
In Silico Investigation of Mineralocorticoid Receptor Antagonists: Insights into Binding Mechanisms and Structural Dynamics
 , ,
, ,  , and
, and
Abstract
1. Introduction
2. Results and Discussion
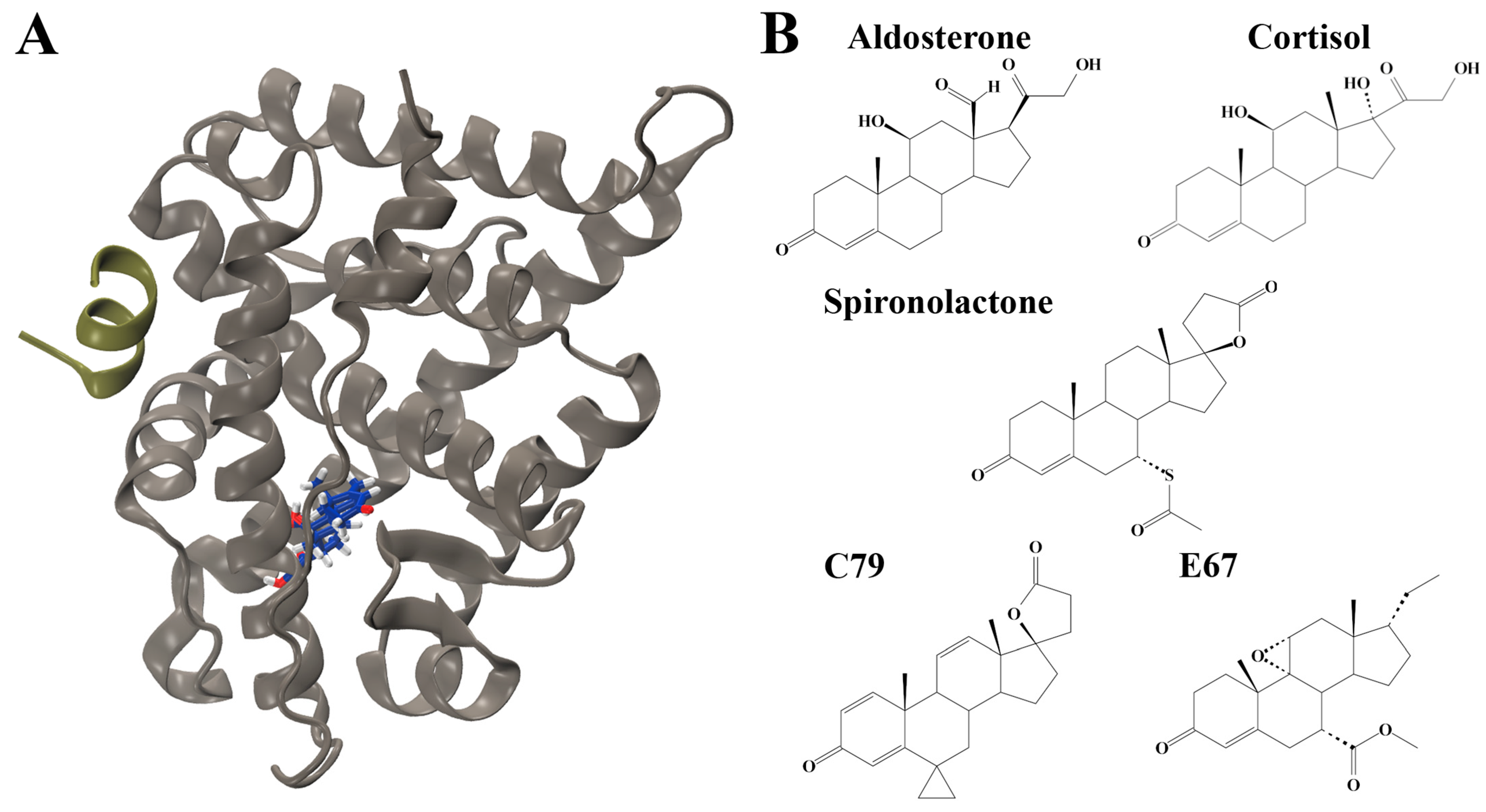
2.1. Molecular Docking to Identify Novel MR Antagonists
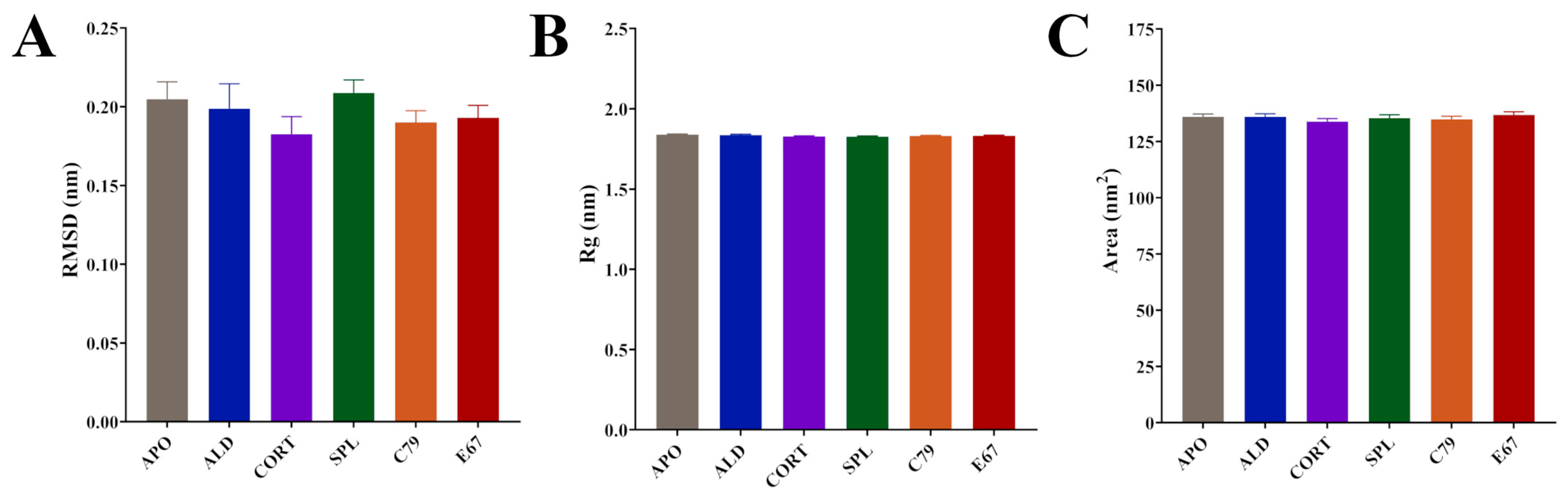
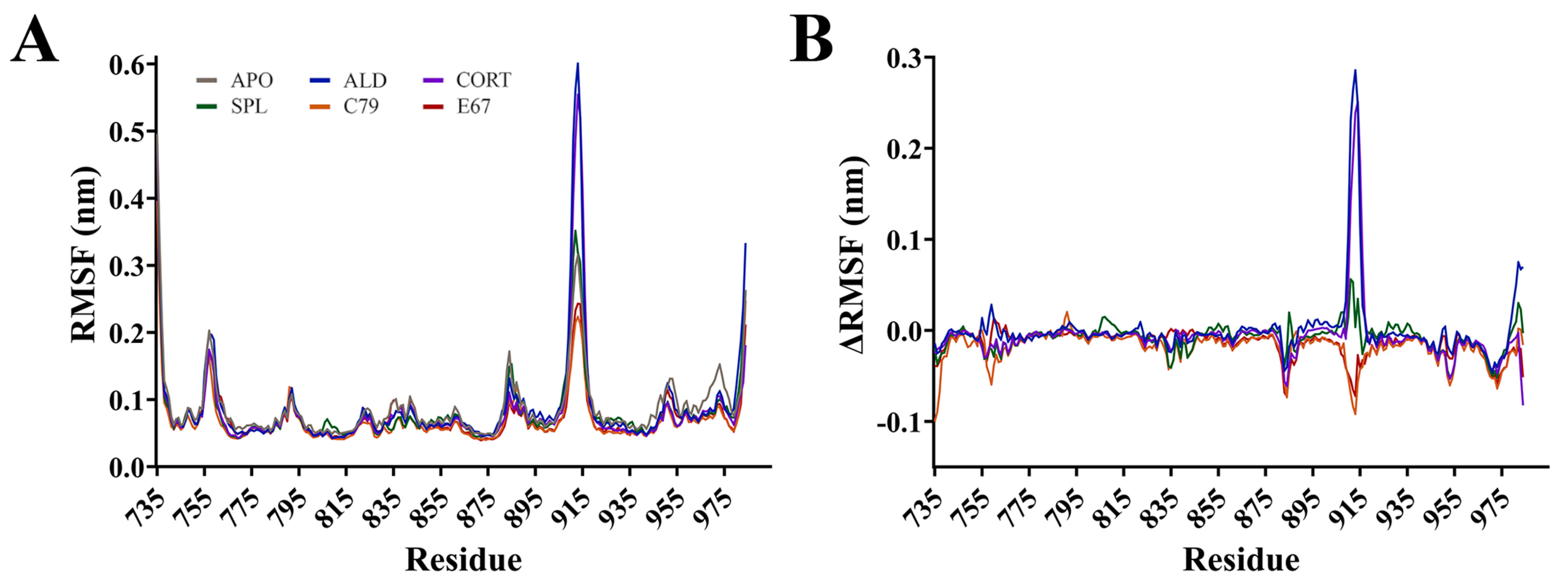
2.2. Protein Dynamics of the MR LBD
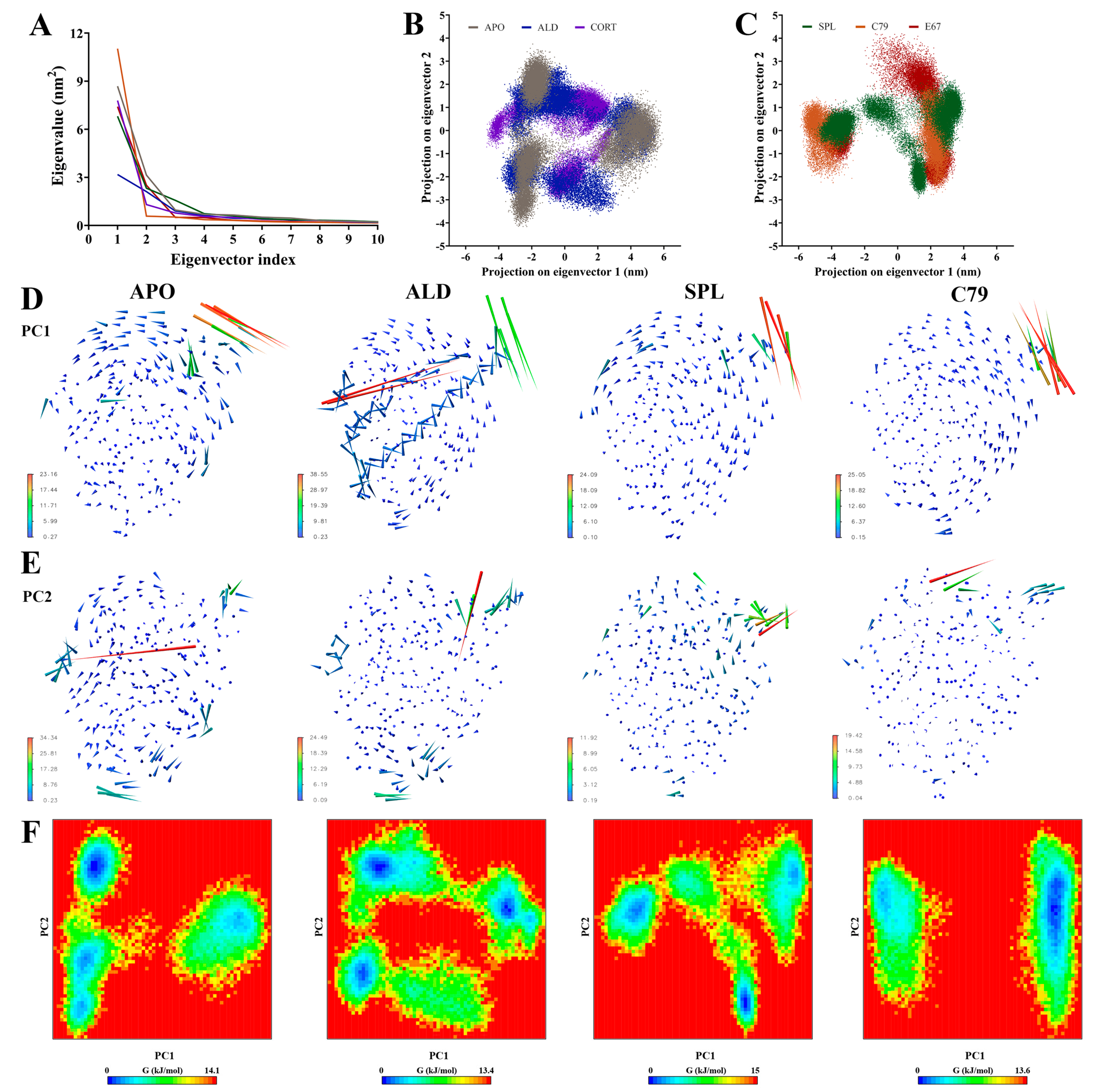
2.3. Principal Components Analysis of the MR LBD
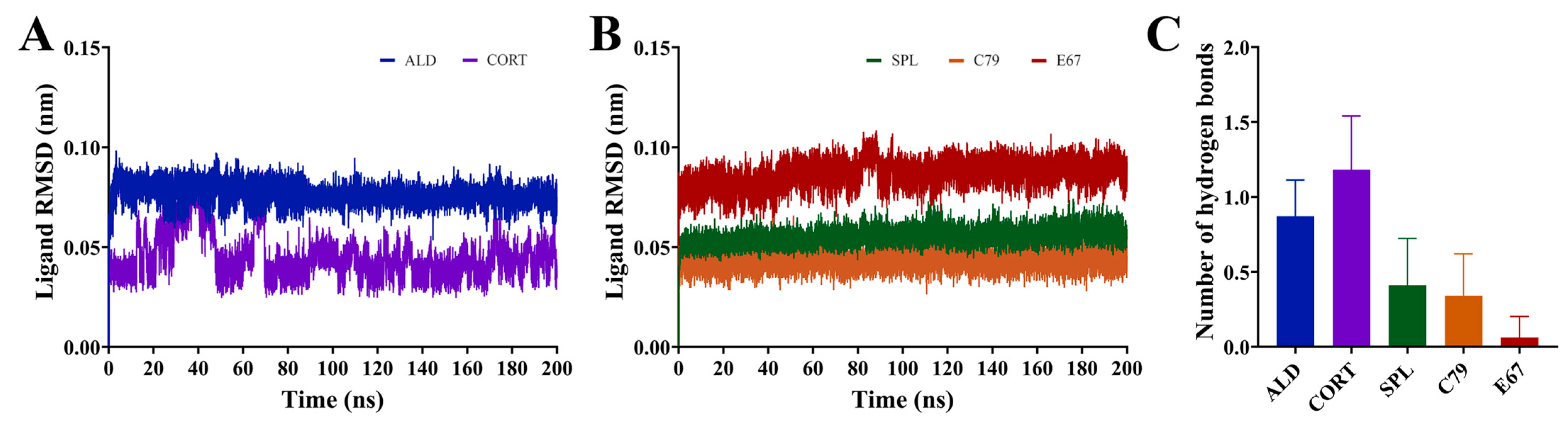
2.4. Dynamic Interactions of Ligands to the MR LBD
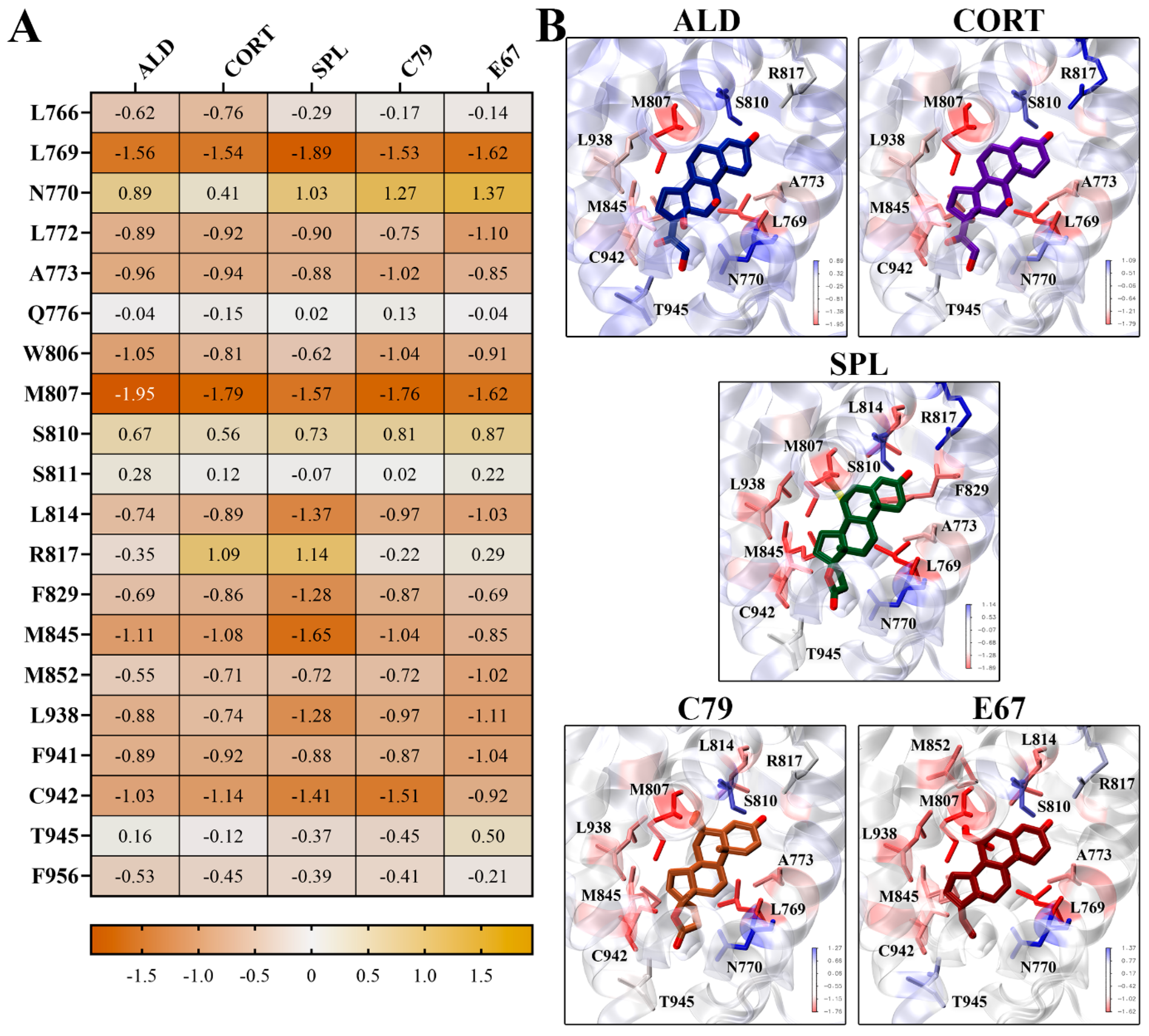
2.5. Binding Free Energy Calculations of Ligands to MR
3. Methods
3.1. Protein and Ligand Structure Preparation
3.2. Molecular Docking
3.3. Molecular Dynamics Simulations
3.4. Analysis of Simulation Trajectories
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Viengchareun, S.; Le Menuet, D.; Martinerie, L.; Munier, M.; Pascual-Le Tallec, L.; Lombès, M. The mineralocorticoid receptor: Insights into its molecular and (patho)physiological biology. Nucl. Recept. Signal 2007, 5, e012. [Google Scholar] [CrossRef] [PubMed]
- Fuller, P.J.; Young, M.J. Mechanisms of mineralocorticoid action. Hypertension 2005, 46, 1227–1235. [Google Scholar] [CrossRef]
- Hellal-Levy, C.; Fagart, J.; Souque, A.; Rafestin-Oblin, M.E. Mechanistic aspects of mineralocorticoid receptor activation. Kidney Int. 2000, 57, 1250–1255. [Google Scholar] [CrossRef] [PubMed]
- Fuller, P.J.; Yao, Y.; Yang, J.; Young, M.J. Mechanisms of ligand specificity of the mineralocorticoid receptor. J. Endocrinol. 2012, 213, 15–24. [Google Scholar] [CrossRef]
- Corvol, P.; Claire, M.; Oblin, M.E.; Geering, K.; Rossier, B. Mechanism of the antimineralocorticoid effects of spirolactones. Kidney Int. 1981, 20, 1–6. [Google Scholar] [CrossRef]
- Pitt, B.; Remme, W.; Zannad, F.; Neaton, J.; Martinez, F.; Roniker, B.; Bittman, R.; Hurley, S.; Kleiman, J.; Gatlin, M. Eplerenone, a selective aldosterone blocker, in patients with left ventricular dysfunction after myocardial infarction. N. Engl. J. Med. 2003, 348, 1309–1321. [Google Scholar] [CrossRef] [PubMed]
- Couette, B.; Fagart, J.; Jalaguier, S.; Lombes, M.; Souque, A.; Rafestin-Oblin, M.E. Ligand-induced conformational change in the human mineralocorticoid receptor occurs within its hetero-oligomeric structure. Biochem. J. 1996, 315 Pt 2, 421–427. [Google Scholar] [CrossRef]
- Lainscak, M.; Pelliccia, F.; Rosano, G.; Vitale, C.; Schiariti, M.; Greco, C.; Speziale, G.; Gaudio, C. Safety profile of mineralocorticoid receptor antagonists: Spironolactone and eplerenone. Int. J. Cardiol. 2015, 200, 25–29. [Google Scholar] [CrossRef]
- Koca, D.; Lother, A. Molecular pharmacology of mineralocorticoid receptor antagonists: The role of co-regulators. Steroids 2023, 199, 109291. [Google Scholar] [CrossRef]
- Pérez-Gordillo, F.L.; Serrano-Morillas, N.; Acosta-García, L.M.; Aranda, M.T.; Passeri, D.; Pellicciari, R.; Pérez de Vega, M.J.; González-Muñiz, R.; Alvarez de la Rosa, D.; Martín-Martínez, M. Novel 1,4-Dihydropyridine Derivatives as Mineralocorticoid Receptor Antagonists. Int. J. Mol. Sci. 2023, 24, 2439. [Google Scholar] [CrossRef]
- Nashev, L.G.; Vuorinen, A.; Praxmarer, L.; Chantong, B.; Cereghetti, D.; Winiger, R.; Schuster, D.; Odermatt, A. Virtual screening as a strategy for the identification of xenobiotics disrupting corticosteroid action. PLoS ONE 2012, 7, e46958. [Google Scholar] [CrossRef] [PubMed]
- Fuller, P.J.; Yao, Y.Z.; Jin, R.; He, S.; Martín-Fernández, B.; Young, M.J.; Smith, B.J. Molecular evolution of the switch for progesterone and spironolactone from mineralocorticoid receptor agonist to antagonist. Proc. Natl. Acad. Sci. USA 2019, 116, 18578–18583. [Google Scholar] [CrossRef] [PubMed]
- Edman, K.; Hosseini, A.; Bjursell, M.K.; Aagaard, A.; Wissler, L.; Gunnarsson, A.; Kaminski, T.; Köhler, C.; Bäckström, S.; Jensen, T.J.; et al. Ligand Binding Mechanism in Steroid Receptors: From Conserved Plasticity to Differential Evolutionary Constraints. Structure 2015, 23, 2280–2290. [Google Scholar] [CrossRef] [PubMed]
- Bledsoe, R.K.; Madauss, K.P.; Holt, J.A.; Apolito, C.J.; Lambert, M.H.; Pearce, K.H.; Stanley, T.B.; Stewart, E.L.; Trump, R.P.; Willson, T.M.; et al. A ligand-mediated hydrogen bond network required for the activation of the mineralocorticoid receptor. J. Biol. Chem. 2005, 280, 31283–31293. [Google Scholar] [CrossRef]
- Huyet, J.; Pinon, G.M.; Fay, M.R.; Rafestin-Oblin, M.E.; Fagart, J. Structural determinants of ligand binding to the mineralocorticoid receptor. Mol. Cell Endocrinol. 2012, 350, 187–195. [Google Scholar] [CrossRef]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert. Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef]
- Bamberg, K.; Johansson, U.; Edman, K.; William-Olsson, L.; Myhre, S.; Gunnarsson, A.; Geschwindner, S.; Aagaard, A.; Björnson Granqvist, A.; Jaisser, F.; et al. Preclinical pharmacology of AZD9977: A novel mineralocorticoid receptor modulator separating organ protection from effects on electrolyte excretion. PLoS ONE 2018, 13, e0193380. [Google Scholar] [CrossRef]
- Eswar, N.; Webb, B.; Marti-Renom, M.A.; Madhusudhan, M.S.; Eramian, D.; Shen, M.Y.; Pieper, U.; Sali, A. Comparative protein structure modeling using Modeller. In Current Protocols in Bioinformatics; Wiley: New York, NY, USA, 2006; Chapter 5, Unit 5–6. [Google Scholar]
- Fiser, A.; Do, R.K.; Sali, A. Modeling of loops in protein structures. Protein Sci. 2000, 9, 1753–1773. [Google Scholar] [CrossRef]
- Schrödinger, L.L.C. The PyMOL Molecular Graphics System, version 2.0; PyMOL: New York, NY, USA, 2015.
- Laskowski, R.A.; Rullmannn, J.A.; MacArthur, M.W.; Kaptein, R.; Thornton, J.M. AQUA and PROCHECK-NMR: Programs for checking the quality of protein structures solved by NMR. J. Biomol. NMR 1996, 8, 477–486. [Google Scholar] [CrossRef]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B.; et al. PubChem 2023 update. Nucleic Acids Res. 2023, 51, D1373–D1380. [Google Scholar] [CrossRef]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An open chemical toolbox. J. Cheminfor. 2011, 3, 33. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; van der Spoel, D.; van Drunen, R. GROMACS: A message-passing parallel molecular dynamics implementation. Comput. Phys. Commun. 1995, 91, 43–56. [Google Scholar] [CrossRef]
- Best, R.B.; Zhu, X.; Shim, J.; Lopes, P.E.; Mittal, J.; Feig, M.; Mackerell, A.D., Jr. Optimization of the additive CHARMM all-atom protein force field targeting improved sampling of the backbone φ, ψ and side-chain χ1 and χ2 dihedral angles. J. Chem. Theory Comput. 2012, 8, 3257–3273. [Google Scholar] [CrossRef]
- Vanommeslaeghe, K.; Hatcher, E.; Acharya, C.; Kundu, S.; Zhong, S.; Shim, J.; Darian, E.; Guvench, O.; Lopes, P.; Vorobyov, I.; et al. CHARMM general force field: A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J. Comput. Chem. 2010, 31, 671–690. [Google Scholar] [CrossRef]
- Price, D.J.; Brooks, C.L., III. A modified TIP3P water potential for simulation with Ewald summation. J. Chem. Phys. 2004, 121, 10096–10103. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; Postma, J.P.M.; Gunsteren, W.F.v.; DiNola, A.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef]
- Parrinello, M.; Rahman, A. Crystal Structure and Pair Potentials: A Molecular-Dynamics Study. Phys. Rev. Lett. 1980, 45, 1196–1199. [Google Scholar] [CrossRef]
- Hess, B.; Bekker, H.; Berendsen, H.J.; Fraaije, J.G. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N·log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38, 27–38. [Google Scholar] [CrossRef] [PubMed]
- Kumari, R.; Kumar, R.; Lynn, A. g_mmpbsa—A GROMACS Tool for High-Throughput MM-PBSA Calculations. J. Chem. Inf. Model. 2014, 54, 1951–1962. [Google Scholar] [CrossRef]
- Robert, K.; Nathan, A.B.; McCammon, J.A. iAPBS: A programming interface to the adaptive Poisson–Boltzmann solver. Comput. Sci. Discov. 2012, 5, 015005. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Residue | ALD | CORT | SPL | C79 | E67 |
|---|---|---|---|---|---|
| S767 | 1 | 1 | |||
| N770 | 5 | 5 | 1 | 1 | |
| Q776 | 1 | 1 | 1 | 1 | 1 |
| S810 | 1 | 1 | 1 | 1 | 1 |
| S811 | 1 | ||||
| R817 | 1 | 1 | 2 | 1 | |
| A844 | 1 | ||||
| M845 | 1 | ||||
| T945 | 3 | 2 | 1 | 1 |
| Energy Terms | ALD | CORT | SPL | C79 | E67 |
|---|---|---|---|---|---|
| ΔEvdW | −48.1 ± 0.1 | −48.9 ± 0.8 | −56.3 ± 0.3 | −50.6 ± 0.8 | −48.3 ± 0.2 |
| ΔEelec | −10.4 ± 0.2 | −9.9 ± 0.5 | −6.9 ± 0.2 | −3.9 ± 0.2 | −5.4 ± 0.4 |
| ΔGpolar | 31.1 ± 0.2 | 33.7 ± 1.5 | 37.4 ± 0.9 | 27.1 ± 0.3 | 26.2 ± 2.7 |
| ΔGnonpolar | −4.5 ± 0.0 | −4.5 ± 0.0 | −5.1 ± 0.0 | −4.6 ± 0.0 | −4.9 ± 0.0 |
| ΔGbinding | −31.9 ± 0.3 | −29.7 ± 1.4 | −30.9 ± 1.2 | −31.9 ± 0.5 | −32.4 ± 2.4 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liang, J.J.; Cao, S.; Hung, A.; El-Osta, A.; Karagiannis, T.C.; Young, M.J. In Silico Investigation of Mineralocorticoid Receptor Antagonists: Insights into Binding Mechanisms and Structural Dynamics. Molecules 2025, 30, 1226. https://doi.org/10.3390/molecules30061226
Liang JJ, Cao S, Hung A, El-Osta A, Karagiannis TC, Young MJ. In Silico Investigation of Mineralocorticoid Receptor Antagonists: Insights into Binding Mechanisms and Structural Dynamics. Molecules. 2025; 30(6):1226. https://doi.org/10.3390/molecules30061226
Chicago/Turabian StyleLiang, Julia J., Sara Cao, Andrew Hung, Assam El-Osta, Tom C. Karagiannis, and Morag J. Young. 2025. "In Silico Investigation of Mineralocorticoid Receptor Antagonists: Insights into Binding Mechanisms and Structural Dynamics" Molecules 30, no. 6: 1226. https://doi.org/10.3390/molecules30061226
APA StyleLiang, J. J., Cao, S., Hung, A., El-Osta, A., Karagiannis, T. C., & Young, M. J. (2025). In Silico Investigation of Mineralocorticoid Receptor Antagonists: Insights into Binding Mechanisms and Structural Dynamics. Molecules, 30(6), 1226. https://doi.org/10.3390/molecules30061226