
Structural Insights into Salinosporamide a Mediated Inhibition of the Human 20S Proteasome


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
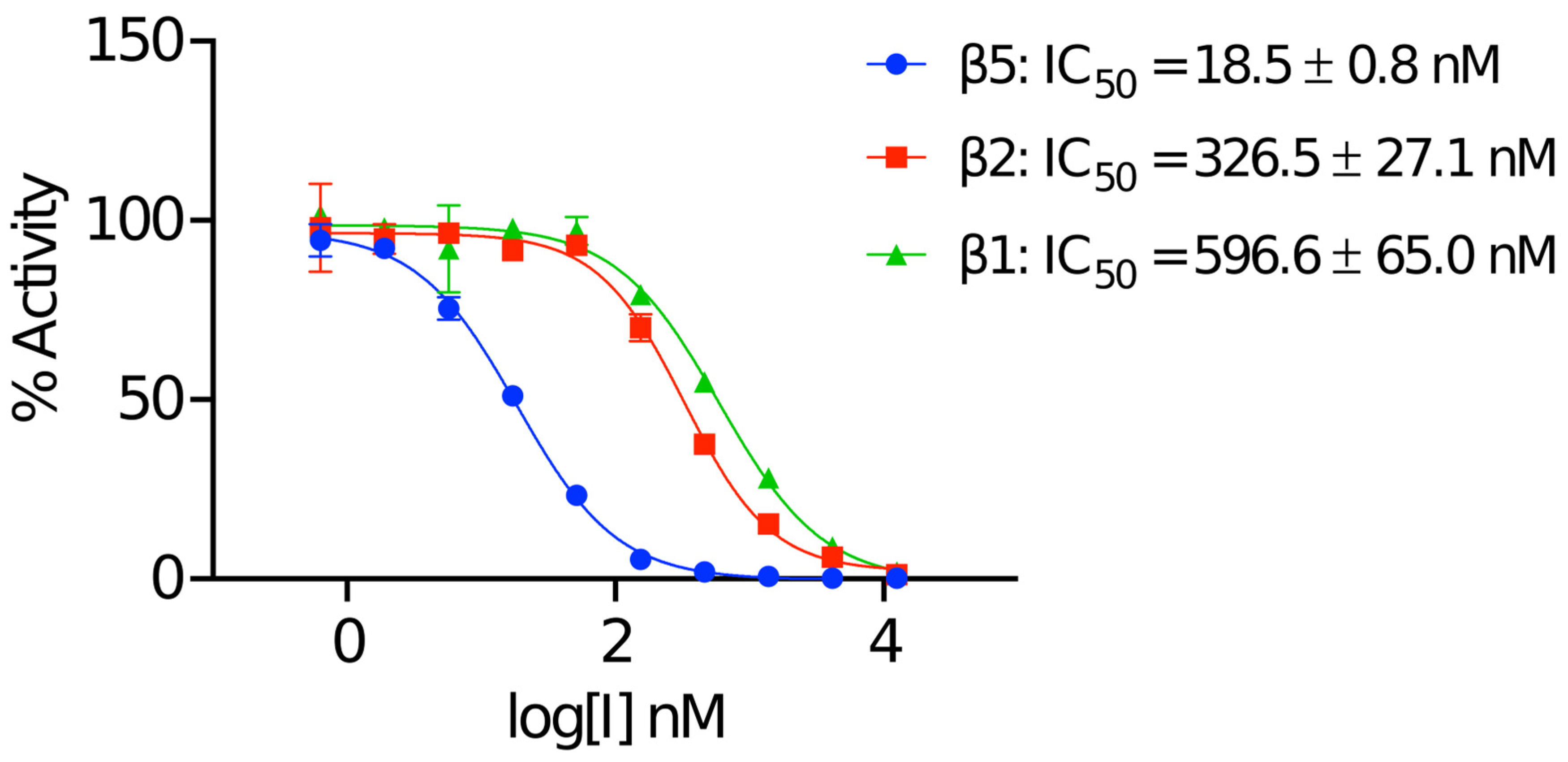
2.1. Biochemical Validation of the Human 20S Proteasome
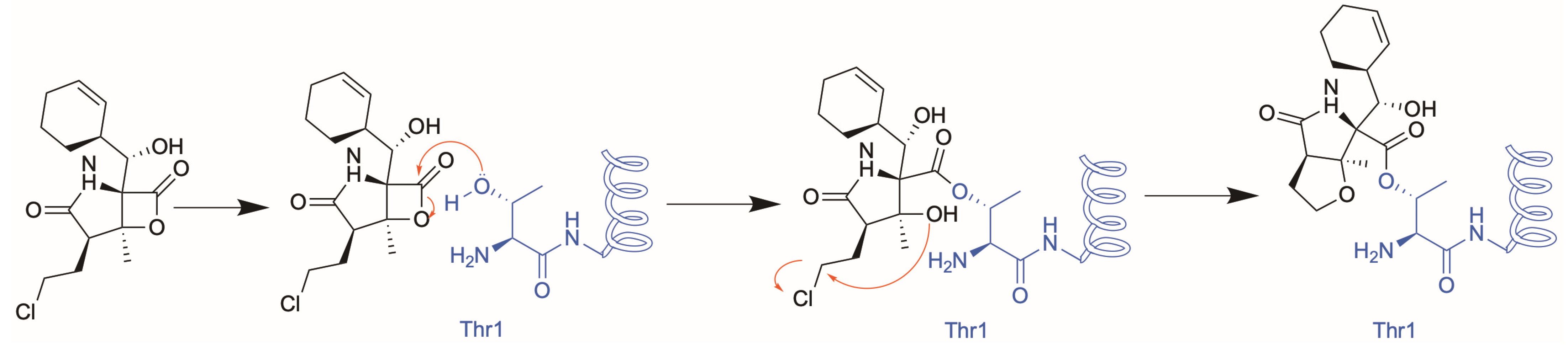
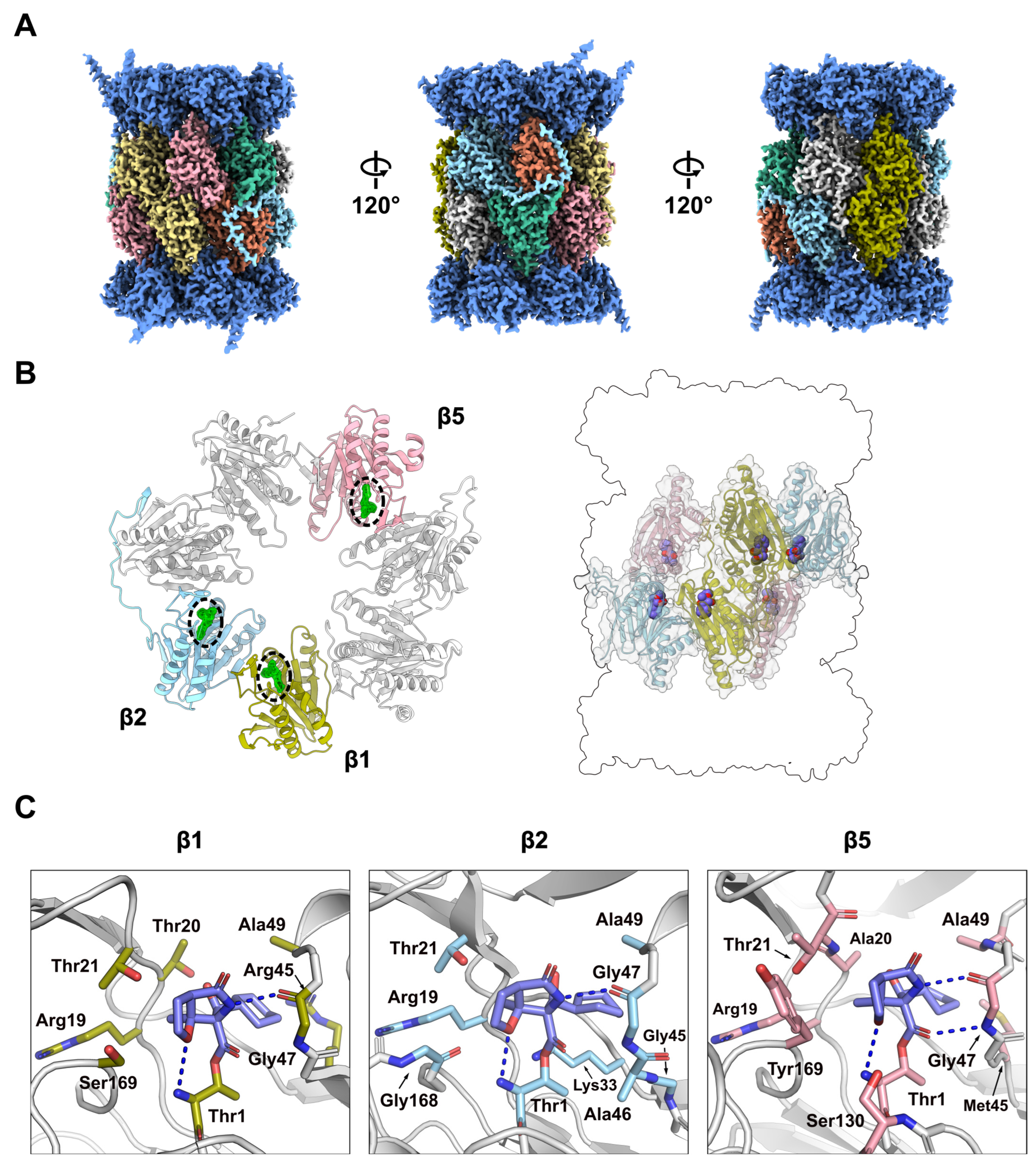
2.2. Structural Characterisation of the h20S Proteasome
2.3. Binding of MZB to Catalytic Subunits Β1, Β2 and Β5
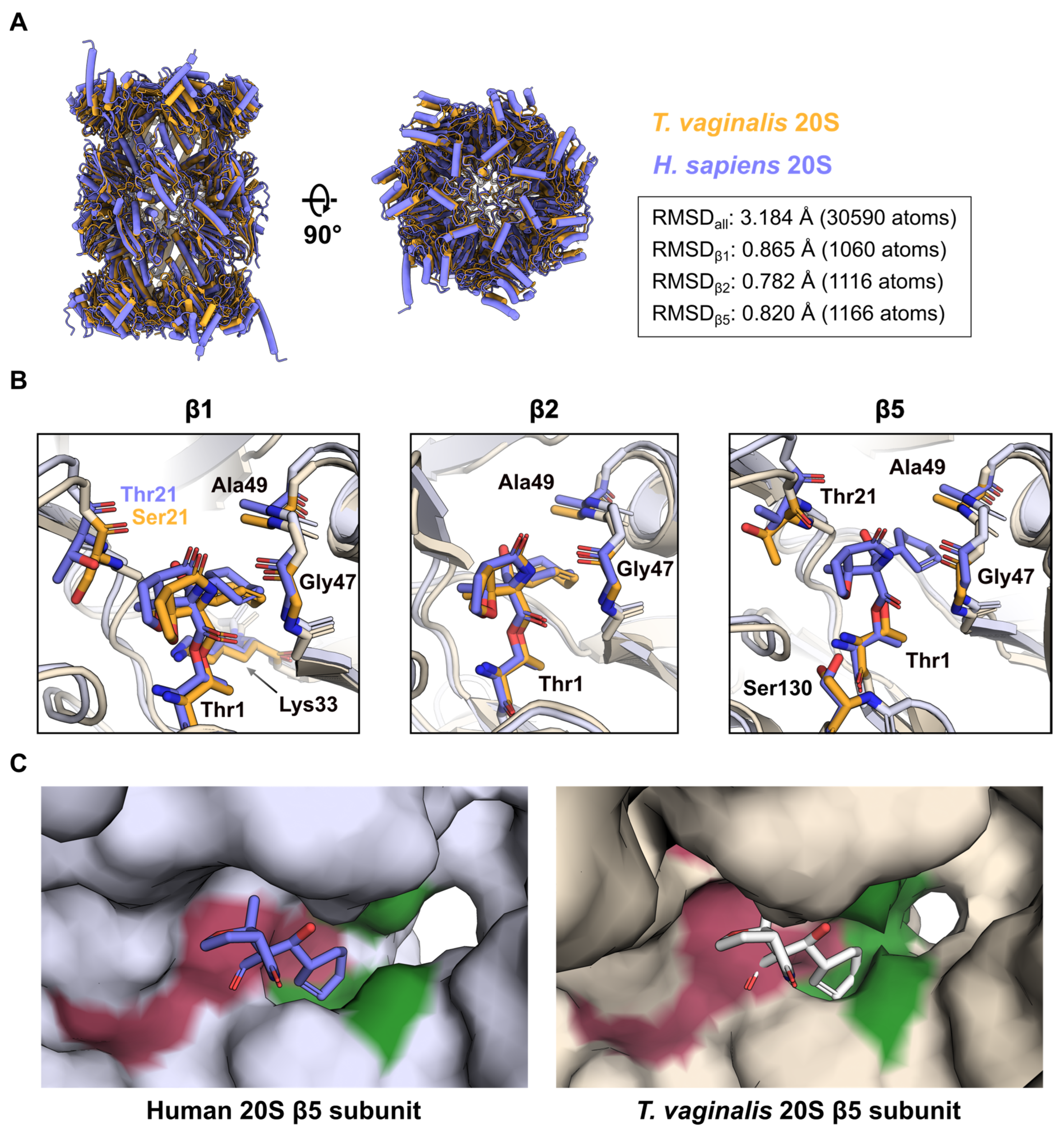
2.4. Comparison of MZB Binding to the Active Proteasome Sites of Human and T. vaginalis
3. Discussion
4. Materials and Methods
4.1. Half-Maximal Inhibitory Concentration (IC50) Assays
4.2. Preparation of Cryo-EM Grids and Data Acquisition
4.3. Cryo-EM Data Processing
4.4. Modelling and Refinement
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Feling, R.H.; Buchanan, G.O.; Mincer, T.J.; Kauffman, C.A.; Jensen, P.R.; Fenical, W. Salinosporamide A: A highly cytotoxic proteasome inhibitor from a novel microbial source, a marine bacterium of the new genus Salinospora. Angew. Chem. Int. Ed. 2003, 42, 355–357. [Google Scholar] [CrossRef]
- Mincer, T.J.; Jensen, P.R.; Kauffman, C.A.; Fenical, W. Widespread and Persistent Populations of a Major New Marine Actinomycete Taxon in Ocean Sediments. Appl. Environ. Microbiol. 2002, 68, 5005–5011. [Google Scholar] [CrossRef] [PubMed]
- Roth, P.; Gorlia, T.; Reijneveld, J.C.; de Vos, F.; Idbaih, A.; Frenel, J.-S.; Le Rhun, E.; Sepulveda, J.M.; Perry, J.; Masucci, G.L.; et al. Marizomib for patients with newly diagnosed glioblastoma: A randomized phase 3 trial. Neuro-Oncology 2024, 26, 1670–1682. [Google Scholar] [CrossRef]
- Di, K.; Lloyd, G.K.; Abraham, V.; MacLaren, A.; Burrows, F.J.; Desjardins, A.; Trikha, M.; Bota, D.A. Marizomib activity as a single agent in malignant gliomas: Ability to cross the blood-brain barrier. Neuro-Oncology 2015, 18, 840–848. [Google Scholar] [CrossRef]
- Manton, C.A.; Johnson, B.; Singh, M.; Bailey, C.P.; Bouchier-Hayes, L.; Chandra, J. Induction of cell death by the novel proteasome inhibitor marizomib in glioblastoma in vitro and in vivo. Sci. Rep. 2016, 6, 18953. [Google Scholar] [CrossRef]
- Manasanch, E.E.; Korde, N.; Zingone, A.; Tageja, N.; de Larrea, C.F.; Bhutani, M.; Wu, P.; Roschewski, M.; Landgren, O. The proteasome: Mechanisms of biology and markers of activity and response to treatment in multiple myeloma. Leuk. Lymphoma 2014, 55, 1707–1714. [Google Scholar] [CrossRef]
- Eisenberg-Lerner, A.; Benyair, R.; Hizkiahou, N.; Nudel, N.; Maor, R.; Kramer, M.P.; Shmueli, M.D.; Zigdon, I.; Lev, M.C.; Ulman, A.; et al. Golgi organization is regulated by proteasomal degradation. Nat. Commun. 2020, 11, 409. [Google Scholar] [CrossRef]
- Tanaka, K. The proteasome: Overview of structure and functions. Proc. Jpn. Acad Ser. B Phys. Biol. Sci. 2009, 85, 12–36. [Google Scholar] [CrossRef]
- Coux, O.; Tanaka, K.; Goldberg, A.L. STRUCTURE AND FUNCTIONS OF THE 20S AND 26S PROTEASOMES. Annu. Rev. Biochem. 1996, 65, 801–847. [Google Scholar] [CrossRef]
- Lupas, A.; Zwickl, P.; Wenzel, T.; Seemuller, E.; Baumeister, W. Structure and Function of the 20S Proteasome and of Its Regulatory Complexes. Cold Spring Harb. Symp. Quant. Biol. 1995, 60, 515–524. [Google Scholar] [CrossRef]
- Löwe, J.; Stock, D.; Jap, B.; Zwickl, P.; Baumeister, W.; Huber, R. Crystal Structure of the 20S Proteasome from the Archaeon T. acidophilum at 3.4 Å Resolution. Science 1995, 268, 533–539. [Google Scholar] [CrossRef] [PubMed]
- Orlowski, M.; Cardozo, C.; Michaud, C. Evidence for the presence of five distinct proteolytic components in the pituitary multicatalytic proteinase complex. Properties of two components cleaving bonds on the carboxyl side of branched chain and small neutral amino acids. Biochemistry 1993, 32, 1563–1572. [Google Scholar] [CrossRef]
- Mishra, R.; Upadhyay, A.; Prajapati, V.K.; Mishra, A. Proteasome-mediated proteostasis: Novel medicinal and pharmacological strategies for diseases. Med. Res. Rev. 2018, 38, 1916–1973. [Google Scholar] [CrossRef]
- Schmidt, M.; Finley, D. Regulation of proteasome activity in health and disease. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2014, 1843, 13–25. [Google Scholar] [CrossRef]
- Fenteany, G.; Standaert, R.F.; Lane, W.S.; Choi, S.; Corey, E.J.; Schreiber, S.L. Inhibition of Proteasome Activities and Subunit-Specific Amino-Terminal Threonine Modification by Lactacystin. Science 1995, 268, 726–731. [Google Scholar] [CrossRef]
- Groll, M.; Potts, B.C. Proteasome Structure, Function, and Lessons Learned from Beta-Lactone Inhibitors. Curr. Top. Med. Chem. 2011, 11, 2850–2878. [Google Scholar] [CrossRef]
- Macherla, V.R.; Mitchell, S.S.; Manam, R.R.; Reed, K.A.; Chao, T.-H.; Nicholson, B.; Deyanat-Yazdi, G.; Mai, B.; Jensen, P.R.; Fenical, W.F.; et al. Structure–Activity Relationship Studies of Salinosporamide A (NPI-0052), a Novel Marine Derived Proteasome Inhibitor. J. Med. Chem. 2005, 48, 3684–3687. [Google Scholar] [CrossRef]
- Groll, M.; Huber, R.; Potts, B.C.M. Crystal Structures of Salinosporamide A (NPI-0052) and B (NPI-0047) in Complex with the 20S Proteasome Reveal Important Consequences of β-Lactone Ring Opening and a Mechanism for Irreversible Binding. J. Am. Chem. Soc. 2006, 128, 5136–5141. [Google Scholar] [CrossRef]
- Silhan, J.; Silhan, J.; Fajtova, P.; Fajtova, P.; Bartosova, J.; Bartosova, J.; Hurysz, B.M.; Hurysz, B.M.; Almaliti, J.; Almaliti, J.; et al. Structural elucidation of recombinant Trichomonas vaginalis 20S proteasome bound to covalent inhibitors. Nat. Commun. 2024, 15, 8621. [Google Scholar] [CrossRef]
- Prudhomme, J.; McDaniel, E.; Ponts, N.; Bertani, S.; Fenical, W.; Jensen, P.; Le Roch, K. Marine Actinomycetes: A New Source of Compounds against the Human Malaria Parasite. PLoS ONE 2008, 3, e2335. [Google Scholar] [CrossRef]
- Afonine, P.V.; Klaholz, B.P.; Moriarty, N.W.; Poon, B.K.; Sobolev, O.V.; Terwilliger, T.C.; Adams, P.D.; Urzhumtsev, A. New tools for the analysis and validation of cryo-EM maps and atomic models. Acta Crystallogr. Sect. D Struct. Biol. 2018, 74, 814–840. [Google Scholar] [CrossRef]
- He, J.; Li, T.; Huang, S.-Y. Improvement of cryo-EM maps by simultaneous local and non-local deep learning. Nat. Commun. 2023, 14, 3217. [Google Scholar] [CrossRef]
- Borissenko, L.; Groll, M. 20S Proteasome and Its Inhibitors: Crystallographic Knowledge for Drug Development. Chem. Rev. 2007, 107, 687–717. [Google Scholar] [CrossRef] [PubMed]
- Fajtova, P.; Hurysz, B.M.; Miyamoto, Y.; Serafim, M.S.M.; Jiang, Z.; Vazquez, J.M.; Trujillo, D.F.; Liu, L.J.; Somani, U.; Almaliti, J.; et al. Distinct substrate specificities of the three catalytic subunits of the Trichomonas vaginalis proteasome. Protein Sci. 2024, 33, e5225. [Google Scholar] [CrossRef]
- Ciechanover, A. The ubiquitin-proteasome proteolytic pathway. Cell 1994, 79, 13–21. [Google Scholar] [CrossRef]
- Chauhan, D.; Catley, L.; Li, G.; Podar, K.; Hideshima, T.; Velankar, M.; Mitsiades, C.; Mitsiades, N.; Yasui, H.; Letai, A.; et al. A novel orally active proteasome inhibitor induces apoptosis in multiple myeloma cells with mechanisms distinct from Bortezomib. Cancer Cell 2005, 8, 407–419. [Google Scholar] [CrossRef]
- Nunes, A.T.; Annunziata, C.M. Proteasome inhibitors: Structure and function. Semin. Oncol. 2017, 44, 377–380. [Google Scholar] [CrossRef]
- Park, J.E.; Miller, Z.; Jun, Y.; Lee, W.; Kim, K.B. Next-generation proteasome inhibitors for cancer therapy. Transl. Res. 2018, 198, 1–16. [Google Scholar] [CrossRef]
- Kegyes, D.; Gulei, D.; Drula, R.; Cenariu, D.; Tigu, B.; Dima, D.; Tanase, A.; Badelita, S.; Buzoianu, A.-D.; Ciurea, S.; et al. Proteasome inhibition in combination with immunotherapies: State-of-the-Art in multiple myeloma. Blood Rev. 2023, 61, 101100. [Google Scholar] [CrossRef]
- Raninga, P.V.; Lee, A.; Sinha, D.; Dong, L.-F.; Datta, K.K.; Lu, X.; Croft, P.K.-D.; Dutt, M.; Hill, M.; Pouliot, N.; et al. Marizomib suppresses triple-negative breast cancer via proteasome and oxidative phosphorylation inhibition. Theranostics 2020, 10, 5259–5275. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhang, S.; Lin, B.; Wang, Q.; Nie, X.; Shi, Y. Combined treatment of marizomib and cisplatin modulates cervical cancer growth and invasion and enhances antitumor potential in vitro and in vivo. Front. Oncol. 2022, 12, 974573. [Google Scholar] [CrossRef]
- Kaplan, G.S.; Torcun, C.C.; Grune, T.; Ozer, N.K.; Karademir, B. Proteasome inhibitors in cancer therapy: Treatment regimen and peripheral neuropathy as a side effect. Free Radic. Biol. Med. 2017, 103, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Robbertse, L.; Fajtová, P.; Šnebergerová, P.; Jalovecká, M.; Levytska, V.; da Silva, E.B.; Sharma, V.; Pachl, P.; Almaliti, J.; Al-Hindy, M.; et al. Evaluating Antimalarial Proteasome Inhibitors for Efficacy in Babesia Blood Stage Cultures. ACS Omega 2024, 9, 44989–44999. [Google Scholar] [CrossRef] [PubMed]
- Eadsforth, T.C.; Torrie, L.S.; Rowland, P.; Edgar, E.V.; MacLean, L.M.; Paterson, C.; Robinson, D.A.; Shepherd, S.M.; Thomas, J.; Thomas, M.G.; et al. Pharmacological and structural understanding of the Trypanosoma cruzi proteasome provides key insights for developing site-specific inhibitors. J. Biol. Chem. 2024, 301, 108049. [Google Scholar] [CrossRef]
- Miller, C.P.; Manton, C.A.; Hale, R.; DeBose, L.; Macherla, V.R.; Potts, B.C.; Palladino, M.A.; Chandra, J. Specific and prolonged proteasome inhibition dictates apoptosis induction by marizomib and its analogs. Chem. Interact. 2011, 194, 58–68. [Google Scholar] [CrossRef]
- Punjani, A.; Rubinstein, J.L.; Fleet, D.J.; A Brubaker, M. cryoSPARC: Algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 2017, 14, 290–296. [Google Scholar] [CrossRef]
- Sahu, I.; Mali, S.M.; Sulkshane, P.; Xu, C.; Rozenberg, A.; Morag, R.; Sahoo, M.P.; Singh, S.K.; Ding, Z.; Wang, Y.; et al. The 20S as a stand-alone proteasome in cells can degrade the ubiquitin tag. Nat. Commun. 2021, 12, 6173. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Meng, E.C.; Couch, G.S.; Croll, T.I.; Morris, J.H.; Ferrin, T.E. UCSF ChimeraX: Structure visualization for researchers, educators, and developers. Protein Sci. 2020, 30, 70–82. [Google Scholar] [CrossRef]
- Afonine, P.V.; Poon, B.K.; Read, R.J.; Sobolev, O.V.; Terwilliger, T.C.; Urzhumtsev, A.; Adams, P.D. Real-space refinement in PHENIX for cryo-EM and crystallography. Acta Crystallogr. Sect. D Struct. Biol. 2018, 74, 531–544. [Google Scholar] [CrossRef]
- Liebschner, D.; Afonine, P.V.; Baker, M.L.; Bunkóczi, G.; Chen, V.B.; Croll, T.I.; Hintze, B.; Hung, L.-W.; Jain, S.; McCoy, A.J.; et al. Macromolecular structure determination using X-rays, neutrons and electrons: Recent developments in Phenix. Acta Crystallogr. Sect. D Struct. Biol. 2019, 75, 861–877. [Google Scholar] [CrossRef]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and development of Coot. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 486–501. [Google Scholar] [CrossRef] [PubMed]
- Croll, T.I. ISOLDE: A physically realistic environment for model building into low-resolution electron-density maps. Acta Crystallogr. Sect. D Struct. Biol. 2018, 74, 519–530. [Google Scholar] [CrossRef] [PubMed]
- Lebedev, A.A.; Young, P.; Isupov, M.N.; Moroz, O.V.; Vagin, A.A.; Murshudov, G.N. JLigand: A graphical tool for the CCP4 template-restraint library. Acta Crystallogr. Sect. D Struct. Biol. 2012, 68, 431–440. [Google Scholar] [CrossRef] [PubMed]
- Williams, C.J.; Headd, J.J.; Moriarty, N.W.; Prisant, M.G.; Videau, L.L.; Deis, L.N.; Verma, V.; Keedy, D.A.; Hintze, B.J.; Chen, V.B.; et al. MolProbity: More and better reference data for improved all-atom structure validation. Protein Sci. 2018, 27, 293–315. [Google Scholar] [CrossRef]
- Laskowski, R.A.; Swindells, M.B. LigPlot+: Multiple ligand–protein interaction diagrams for drug discovery. J. Chem. Inf. Model. 2011, 51, 2778–2786. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sülzen, H.; Fajtova, P.; O’Donoghue, A.J.; Silhan, J.; Boura, E. Structural Insights into Salinosporamide a Mediated Inhibition of the Human 20S Proteasome. Molecules 2025, 30, 1386. https://doi.org/10.3390/molecules30061386
Sülzen H, Fajtova P, O’Donoghue AJ, Silhan J, Boura E. Structural Insights into Salinosporamide a Mediated Inhibition of the Human 20S Proteasome. Molecules. 2025; 30(6):1386. https://doi.org/10.3390/molecules30061386
Chicago/Turabian StyleSülzen, Hagen, Pavla Fajtova, Anthony J. O’Donoghue, Jan Silhan, and Evzen Boura. 2025. "Structural Insights into Salinosporamide a Mediated Inhibition of the Human 20S Proteasome" Molecules 30, no. 6: 1386. https://doi.org/10.3390/molecules30061386
APA StyleSülzen, H., Fajtova, P., O’Donoghue, A. J., Silhan, J., & Boura, E. (2025). Structural Insights into Salinosporamide a Mediated Inhibition of the Human 20S Proteasome. Molecules, 30(6), 1386. https://doi.org/10.3390/molecules30061386