén, J.; Cole, M.D.; Finnon, Y.; Huizer, H.; Jalava, K.; Kaa, E.; Lopes, A.; et al. Development of a Harmonised Method for the Profiling of Amphetamines V. Forensic Sci. Int. 2007, 169, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Metsalu, T.; Vilo, J. ClustVis: A Web Tool for Visualizing Clustering of Multivariate Data Using Principal Component Analysis and Heatmap. Nucleic Acids Res. 2015, 43, W566–W570. [Google Scholar] [CrossRef]
- Global Initiative Against Transnational Organized Crime. The Organized Crime Index—Netherlands; Global Initiative Against Transnational Organized Crime: Geneva, Switzerland, 2023. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Carrier gas type and flow | helium, 1 mL/min |
| Volume of sample injected | 2 μL |
| Injector type | 1:10 split mode |
| Injector temperature | 250 °C |
| Column temperature program | 0.0–0.2 min–75 °C (0 °C/min) |
| 0.2–0.5 min–90 °C (10 °C/min) | |
| 0.5–1.0 min–325 °C (25 °C/min) | |
| Transfer line temperature | 310 °C |
| Ion source temperature | 230 °C |
| Electron bombardment energy | 70 eV |
| Mass scanning range | 50–550 amu |
| The average concentration of amphetamine in the analyzed samples | 7.6% |
| The median concentration of amphetamine in the analyzed samples | 6.2% |
| Samples with a dominant amount of amphetamine | 84.4% |
| The total mass of the analyzed evidence material containing amphetamine [g] | 8113 |
| The estimated total market value of the analyzed evidence [EUR] | 81,132 |
| Peak Number | tR [min] | Retention Indices | m/z for Four Most Intense Signals in Mass Spectrum | Proposed Identity | Score [%] |
|---|---|---|---|---|---|
| 1 | 1.8 | 1042 | 135, 91, 65, 58 | N,N-dimethylbenzylamine, ISTD | 90 |
| 2 | 2.4 | 1128 | 134, 92, 91, 65 | Benzyl methyl ketone, BMK | 59 |
| 3 | 2.5 | 1171 | 120, 91, 65, 51 | Amphetamine, AMF | 75 |
| 4 | 4.7 | 1468 | 170, 169, 115, 102 | 4-methyl-5-phenyl pyrimidine | 60 |
| 5 | 4.8 | 1415 | 118, 91, 72, 51 | N-formyl amphetamine | 62 |
| 6 | 4.8 | 1454 | 170, 169, 91, 72 | 4-benzylpyrimidine | 68 |
| 7 | 5.4 | 1663 | 91, 71, 56, 42 | 4-methylphenmetrazine | 58 |
| 8 | 6.2 | 1896 | 132, 105, 91, 77 | N-(-phenylisopropyl)benzaldimine | 65 |
| 9 | 7.1 | 1795 | 194, 109, 82, 67 | Caffeine | 99 |
| 10 | 8.6 | - | 280, 221, 208, 194 | Unknown, UN | - |
| 11 | 8.8 | - | 281, 235, 194, 150 | Unknown, UN | - |
| Total Number of Packages | Number of Packages Taken for Testing |
|---|---|
| n < 10 | n |
| 10 < n < 100 | 10 |
| n > 100 | √n |
| Time | Curve’s Equation | R2 | CAMF [μg/mL] | STD [μg/mL] | CV [%] |
|---|---|---|---|---|---|
| 11 May 2020 | y = 0.0115x + 0.0253 | 0.9994 | 5 | 0.0026 | 3.3 |
| 10 | 0.0038 | 2.9 | |||
| 50 | 0.0108 | 1.8 | |||
| 100 | 0.0268 | 2.2 | |||
| 200 | 0.0281 | 1.2 | |||
| 1 June 2020 | y = 0.0119x − 0.0214 | 0.9950 | 5 | 0.0007 | 1.6 |
| 10 | 0.0023 | 2.7 | |||
| 50 | 0.0005 | 0.1 | |||
| 100 | 0.0118 | 1.0 | |||
| 200 | 0.0311 | 1.2 | |||
| 7 July 2020 | y = 0.0166x + 0.0022 | 0.9954 | 5 | 0.0005 | 0.8 |
| 10 | 0.0023 | 1.7 | |||
| 50 | 0.0031 | 0.4 | |||
| 100 | 0.0087 | 0.5 | |||
| 200 | 0.1404 | 4.3 | |||
| 30 July 2020 | y = 0.0115x + 0.0154 | 0.9983 | 5 | 0.0110 | 15 |
| 10 | 0.0007 | 0.6 | |||
| 50 | 0.0191 | 3.1 | |||
| 100 | 0.0687 | 5.9 | |||
| 200 | 0.0092 | 0.4 | |||
| 15 September 2020 | y = 0.0132x − 0.0379 | 0.9967 | 10 | 0.0025 | 2.7 |
| 25 | 0.0072 | 2.5 | |||
| 50 | 0.0073 | 1.2 | |||
| 100 | 0.0212 | 1.6 | |||
| 200 | 0.1385 | 5.3 | |||
| 1 October 2020 | y = 0.0123x + 0.0330 | 0.9960 | 10 | 0.0034 | 3.9 |
| 25 | 0.0011 | 0.4 | |||
| 50 | 0.0037 | 0.6 | |||
| 100 | 0.0364 | 3.0 | |||
| 200 | 0.0106 | 0.5 | |||
| 11 January 2021 | y = 0.0140x + 0.0732 | 0.9986 | 10 | 0.0013 | 0.7 |
| 25 | 0.0381 | 8.5 | |||
| 50 | 0.0270 | 3.3 | |||
| 100 | 0.0017 | 0.1 | |||
| 200 | 0.0004 | 0.1 | |||
| 3 March 2021 | y = 0.0103x − 0.0436 | 0.9940 | 10 | 0.0045 | 7.3 |
| 25 | 0.0053 | 2.6 | |||
| 50 | 0.0356 | 7.9 | |||
| 100 | 0.0302 | 3.0 | |||
| 200 | 0.0958 | 4.5 | |||
| 23 March 2021 | y = 0.0124x − 0.0707 | 0.9941 | 10 | 0.0042 | 7.2 |
| 25 | 0.0086 | 3.9 | |||
| 50 | 0.0223 | 4.2 | |||
| 100 | 0.0418 | 3.39 | |||
| 200 | 0.0886 | 3.55 |
| Weight [g] | Amphetamine Concentration [%] | Mean Amphetamine Concentration [%] | SD | RSD [%] |
|---|---|---|---|---|
| 0.2025 | 5.94 | 6.56 | 0.86 (0.50) | 13.09 (6.60) |
| 0.2044 | 6.88 | |||
| 0.2017 | 6.46 | |||
| 0.2017 | 6.88 | |||
| 0.2019 | 6.64 |
| Task | Frequency |
|---|---|
| Replacing the septa | Every week |
| Replacing the inlet liner | Every week |
| Cleaning of interface components | Every three months |
| Replacing the filament | Every three months |
| Replacing the chromatographic column | Every year After 3000 samples |
| Checking the sensitivity of the instrument by means of a QC sample | Before each sequence on the day of analysis |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Czyż, A.; Pawlak, K.; Waraksa, E.; Bieńkowski, T. Comprehensive Profiling of Illicit Amphetamines Seized in Poland: Insights from Gas Chromatography–Mass Spectrometry and Chemometric Analysis. Molecules 2025, 30, 579. https://doi.org/10.3390/molecules30030579
Czyż A, Pawlak K, Waraksa E, Bieńkowski T. Comprehensive Profiling of Illicit Amphetamines Seized in Poland: Insights from Gas Chromatography–Mass Spectrometry and Chemometric Analysis. Molecules. 2025; 30(3):579. https://doi.org/10.3390/molecules30030579
Chicago/Turabian StyleCzyż, Anna, Katarzyna Pawlak, Emilia Waraksa, and Tomasz Bieńkowski. 2025. "Comprehensive Profiling of Illicit Amphetamines Seized in Poland: Insights from Gas Chromatography–Mass Spectrometry and Chemometric Analysis" Molecules 30, no. 3: 579. https://doi.org/10.3390/molecules30030579
APA StyleCzyż, A., Pawlak, K., Waraksa, E., & Bieńkowski, T. (2025). Comprehensive Profiling of Illicit Amphetamines Seized in Poland: Insights from Gas Chromatography–Mass Spectrometry and Chemometric Analysis. Molecules, 30(3), 579. https://doi.org/10.3390/molecules30030579