
Studies on the Synthesis Process of Plant-Derived Ursodeoxycholic Acid Intermediates

Abstract
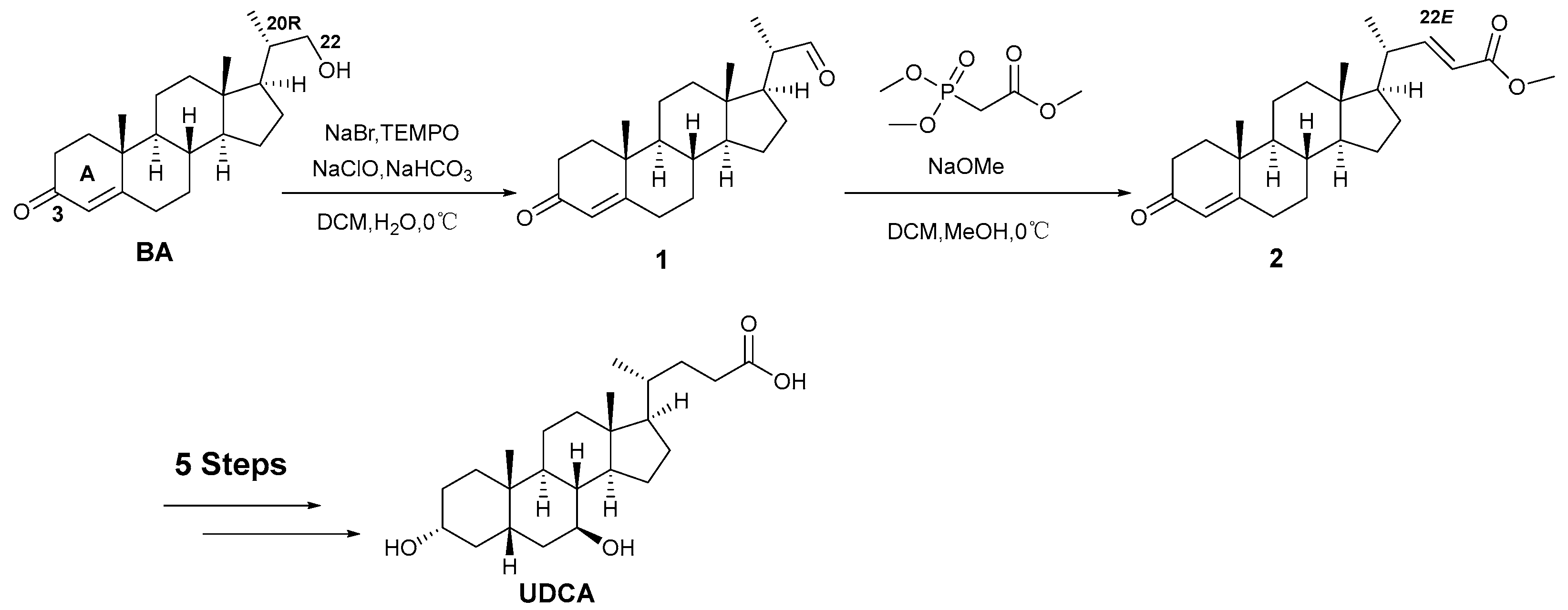
:1. Introduction
2. Results and Discussion
2.1. Study on the Process of the Hydroxyl Oxidation Reaction
2.1.1. Effect of NaClO Equivalent
2.1.2. Effect of Each Reagent in Hydroxyl Oxidation Reaction
2.1.3. Effect of Reaction Temperature
2.2. Study on the Process of the Horner–Wadsworth–Emmons Reaction
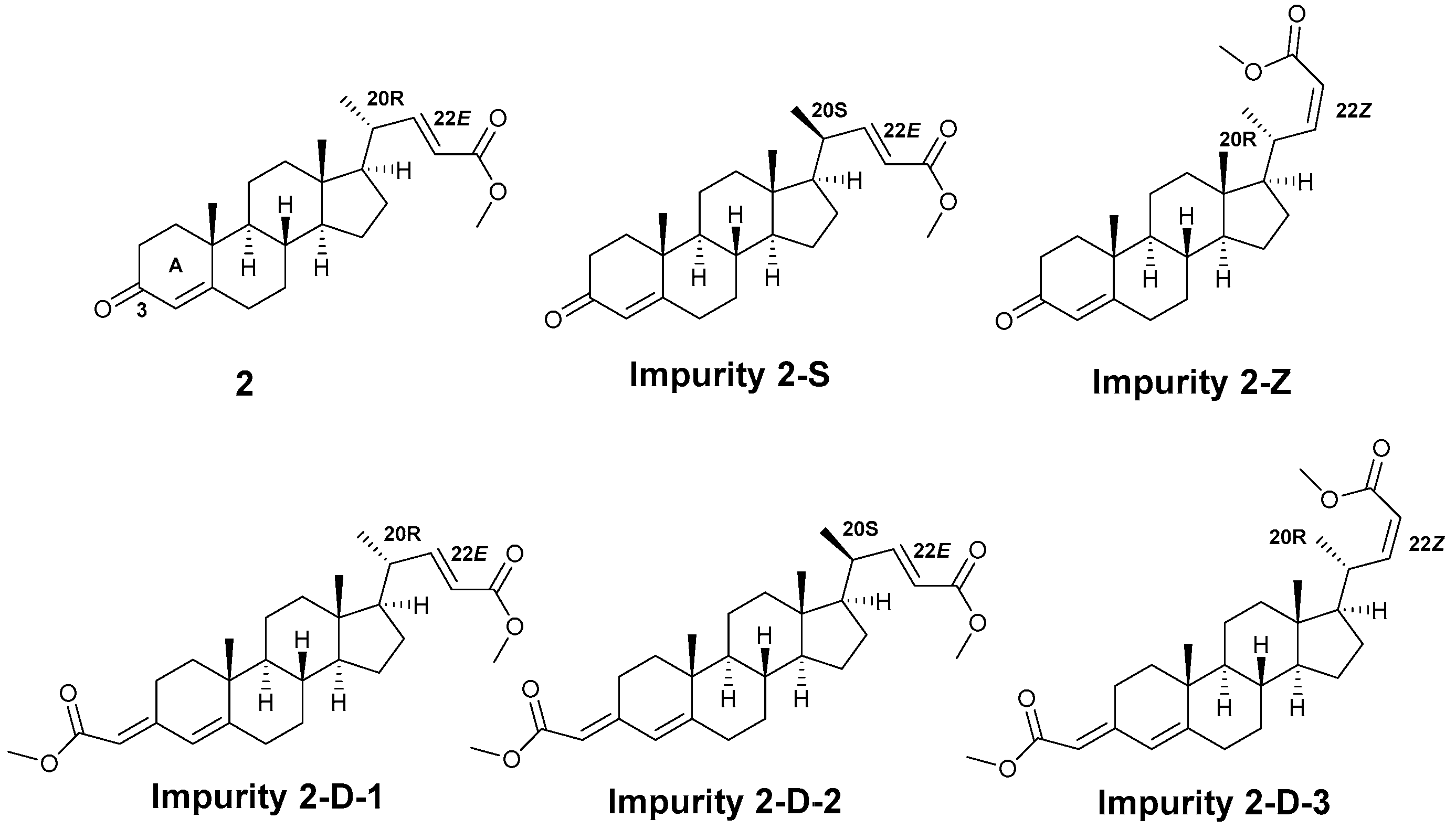
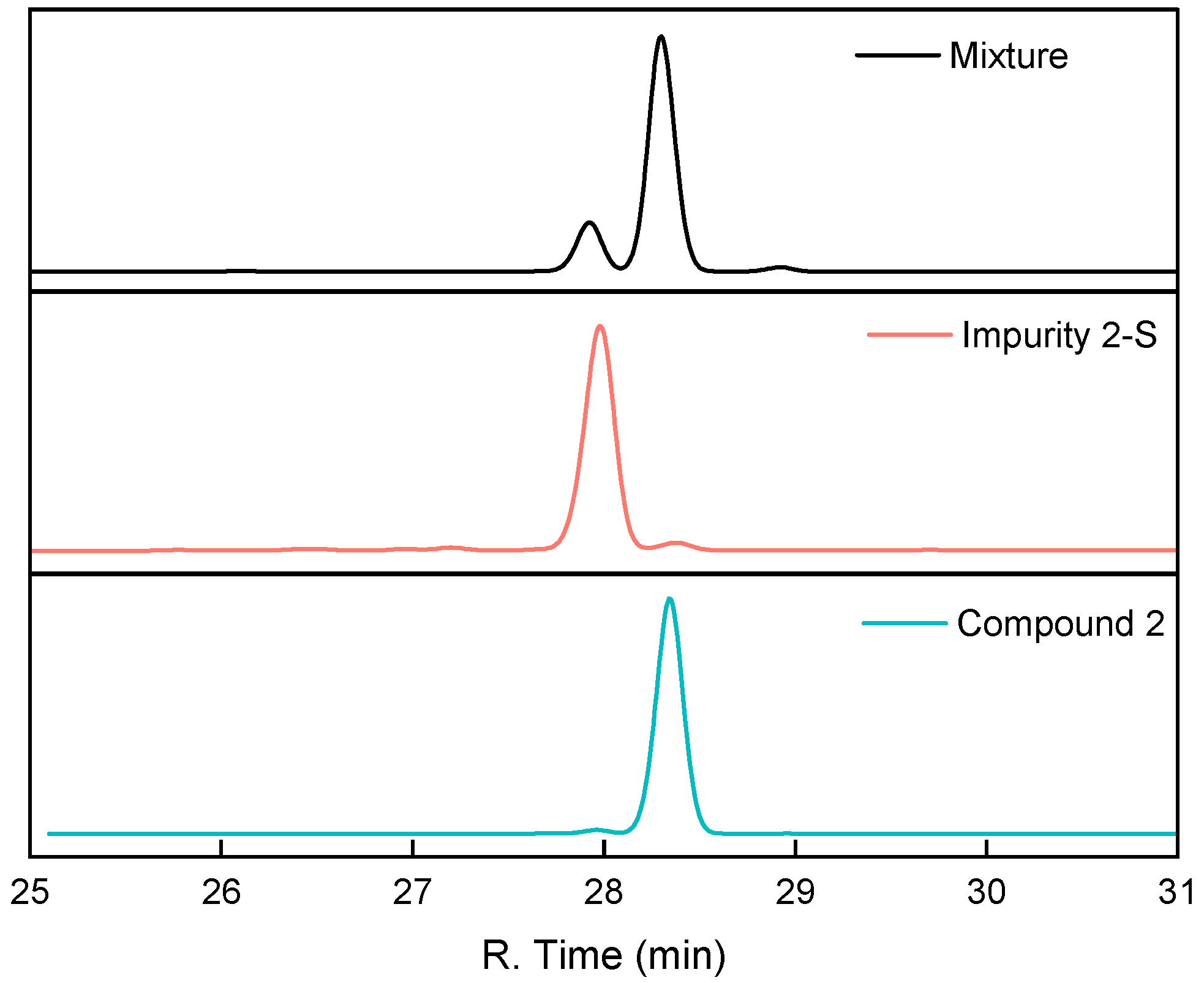
2.2.1. Synthesis, Characterization, and Validation of Impurities in the Horner–Wadsworth–Emmons Reaction
2.2.2. Process Optimization for Impurity 2-S
3. Materials and Methods
3.1. Synthesis of (20R)-3-Oxopregna-4-en-22-al (1)
3.2. Synthesis of (20R,22E)-3-Oxopregna-4,22-dien-24-oic Acid Methyl Ester (2)
3.3. Synthesis of (20R)-3-Oxopregna-4-en-22-oic Acid (1-A)
3.4. Synthesis of (20S,22E)-3-Oxopregna-4,22-dien-24-oic Acid Methyl Ester (2-S)
3.5. Synthesis of (20R,22Z)-3-Oxopregna-4,22-dien-24-oic Acid Methyl Ester (2-Z)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Paumgartner, G.; Beuers, U. Ursodeoxycholic acid in cholestatic liver disease: Mechanisms of action and therapeutic use revisited. Hepatology 2002, 36, 525–531. [Google Scholar] [CrossRef] [PubMed]
- Ikegami, T.; Matsuzaki, Y. Ursodeoxycholic acid: Mechanism of action and novel clinical applications. Hepatol. Res. Off. J. Jpn. Soc. Hepatol. 2008, 38, 123–131. [Google Scholar] [CrossRef]
- Jiang, H.X.; Hu, J.G.; Xiao, F.; Wang, Z.M. Research Progress of Ursodeoxycholic Acid in the Treatment of Gallstones and Chronic Cholecystitis. Adv. Clin. Med. 2021, 11, 1361–1367. [Google Scholar] [CrossRef]
- Roda, E.; Bazzoli, F.; Labate, A.M.; Mazzella, G.; Roda, A.; Sama, C.; Festi, D.; Aldini, R.; Taroni, F.; Barbara, L. Ursodeoxycholic acid vs. chenodeoxycholic acid as cholesterol gallstone-dissolving agents: A comparative randomized study. Hepatology 1982, 2, 804–810. [Google Scholar] [CrossRef] [PubMed]
- Salen, G.; Colalillo, A.; Verga, D.; Bagan, E.; Tint, G.S.; Shefer, S. Effect of high and low doses of ursodeoxycholic acid on gallstone dissolution in humans. Gastroenterology 1980, 78, 1412–1418. [Google Scholar] [PubMed]
- Amaral, J.D.; Viana, R.J.; Ramalho, R.M.; Steer, C.J.; Rodrigues, C.M. Bile acids: Regulation of apoptosis by ursodeoxycholic acid. J. Lipid Res. 2009, 50, 1721–1734. [Google Scholar] [CrossRef] [PubMed]
- Ko, W.K.; Kim, S.J.; Jo, M.J.; Choi, H.; Lee, D.; Kwon, I.K.; Lee, S.H.; Han, I.B.; Sohn, S. Ursodeoxycholic Acid Inhibits Inflammatory Responses and Promotes Functional Recovery After Spinal Cord Injury in Rats. Mol. Neurobiol. 2019, 56, 267–277. [Google Scholar] [CrossRef] [PubMed]
- Tsuchida, T.; Shiraishi, M.; Ohta, T.; Sakai, K.; Ishii, S. Ursodeoxycholic acid improves insulin sensitivity and hepatic steatosis by inducing the excretion of hepatic lipids in high-fat diet-fed KK-Ay mice. Metab. Clin. Exp. 2012, 61, 944–953. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.C.; Duong, H.Q.; Parajuli, K.R.; Han, S.I. Pro-apoptotic role of the MEK/ERK pathway in ursodeoxycholic acid-induced apoptosis in SNU601 gastric cancer cells. Oncol. Rep. 2012, 28, 1429–1434. [Google Scholar] [CrossRef] [PubMed]
- Bell, S.M.; Barnes, K.; Clemmens, H.; Al-Rafiah, A.R.; Al-Ofi, E.A.; Leech, V.; Bandmann, O.; Shaw, P.J.; Blackburn, D.J.; Ferraiuolo, L.; et al. Ursodeoxycholic Acid Improves Mitochondrial Function and Redistributes Drp1 in Fibroblasts from Patients with Either Sporadic or Familial Alzheimer’s Disease. J. Mol. Biol. 2018, 430, 3942–3953. [Google Scholar] [CrossRef] [PubMed]
- Tonin, F.; Arends, I. Latest development in the synthesis of ursodeoxycholic acid (UDCA): A critical review. Beilstein J. Org. Chem. 2018, 14, 470–483. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.Z.; Shi, X.Y. Progress on the synthesis of ursodeoxycholic acid from hyodeoxycholic acid. Chin. J. Med. Chem. 2019, 29, 224–233. [Google Scholar] [CrossRef]
- Chen, W.; Liu, G.G. Progress in the synthesis of ursodeoxycholic acid from plant-source materials. Chin. J. Med. Chem. 2023, 33, 49–58. [Google Scholar] [CrossRef]
- Xu, L.Q.; Liu, Y.J.; Yao, K.; Liu, H.H.; Tao, X.Y.; Wang, F.Q.; Wei, D.Z. Unraveling and engineering the production of 23,24-bisnorcholenic steroids in sterol metabolism. Sci. Rep. 2016, 6, 21928. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Gu, X.Z.; He, L.M.; Li, C.C.; Qiu, W.W. Synthesis of ursodeoxycholic acid from plant-source (20S)-21-hydroxy-20-methylpregn-4-en-3-one. Steroids 2020, 157, 108600. [Google Scholar] [CrossRef] [PubMed]
- Miyashita, M.; Sasaki, M.; Hattori, I.; Sakai, M.; Tanino, K. Total Synthesis of Norzoanthamine. Science 2004, 305, 495–499. [Google Scholar] [CrossRef] [PubMed]
- Bergmann, E.D.; Solomonovici, A. Application of the wittig-horner reaction to steroid ketones. Steroids 1976, 27, 431–437. [Google Scholar] [CrossRef] [PubMed]
- Linker, M.; Kreiser, W. Synthesis of methyl (20R,22E)- and (20S,22E)-3-oxochola-1,4,22-trien-24-oate. Helv. Chim. Acta 2002, 85, 1096–1101. [Google Scholar] [CrossRef]
- Blakemore, P.R.; Browder, C.C.; Hong, J.; Lincoln, C.M.; Nagornyy, P.A.; Robarge, L.A.; Wardrop, D.J.; White, J.D. Total Synthesis of Polycavernoside A, A Lethal Toxin of the Red Alga Polycavernosa tsudai. J. Org. Chem. 2005, 70, 5449–5460. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | NaClO (Equiv.) | BA (HPLC Area%) | 1-A (HPLC Area%) | Emulsification |
|---|---|---|---|---|
| 1 | 0.9 | 2.69 | 0.12 | NO |
| 2 | 1.0 | 0.85 | 0.23 | NO |
| 3 | 1.1 | 0.33 | 0.31 | NO |
| 4 | 1.2 | 0.12 | 0.86 | YES |
| 5 | 1.4 | 0.12 | 1.20 | YES |
| Entry | Material Addition Condition | Time | BA (HPLC Area%) | Compound 1 (HPLC Area%) | Impurity 1-A (HPLC Area%) |
|---|---|---|---|---|---|
| 1 | Complete material addition | 0.5 h | 0.21 | 97.12 | 0.13 |
| 2 | TEMPO was not added | 12 h | 60.99 | 5.80 | 0.97 |
| 3 | NaBr was not added | 3 h | 0.37 | 95.04 | 2.45 |
| 4 | NaHCO3 was not added | 24 h | 44.27 | 53.07 | 0.09 |
| Entry | Temp. | BA (HPLC Area%) | Compound 1 (HPLC Area%) |
|---|---|---|---|
| 1 | 0 °C | 0.11 | 97.12 |
| 2 | 10 °C | 0.12 | 97.23 |
| 3 | 20 °C | 0.18 | 96.02 |
| 4 | 30 °C | 0.16 | 95.96 |
| 5 | 40 °C | 32.83 | 65.40 |
| Entry | Treatment of Racemization | Compound 2 (HPLC Area%) | Impurity 2-S (HPLC Area%) |
|---|---|---|---|
| 1 | None | 82.51 | 13.64 |
| 2 | Strong acid 1 | 41.39 | 39.20 |
| 3 | Strong base 2 | 51.37 | 42.56 |
| Entry | Compound 1:NaOMe:Trimethyl Phosphonoacetate (Equiv.) | Time | Impurity 2-D (HPLC Area%) |
|---|---|---|---|
| 1 | 1:1.05:1.05 | 1 h | 0.65 |
| 2 | 1:1.05:1.05 | 5 h | 1.80 |
| 3 | 1:3:3 | 5 h | 43.83 |
| Entry | DCM:1 1 | NaOMe 2 | Compound 2 (HPLC Area%) | Impurity 2-S (HPLC Area%) | Impurity 2-Z (HPLC Area%) | Impurity 2-D (HPLC Area%) | Yield |
|---|---|---|---|---|---|---|---|
| 1 | 5 | Solid, Add all at once | 82.51 | 13.64 | 1.21 | 0.65 | 79.1% |
| 2 | 10 | Solid, Add all at once | 84.53 | 9.86 | 1.11 | 0.18 | 82.5% |
| 3 | 30 | Solid, Add all at once | 95.38 | 2.85 | 0.69 | 0.14 | 91.5% |
| 4 | 5 | Methanol solution (w = 20%) | 96.64 | 0.85 | 1.03 | 0.19 | 90.8% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jing, S.; Wang, Z.; Wang, Y.; Yang, Y.; Song, J.; Zhang, B. Studies on the Synthesis Process of Plant-Derived Ursodeoxycholic Acid Intermediates. Molecules 2025, 30, 1454. https://doi.org/10.3390/molecules30071454
Jing S, Wang Z, Wang Y, Yang Y, Song J, Zhang B. Studies on the Synthesis Process of Plant-Derived Ursodeoxycholic Acid Intermediates. Molecules. 2025; 30(7):1454. https://doi.org/10.3390/molecules30071454
Chicago/Turabian StyleJing, Shaoxiong, Zhongyue Wang, Yuan Wang, Yingquan Yang, Jian Song, and Bao Zhang. 2025. "Studies on the Synthesis Process of Plant-Derived Ursodeoxycholic Acid Intermediates" Molecules 30, no. 7: 1454. https://doi.org/10.3390/molecules30071454
APA StyleJing, S., Wang, Z., Wang, Y., Yang, Y., Song, J., & Zhang, B. (2025). Studies on the Synthesis Process of Plant-Derived Ursodeoxycholic Acid Intermediates. Molecules, 30(7), 1454. https://doi.org/10.3390/molecules30071454