

Enhancing Unconditional Molecule Generation via Online Knowledge Distillation of Scaffolds

Abstract

1. Introduction
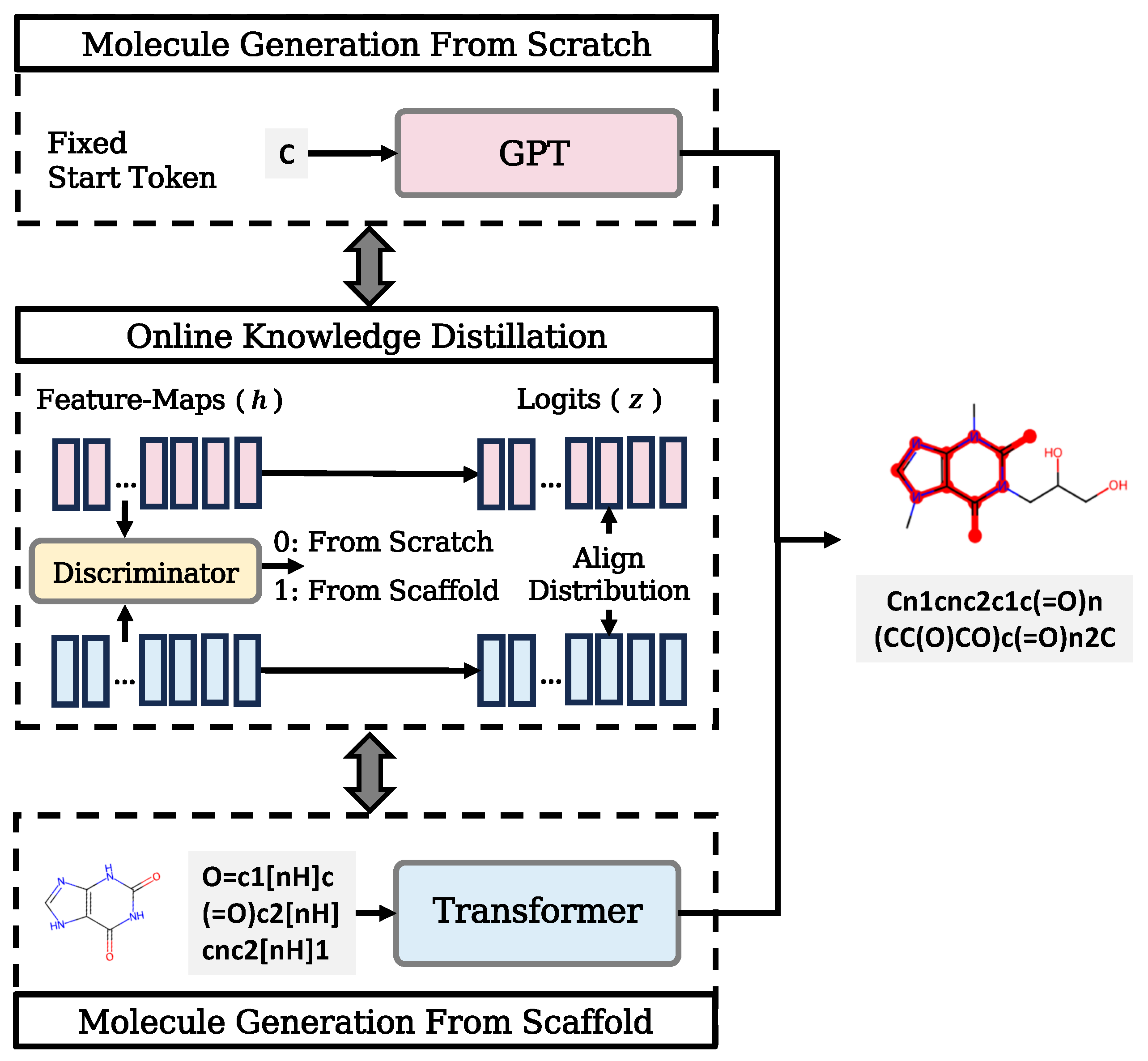
2. Methods
2.1. Online Knowledge Distillation Framework
2.2. Mutual Learning
2.3. Adversarial Distillation
2.4. Training
3. Experiments Configuration
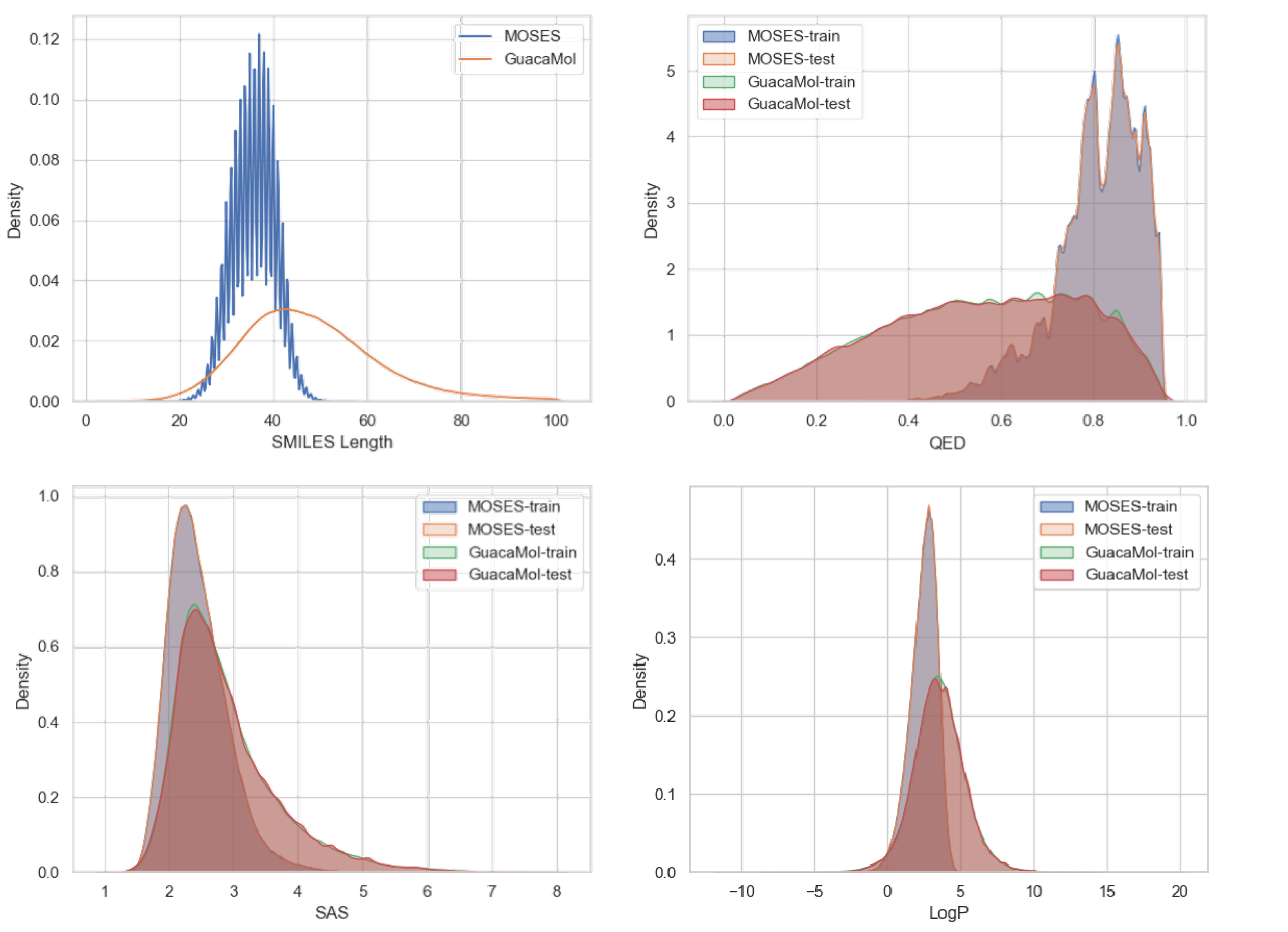
3.1. Datasets
3.2. Evaluation Metrics
- Validity: Validity refers to the proportion of valid molecules among those generated. A molecule is considered valid if it conforms to the rules of molecular structure, such as proper valence and absence of forbidden structures.
- Uniqueness: Uniqueness refers to the proportion of unique molecules among the valid molecules generated. High uniqueness indicates that the model frequently generates distinct molecules, suggesting that it has learned the variance in the distribution well [21].
- Novelty: Novelty refers to the proportion of valid molecules generated that do not appear in the training set. This metric evaluates the model’s ability to generate new molecules that are potentially useful but have not been explored previously.
- Internal Diversity (): This metric assesses the diversity among the molecules generated. It is important to determine whether the model has generated a variety of distinct molecular structures or just similar molecules. Internal diversity is typically calculated using a pairwise similarity measure, most commonly the Tanimoto similarity [30] based on molecular fingerprints. The corresponding formula is as follows:where the terms and represent the molecules in the generated set S. The term represents the power (p) mean of Tanimoto similarity (T). In this work, we report and .
- Scaffold Similarity (Scaff): This metric compares the distribution of Bemis–Murcko scaffolds [31] of molecules in the reference and generated sets. Denoting as the number of times a given scaffold s appears in a set of molecules A, and a set of scaffolds that appear in either the generated set G or the reference set R as S, the metric is defined as a cosine similarity:The purpose of this metric is to show how similar the scaffolds are that are present in the generated and reference datasets [21]. In this paper, we report Scaff/TestSF, where the scaffolds in the test set are selected as the reference set. The score of Scaff/TestSF reveals the model’s capability to discover novel scaffolds.
- Quantitative Estimation of Drug-likeness (QED): QED is a value in the range of [0, 1] that estimates the likelihood of a molecule being a viable candidate for a drug. A higher QED value indicates a greater likelihood of the molecule having drug-like properties.
- Synthetic Accessibility Score (SAS): The Synthetic Accessibility Score (SAS) is a heuristic measure that estimates the difficulty of synthesizing a given molecule. The score ranges from 1 (easy to synthesize) to 10 (hard to synthesize).
- LogP: The logarithm of the partition coefficient (LogP) measures the solubility of a solute between two immiscible solvents at equilibrium. This value is calculated using RDKit’s Crippen [32] estimation method. The ideal LogP value for a drug typically falls within the range of 1 to 5.
- pIC50: The IC50 value represents the molar concentration of an inhibitor (such as a drug) needed to inhibit 50% of a biological function or target, such as an enzyme, cell receptor, or microbe. pIC50 represents the negative logarithm of the IC50, and the high pIC50 value indicates that the tested molecule possesses better biological activity.
3.3. Implementation Settings
4. Results and Discussion
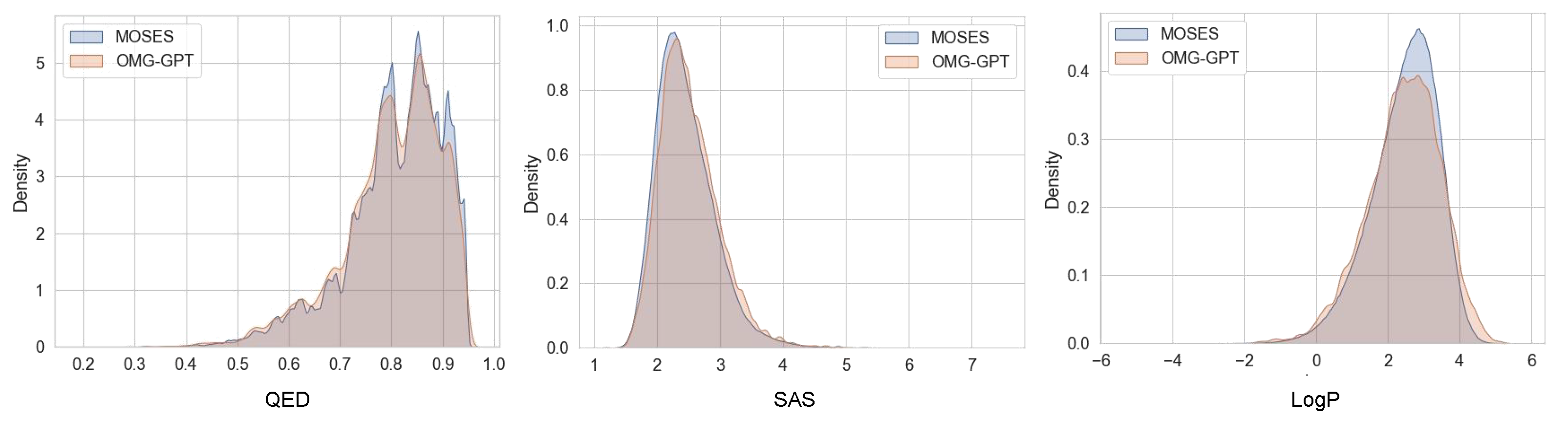
4.1. Performance Comparison on the MOSES Datasets
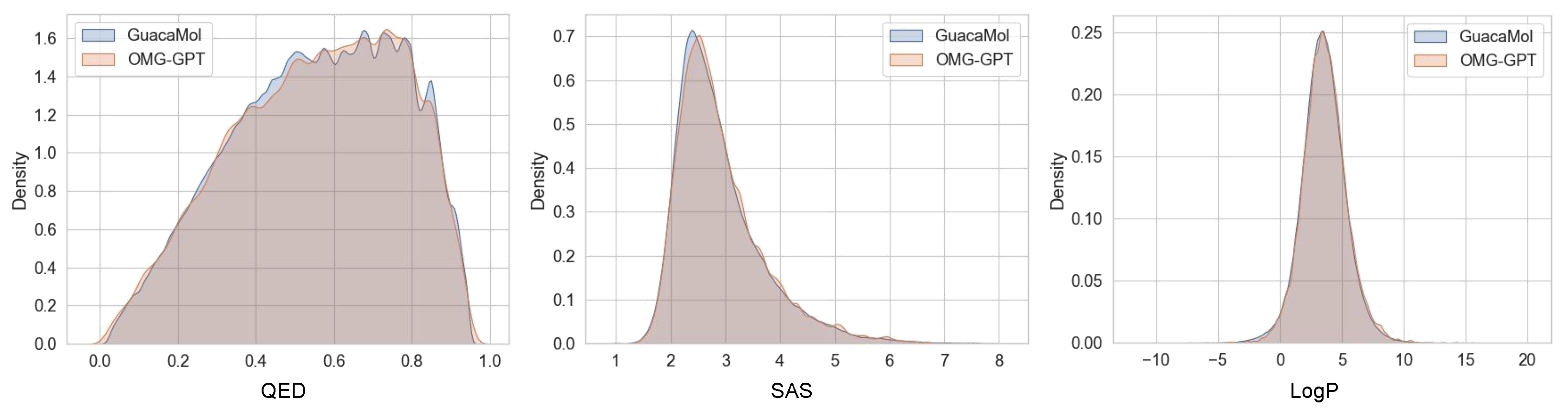
4.2. Performance Comparison on the Guacamol Datasets
4.3. Ablation Experiments
4.3.1. Effects of Individual Knowledge Distillation Modules
4.3.2. Effects of Different Knowledge Distillation Approaches
4.4. Property-Specific Evaluations of the Generated Molecules
4.4.1. Chemical Properties Distribution
4.4.2. Biological Activity Distribution
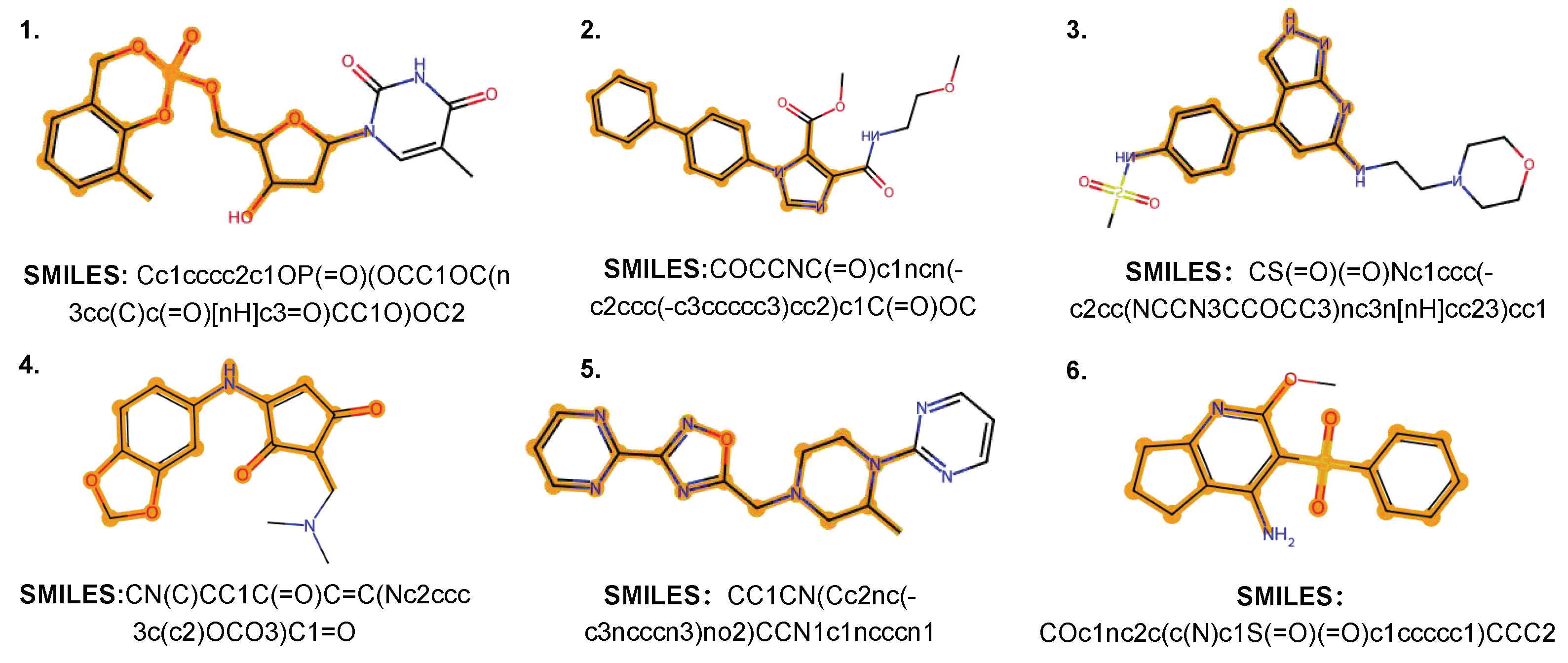
4.5. Case Study
4.6. Limitations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Polishchuk, P.G.; Madzhidov, T.I.; Varnek, A. Estimation of the size of drug-like chemical space based on GDB-17 data. J. Comput. Aided Mol. Des. 2013, 27, 675–679. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Thiessen, P.A.; Bolton, E.; Chen, J.; Fu, G.; Gindulyte, A.; Han, L.; He, J.; He, S.; Shoemaker, B.A.; et al. PubChem Substance and Compound databases. Nucleic Acids Res. 2016, 44, 1202–1213. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Kearnes, S.; Li, L.; Zare, R.N.; Riley, P. Optimization of Molecules via Deep Reinforcement Learning. arXiv 2018, arXiv:1810.08678. [Google Scholar] [CrossRef] [PubMed]
- Mazuz, E.; Shtar, G.; Shapira, B.; Rokach, L. Molecule generation using transformers and policy gradient reinforcement learning. Sci. Rep. 2023, 13, 8799. [Google Scholar] [CrossRef]
- Wang, H.; Fu, T.; Du, Y.; Gao, W.; Huang, K.; Liu, Z.; Chandak, P.; Liu, S.; Van Katwyk, P.; Deac, A.; et al. Scientific discovery in the age of artificial intelligence. Nature 2023, 620, 47–60. [Google Scholar] [CrossRef]
- Schneider, G. De Novo Molecular Design; John Wiley & Sons: Hoboken, NJ, USA, 2013. [Google Scholar]
- Yang, N.; Wu, H.; Yan, J.; Pan, X.; Yuan, Y.; Song, L. Molecule Generation for Drug Design: A Graph Learning Perspective. arXiv 2022, arXiv:2202.09212. [Google Scholar] [CrossRef]
- Kingma, D.P.; Welling, M. Auto-Encoding Variational Bayes. In Proceedings of the 2nd International Conference on Learning Representations, ICLR 2014, Banff, AB, Canada, 14–16 April 2014. [Google Scholar]
- Lippe, P.; Gavves, E. Categorical Normalizing Flows via Continuous Transformations. In Proceedings of the International Conference on Learning Representations, Vienna, Austria, 4 May 2021. [Google Scholar]
- Vignac, C.; Krawczuk, I.; Siraudin, A.; Wang, B.; Cevher, V.; Frossard, P. DiGress: Discrete Denoising diffusion for graph generation. In Proceedings of the Eleventh International Conference on Learning Representations, ICLR 2023, Kigali, Rwanda, 1–5 May 2023. [Google Scholar]
- Schwaller, P.; Gaudin, T.; Lányi, D.; Bekas, C.; Laino, T. “Found in Translation”: Predicting outcomes of complex organic chemistry reactions using neural sequence-to-sequence models. Chem. Sci. 2018, 9, 6091–6098. [Google Scholar] [CrossRef]
- Makhzani, A.; Shlens, J.; Jaitly, N.; Goodfellow, I.J. Adversarial Autoencoders. arXiv 2015, arXiv:1511.05644. [Google Scholar]
- Guimaraes, G.L.; Sánchez-Lengeling, B.; Farias, P.L.C.; Aspuru-Guzik, A. Objective-Reinforced Generative Adversarial Networks (ORGAN) for Sequence Generation Models. arXiv 2017, arXiv:1705.10843. [Google Scholar]
- Zaremba, W.; Sutskever, I.; Vinyals, O. Recurrent Neural Network Regularization. arXiv 2014, arXiv:1409.2329. [Google Scholar]
- Radford, A.; Narasimhan, K.; Salimans, T.; Sutskever, I. Improving Language Understanding by Generative Pre-Training. 2018. Available online: https://www.mikecaptain.com/resources/pdf/GPT-1.pdf (accessed on 7 March 2025).
- Liao, C.; Yu, Y.; Mei, Y.; Wei, Y. From words to molecules: A survey of large language models in chemistry. arXiv 2024, arXiv:2402.01439. [Google Scholar]
- Bagal, V.; Aggarwal, R.; Vinod, P.K.; Priyakumar, U.D. MolGPT: Molecular Generation Using a Transformer-Decoder Model. J. Chem. Inf. Model. 2022, 62, 2064–2076. [Google Scholar] [CrossRef] [PubMed]
- Jhoti, H.; Leach, A.R.; Zhang, K.Y.; Milburn, M.V.; Artis, D.R. Scaffold-based drug discovery. In Structure-Based Drug Discovery; Springer: Dordrecht, The Netherlands, 2007; pp. 129–153. [Google Scholar]
- Arús-Pous, J.; Patronov, A.; Bjerrum, E.J.; Tyrchan, C.; Reymond, J.L.; Chen, H.; Engkvist, O. SMILES-based deep generative scaffold decorator for de-novo drug design. J. Cheminform. 2020, 12, 38. [Google Scholar] [CrossRef] [PubMed]
- Liao, Z.; Xie, L.; Mamitsuka, H.; Zhu, S. Sc2Mol: A scaffold-based two-step molecule generator with variational autoencoder and transformer. Bioinformatics 2023, 39, btac814. [Google Scholar] [CrossRef] [PubMed]
- Polykovskiy, D.; Zhebrak, A.; Sánchez-Lengeling, B.; Golovanov, S.; Tatanov, O.; Belyaev, S.; Kurbanov, R.; Artamonov, A.; Aladinskiy, V.; Veselov, M.; et al. Molecular Sets (MOSES): A Benchmarking Platform for Molecular Generation Models. arXiv 2018, arXiv:1811.12823. [Google Scholar] [CrossRef]
- Brown, N.; Fiscato, M.; Segler, M.H.S.; Vaucher, A.C. GuacaMol: Benchmarking Models for de Novo Molecular Design. J. Chem. Inf. Model. 2019, 59, 1096–1108. [Google Scholar] [CrossRef]
- Zhang, Y.; Xiang, T.; Hospedales, T.M.; Lu, H. Deep Mutual Learning. In Proceedings of the 2018 IEEE Conference on Computer Vision and Pattern Recognition, CVPR 2018, Salt Lake City, UT, USA, 18–22 June 2018; Computer Vision Foundation/IEEE Computer Society: Washington, DC, USA, 2018; pp. 4320–4328. [Google Scholar]
- Chung, I.; Park, S.; Kim, J.; Kwak, N. Feature-map-level Online Adversarial Knowledge Distillation. In Proceedings of the 37th International Conference on Machine Learning, ICML 2020, Virtual Event, 13–18 July 2020; PMLR: 2020; Proceedings of Machine Learning Research. Volume 119, pp. 2006–2015. [Google Scholar]
- Graves, A.; Schmidhuber, J. Framewise phoneme classification with bidirectional LSTM and other neural network architectures. Neural Netw. 2005, 18, 602–610. [Google Scholar] [CrossRef]
- Li, H.; Lu, W. Mixed Cross Entropy Loss for Neural Machine Translation. In Proceedings of the 38th International Conference on Machine Learning, ICML 2021, Virtual Event, 18–24 July 2021; Meila, M., Zhang, T., Eds.; PMLR: 2021; Proceedings of Machine Learning Research. Volume 139, pp. 6425–6436. [Google Scholar]
- Irwin, J.J.; Shoichet, B.K. ZINC—A Free Database of Commercially Available Compounds for Virtual Screening. J. Chem. Inf. Model. 2005, 45, 177–182. [Google Scholar] [CrossRef]
- Gaulton, A.; Bellis, L.J.; Bento, A.P.; Chambers, J.; Davies, M.; Hersey, A.; Light, Y.; McGlinchey, S.; Michalovich, D.; Al-Lazikani, B.; et al. ChEMBL: A large-scale bioactivity database for drug discovery. Nucleic Acids Res. 2012, 40, 1100–1107. [Google Scholar] [CrossRef]
- Landrum, G. RDKit: A software suite for cheminformatics, computational chemistry, and predictive modeling. Greg Landrum 2013, 8, 5281. [Google Scholar]
- Bajusz, D.; Rácz, A.; Héberger, K. Why is Tanimoto index an appropriate choice for fingerprint-based similarity calculations? J. Cheminform. 2015, 7, 20:1–20:13. [Google Scholar] [CrossRef] [PubMed]
- Rogers, D.; Hahn, M. Extended-connectivity fingerprints. J. Chem. Inf. Model. 2010, 50, 742–754. [Google Scholar] [CrossRef] [PubMed]
- Wildman, S.A.; Crippen, G.M. Prediction of Physicochemical Parameters by Atomic Contributions. J. Chem. Inf. Comput. Sci. 1999, 39, 868–873. [Google Scholar] [CrossRef]
- Paszke, A.; Gross, S.; Massa, F.; Lerer, A.; Bradbury, J.; Chanan, G.; Killeen, T.; Lin, Z.; Gimelshein, N.; Antiga, L.; et al. Pytorch: An imperative style, high-performance deep learning library. Adv. Neural Inf. Process. Syst. 2019, 32, 8024–8035. [Google Scholar]
- Mercado, R.; Rastemo, T.; Lindelöf, E.; Klambauer, G.; Engkvist, O.; Chen, H.; Bjerrum, E.J. Graph networks for molecular design. Mach. Learn. Sci. Technol. 2021, 2, 025023. [Google Scholar] [CrossRef]
- Jin, W.; Barzilay, R.; Jaakkola, T.S. Junction Tree Variational Autoencoder for Molecular Graph Generation. arXiv 2018, arXiv:1802.04364. [Google Scholar]
- Kwon, Y.; Lee, D.; Choi, Y.S.; Shin, K.; Kang, S. Compressed graph representation for scalable molecular graph generation. J. Cheminform. 2020, 12, 1–8. [Google Scholar] [CrossRef]
- Segler, M.H.; Kogej, T.; Tyrchan, C.; Waller, M.P. Generating focused molecule libraries for drug discovery with recurrent neural networks. ACS Cent. Sci. 2018, 4, 120–131. [Google Scholar] [CrossRef]
- Prykhodko, O.; Johansson, S.; Kotsias, P.; Arús-Pous, J.; Bjerrum, E.J.; Engkvist, O.; Chen, H. A de novo molecular generation method using latent vector based generative adversarial network. J. Cheminform. 2019, 11, 74. [Google Scholar] [CrossRef]
- Hu, C.; Li, S.; Yang, C.; Chen, J.; Xiong, Y.; Fan, G.; Liu, H.; Hong, L. ScaffoldGVAE: Scaffold generation and hopping of drug molecules via a variational autoencoder based on multi-view graph neural networks. J. Cheminform. 2023, 15, 91. [Google Scholar] [CrossRef]
- Nguyen, T.; Le, H.; Quinn, T.P.; Nguyen, T.; Le, T.D.; Venkatesh, S. GraphDTA: Predicting drug–target binding affinity with graph neural networks. Bioinformatics 2021, 37, 1140–1147. [Google Scholar] [CrossRef] [PubMed]
- Pareto, V. Manuale di Economia Politica; Societa Editrice: Turin, Italy, 1906; Volume 13. [Google Scholar]
- Scott, O.B.; Edith Chan, A. ScaffoldGraph: An open-source library for the generation and analysis of molecular scaffold networks and scaffold trees. Bioinformatics 2020, 36, 3930–3931. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, W.L. The many roles of computation in drug discovery. Science 2004, 303, 1813–1818. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Luo, H.; Qin, R.; Wang, M.; Wan, X.; Fang, M.; Zhang, O.; Gou, Q.; Su, Q.; Shen, C.; et al. 3DSMILES-GPT: 3D molecular pocket-based generation with token-only large language model. Chem. Sci. 2025, 16, 637–648. [Google Scholar] [CrossRef]
- Li, S.; Liu, Z.; Luo, Y.; Wang, X.; He, X.; Kawaguchi, K.; Chua, T.S.; Tian, Q. Towards 3D Molecule-Text Interpretation in Language Models. In Proceedings of the Twelfth International Conference on Learning Representations, Vienna, Austria, 7–11 May 2024. [Google Scholar]
- Eckmann, P.; Sun, K.; Zhao, B.; Feng, M.; Gilson, M.K.; Yu, R. Limo: Latent inceptionism for targeted molecule generation. Proc. Mach. Learn. Res. 2022, 162, 5777. [Google Scholar]
- Fang, Y.; Zhang, N.; Chen, Z.; Guo, L.; Fan, X.; Chen, H. Domain-Agnostic Molecular Generation with Chemical Feedback. In Proceedings of the Twelfth International Conference on Learning Representations, Vienna, Austria, 7–11 May 2024. [Google Scholar]
- Acharya, A.; Yadav, M.; Nagpure, M.; Kumaresan, S.; Guchhait, S.K. Molecular medicinal insights into scaffold hopping-based drug discovery success. Drug Discov. Today 2024, 29, 103845. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Models | Validity | Unique | Novelty | Scaff/TestSF | ||
|---|---|---|---|---|---|---|
| Graph-Based | ||||||
| JT-VAE [35] | 1.0 | 0.999 | 0.914 | 0.855 | 0.849 | 0.101 |
| GraphINVENT [34] | 0.964 | 0.998 | - | 0.857 | 0.851 | 0.127 |
| Digress [10] | 0.857 | 1.0 | 0.936 | 0.855 | 0.849 | 0.118 |
| Sequence-Based | ||||||
| CharRNN [37] | 0.975 | 0.999 | 0.842 | 0.856 | 0.850 | 0.110 |
| AAE [12] | 0.936 | 1.0 | 0.793 | 0.855 | 0.850 | 0.079 |
| VAE [8] | 0.977 | 0.998 | 0.700 | 0.856 | 0.850 | 0.058 |
| LatentGAN [38] | 0.897 | 0.997 | 0.949 | 0.857 | 0.850 | 0.107 |
| MolGPT [17] | 0.988 | 1.0 | 0.821 | 0.855 | 0.849 | 0.085 |
| Sc2Mol [20] | 0.631 | 0.990 | 0.986 | 0.866 | 0.872 | 0.081 |
| OMG-GPT (ours) | 0.990 | 1.0 | 0.903 | 0.855 | 0.849 | 0.134 |
| Models | Validity | Unique | Novelty |
|---|---|---|---|
| Graph-Based | |||
| Digress [10] | 0.852 | 1.0 | 0.996 |
| NAGVAE [36] | 0.929 | 0.955 | 1.0 |
| Sequence-Based | |||
| SMILES LSTM [37] | 0.959 | 1.0 | 0.912 |
| VAE [8] | 0.870 | 0.999 | 0.974 |
| AAE [12] | 0.822 | 1.0 | 0.998 |
| ORGAN [13] | 0.379 | 0.841 | 0.687 |
| MolGPT [17] | 0.979 | 0.999 | 1.0 |
| OMG-GPT (ours) | 0.984 | 1.0 | 0.975 |
| Models | Validity | Unique | Novelty |
|---|---|---|---|
| OMG | 0.990 | 1.0 | 0.903 |
| OMG w/o ML | 0.988 | 1.0 | 0.766 |
| OMG w/o Ad | 0.990 | 1.0 | 0.895 |
| MolGPT | 0.988 | 1.0 | 0.821 |
| OMG-GPT (MF) | 0.983 | 1.0 | 0.893 |
| Dual-GPT | 0.956 | 1.0 | 0.961 |
| Models | Validity | Unique | Novelty |
|---|---|---|---|
| OMG-GPT | 0.990 | 1.0 | 0.903 |
| MolGPT | 0.988 | 1.0 | 0.821 |
| KD-GPT | 0.990 | 1.0 | 0.776 |
| OMG-SG | 0.978 | 1.0 | 0.917 |
| KD-SG | 0.996 | 1.0 | 0.772 |
| Source of Molecules | pIC50 (Mean/Std) | ||||
|---|---|---|---|---|---|
| CDK2 | EGFR | JAK1 | IRRK2 | PIM1 | |
| MOSES | 5.35/0.62 | 5.39/0.28 | 5.22/0.26 | 5.67/0.19 | 5.75/0.34 |
| 5.46/0.59 | 5.37/0.29 | 5.32/0.29 | 5.76/0.19 | 5.91/0.38 | |
| Guacamol | 5.65/0.60 | 5.62/0.40 | 5.34/0.49 | 5.88/0.28 | 6.18/0.64 |
| 5.67/0.64 | 5.60/0.39 | 5.34/0.49 | 5.89/0.28 | 6.23/0.65 | |
| ID Number | LogP | QED ↑ | SAS ↓ | pIC50 ↑ | ||||
|---|---|---|---|---|---|---|---|---|
| CDK2 | EGFR | JAK1 | IRRK2 | PIM1 | ||||
| 1 | 1.54 | 0.71 | 4.21 | 6.64 | 5.13 | 4.80 | 5.14 | 5.27 |
| 2 | 2.70 | 0.50 | 2.24 | 5.07 | 6.49 | 5.29 | 5.16 | 5.18 |
| 3 | 1.74 | 0.54 | 2.76 | 6.00 | 4.78 | 6.62 | 4.80 | 6.77 |
| 4 | 1.04 | 0.84 | 3.29 | 5.48 | 4.98 | 4.99 | 6.98 | 5.53 |
| 5 | 1.03 | 0.69 | 3.04 | 5.70 | 4.16 | 4.87 | 5.43 | 7.33 |
| 6 | 1.99 | 0.94 | 2.46 | 5.08 | 5.45 | 4.72 | 5.31 | 5.26 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, H.; Wang, Z.; Shi, M.; Cheng, Z.; Qian, Y. Enhancing Unconditional Molecule Generation via Online Knowledge Distillation of Scaffolds. Molecules 2025, 30, 1262. https://doi.org/10.3390/molecules30061262
Wang H, Wang Z, Shi M, Cheng Z, Qian Y. Enhancing Unconditional Molecule Generation via Online Knowledge Distillation of Scaffolds. Molecules. 2025; 30(6):1262. https://doi.org/10.3390/molecules30061262
Chicago/Turabian StyleWang, Huibin, Zehui Wang, Minghua Shi, Zixian Cheng, and Ying Qian. 2025. "Enhancing Unconditional Molecule Generation via Online Knowledge Distillation of Scaffolds" Molecules 30, no. 6: 1262. https://doi.org/10.3390/molecules30061262
APA StyleWang, H., Wang, Z., Shi, M., Cheng, Z., & Qian, Y. (2025). Enhancing Unconditional Molecule Generation via Online Knowledge Distillation of Scaffolds. Molecules, 30(6), 1262. https://doi.org/10.3390/molecules30061262