
Electronic–Oxygen Synergy at Ca-Fe Dual-Metal Interfaces for Selective Syngas Regulation in Biomass Chemical Looping Gasification





Abstract
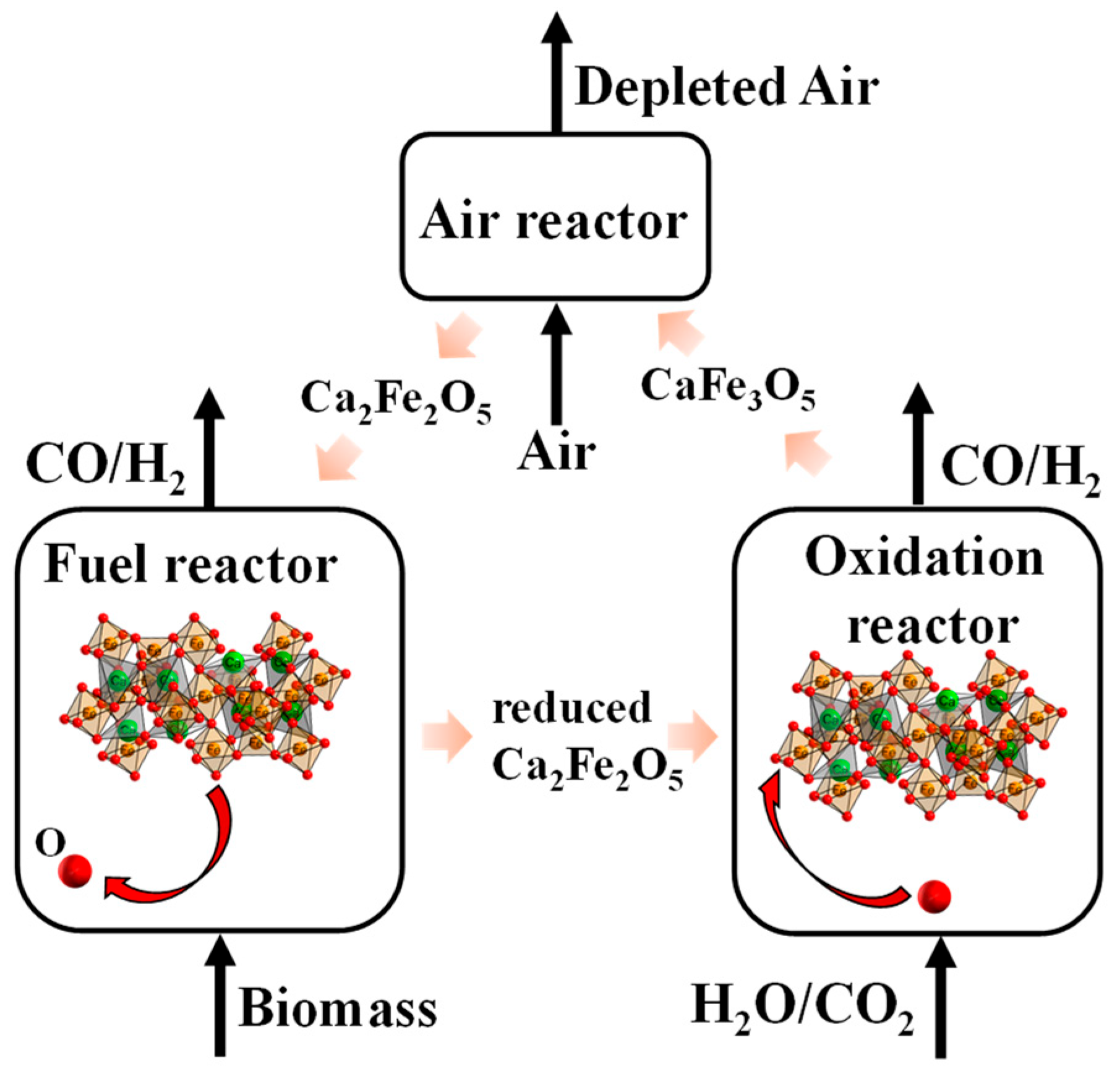
:1. Introduction
2. Results and Discussion
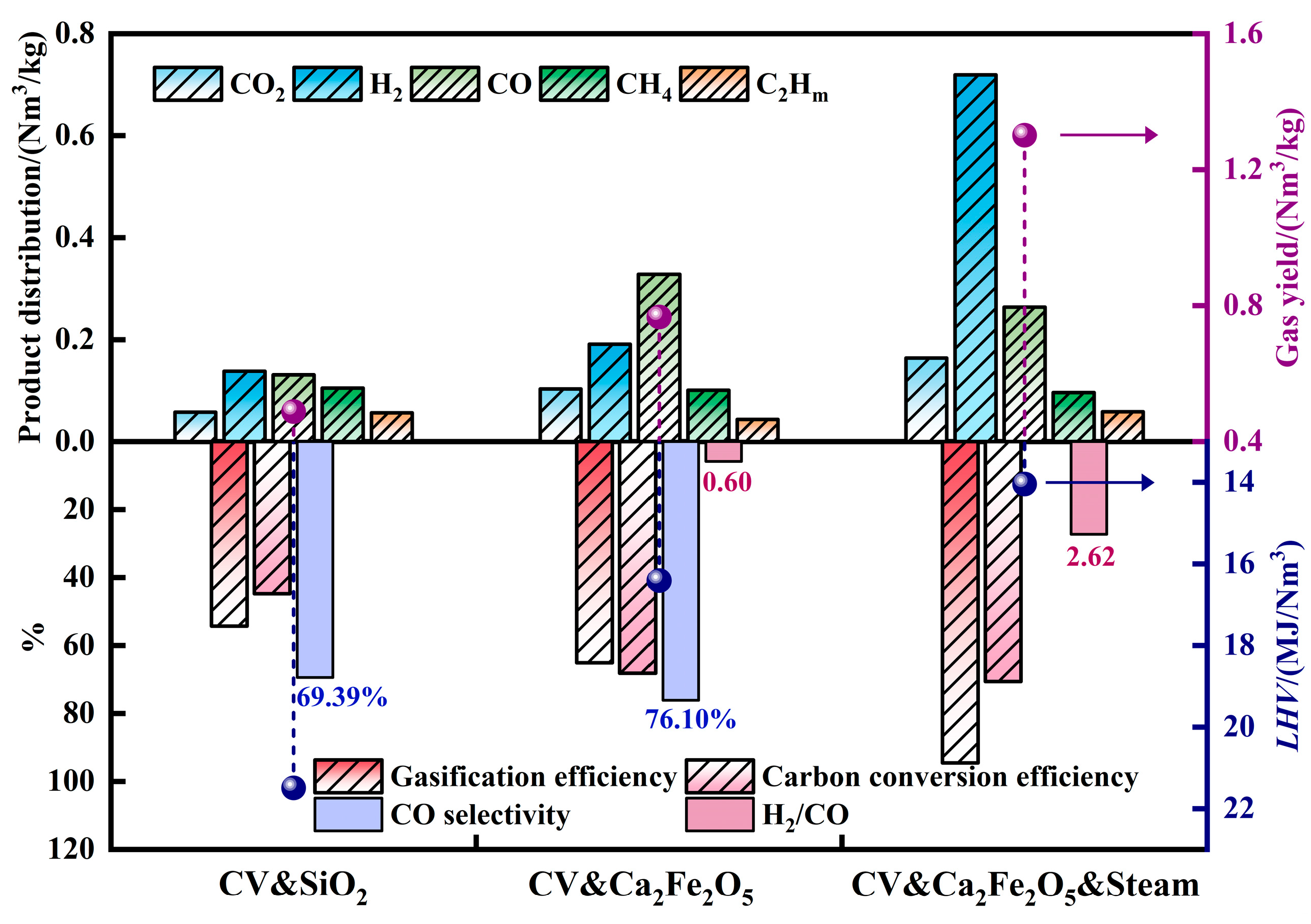
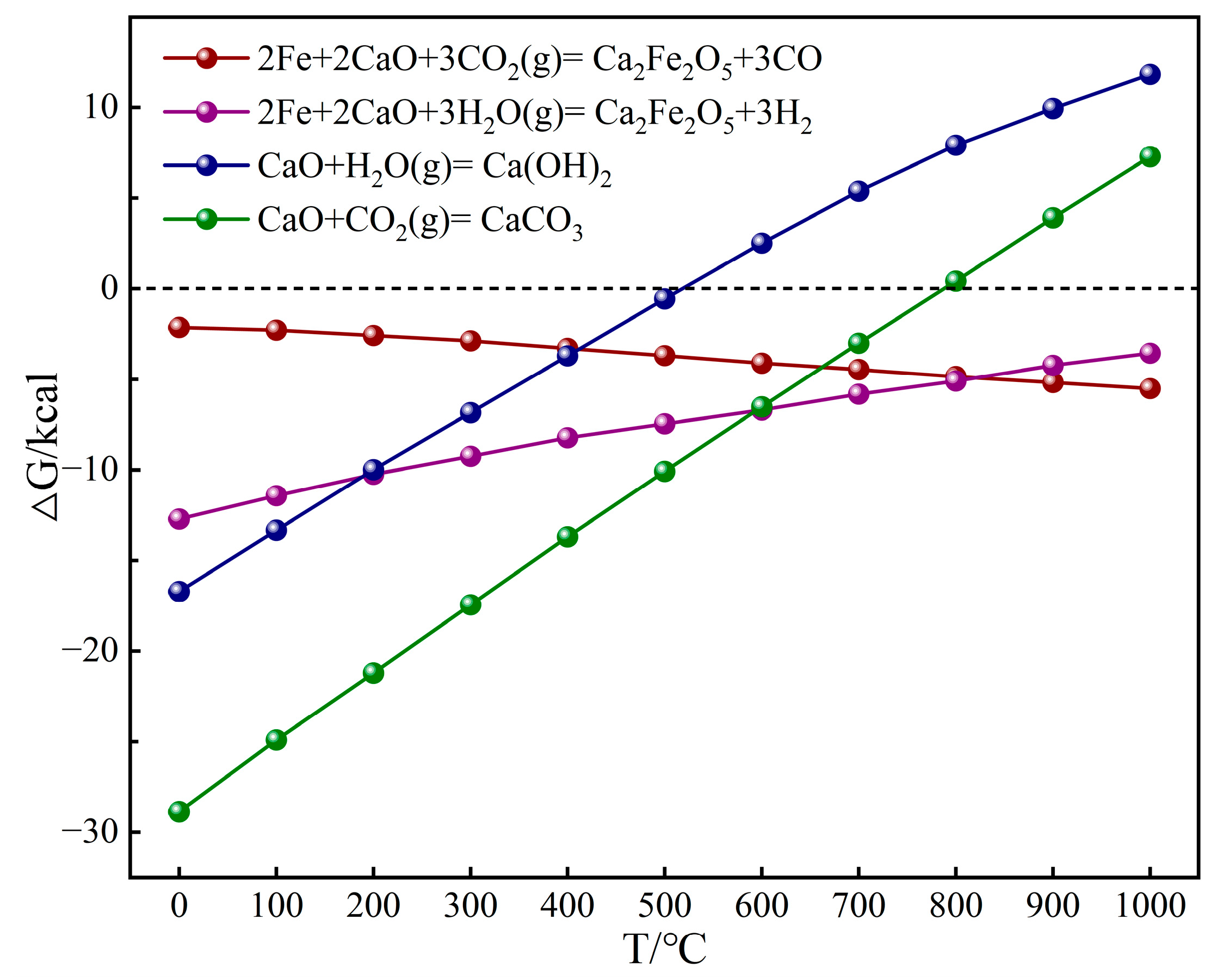
2.1. Chemical Looping Gasification Experimental Results
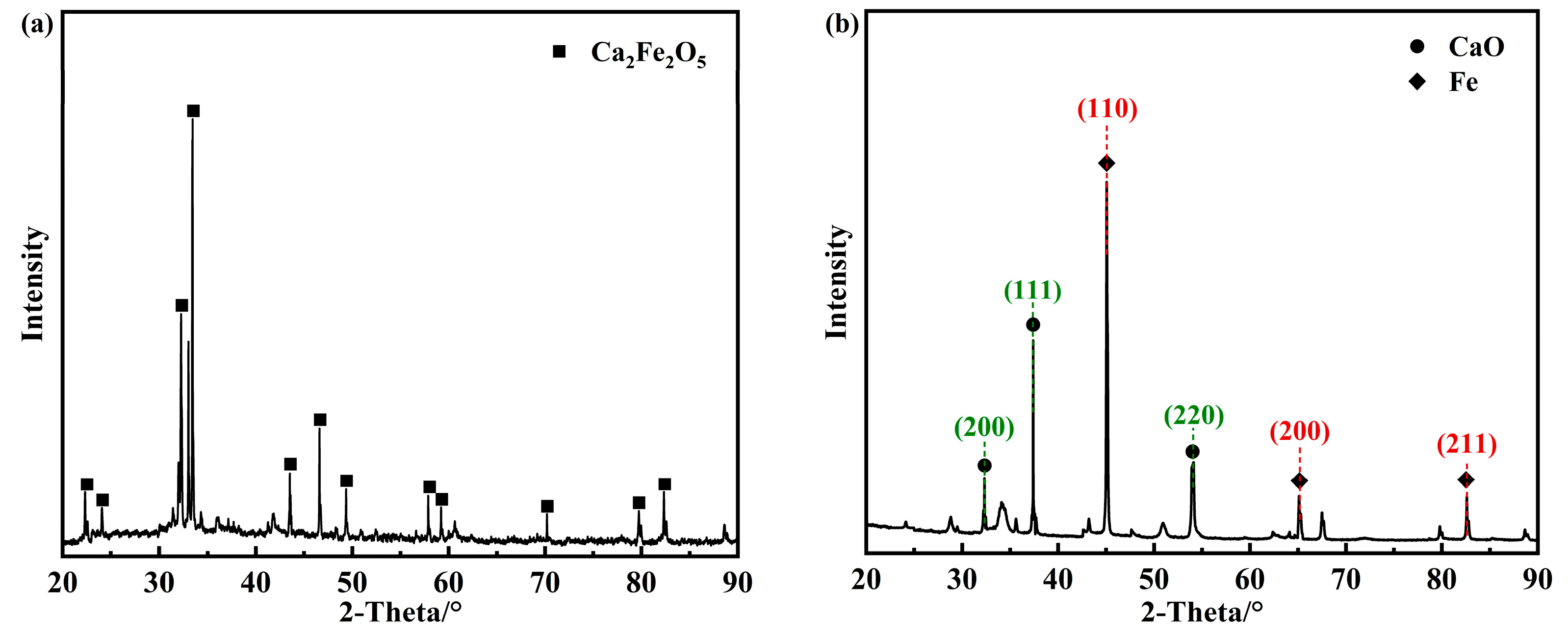
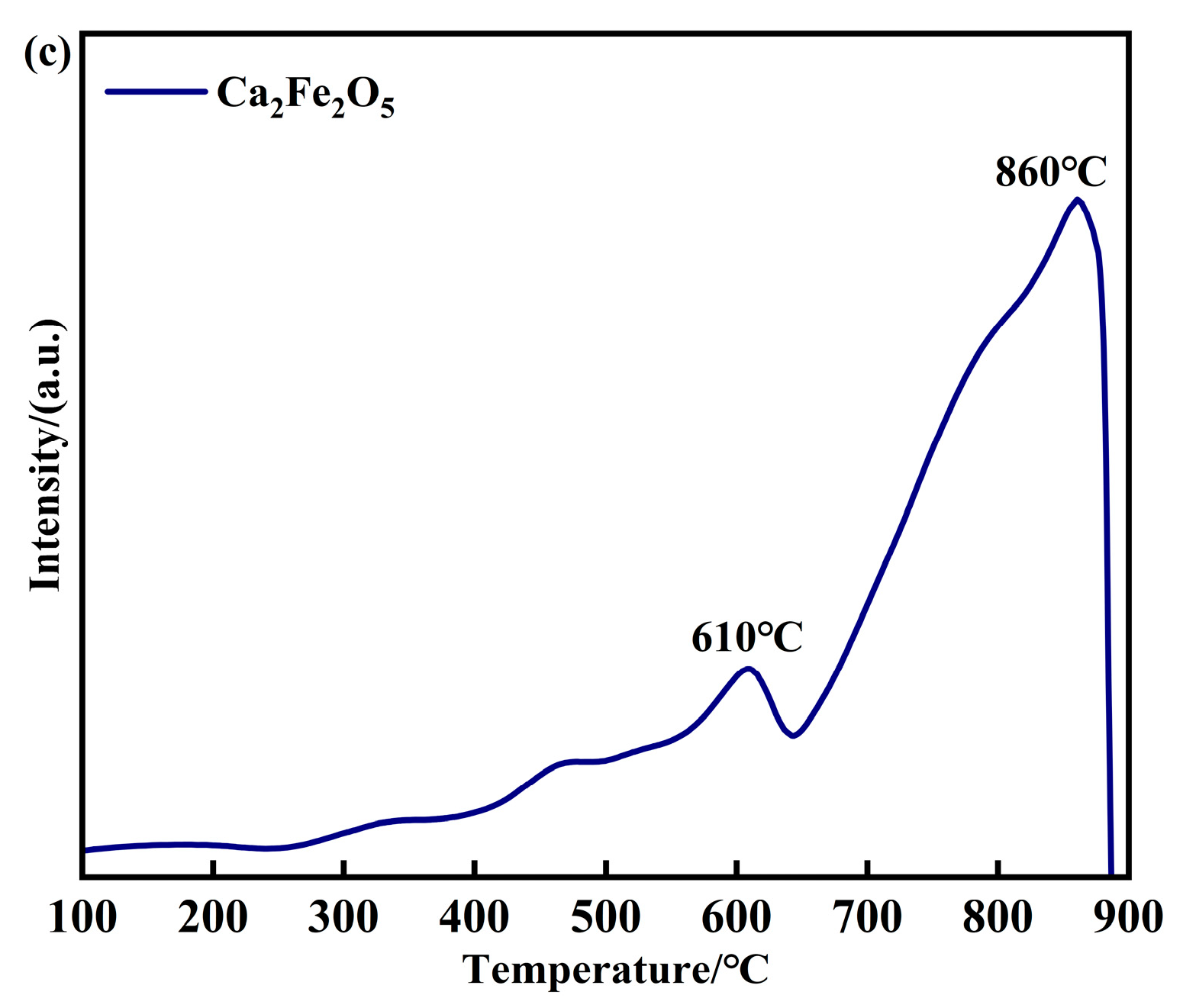
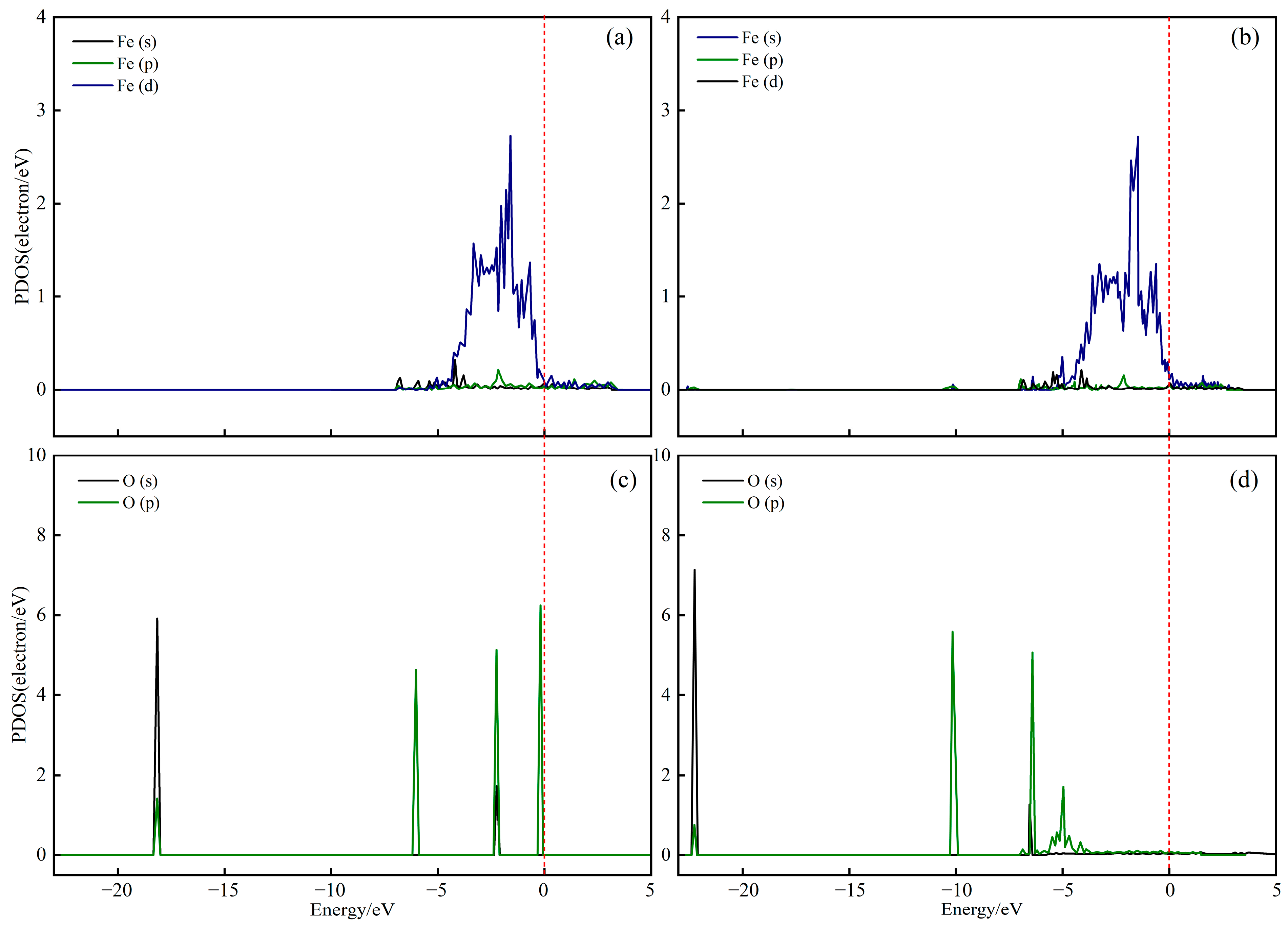
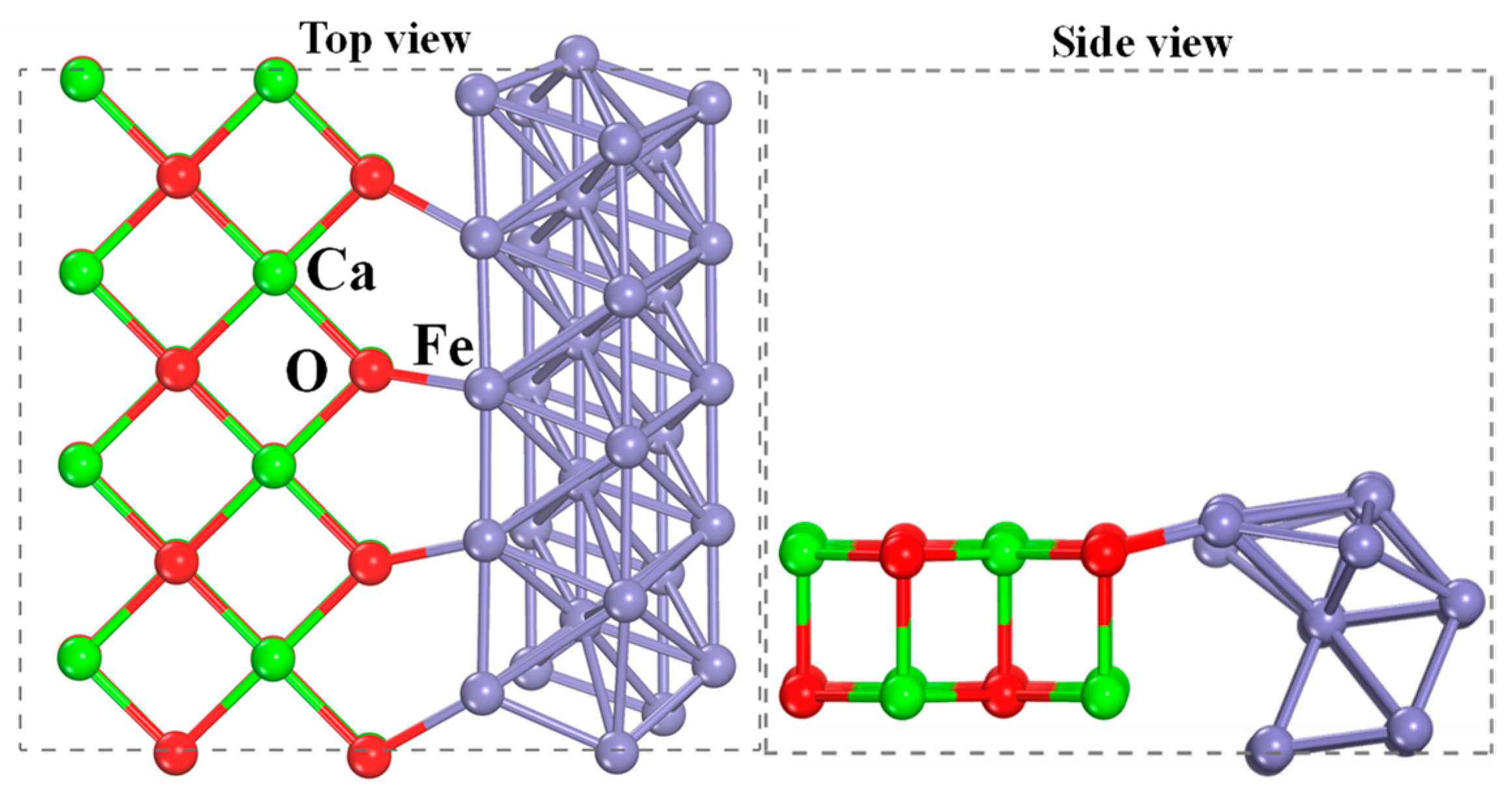
2.2. Characterization of Oxygen Carriers
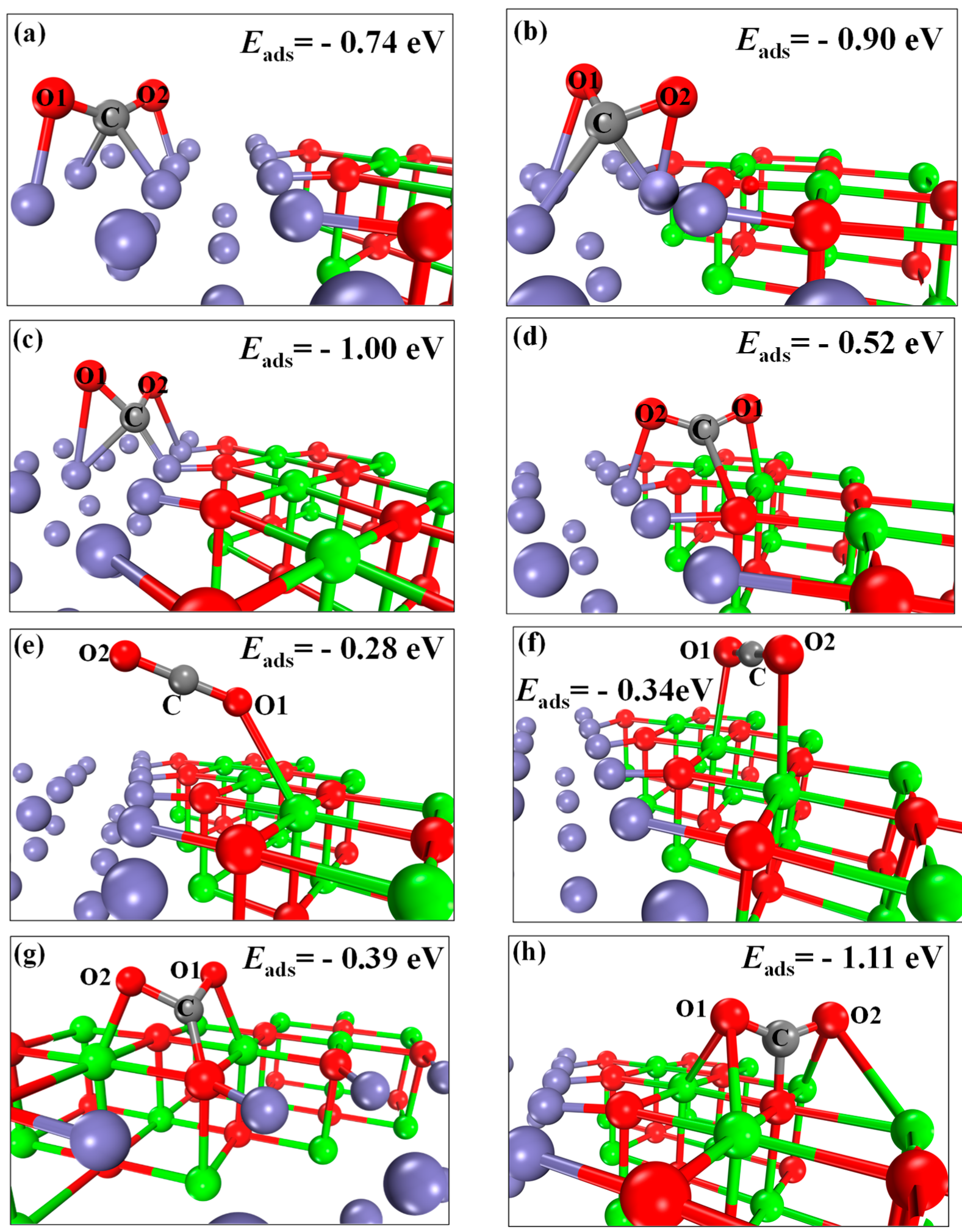
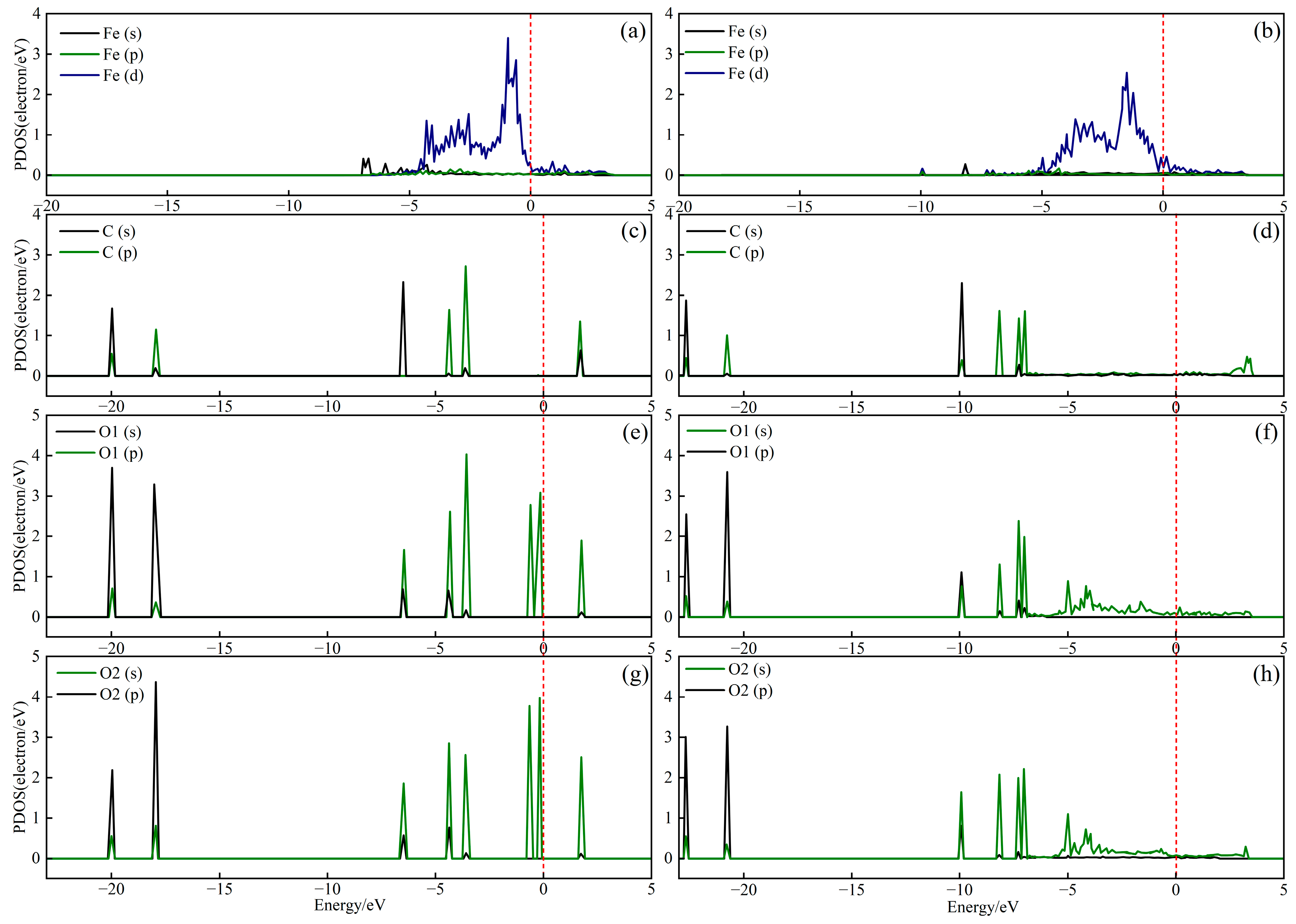
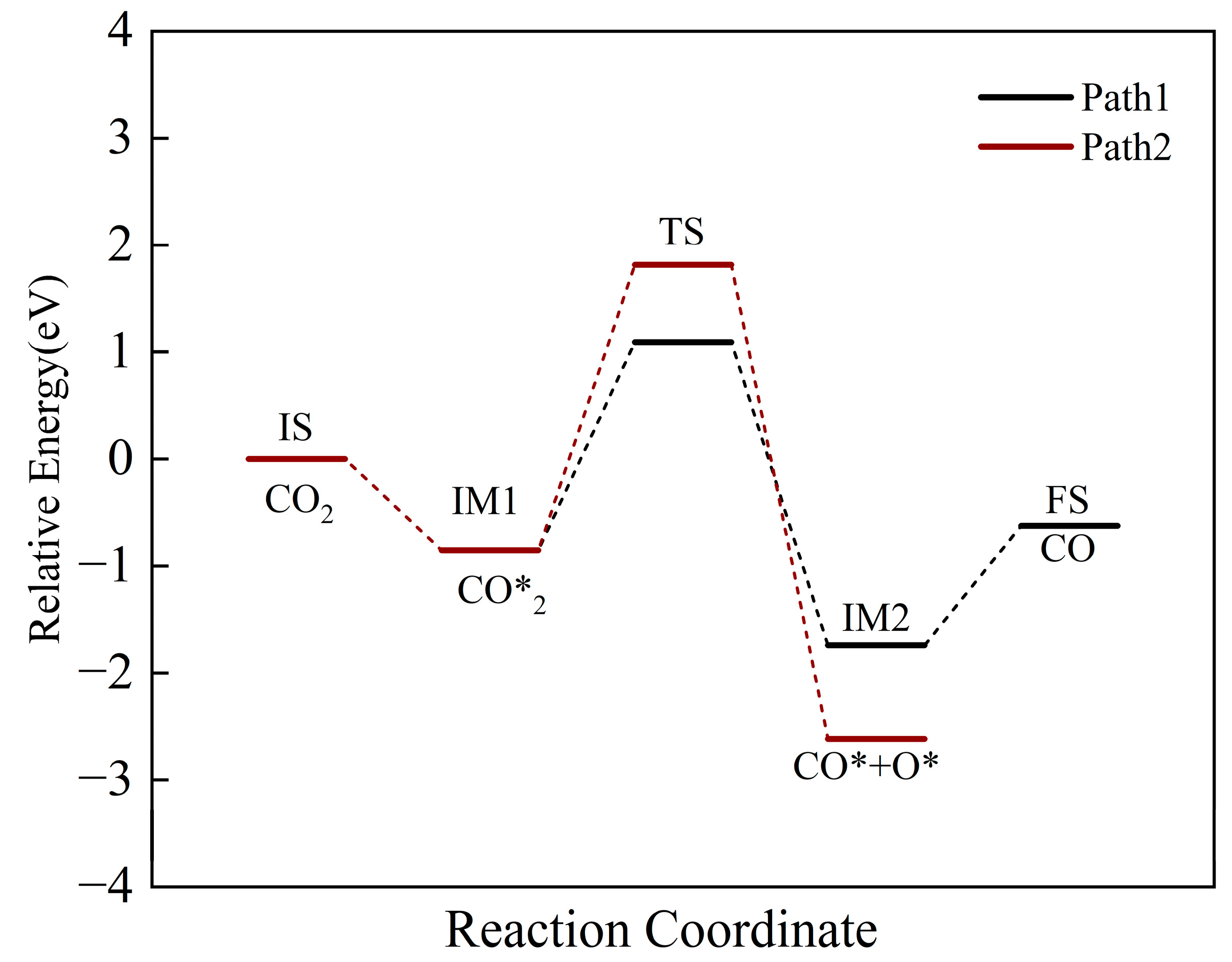
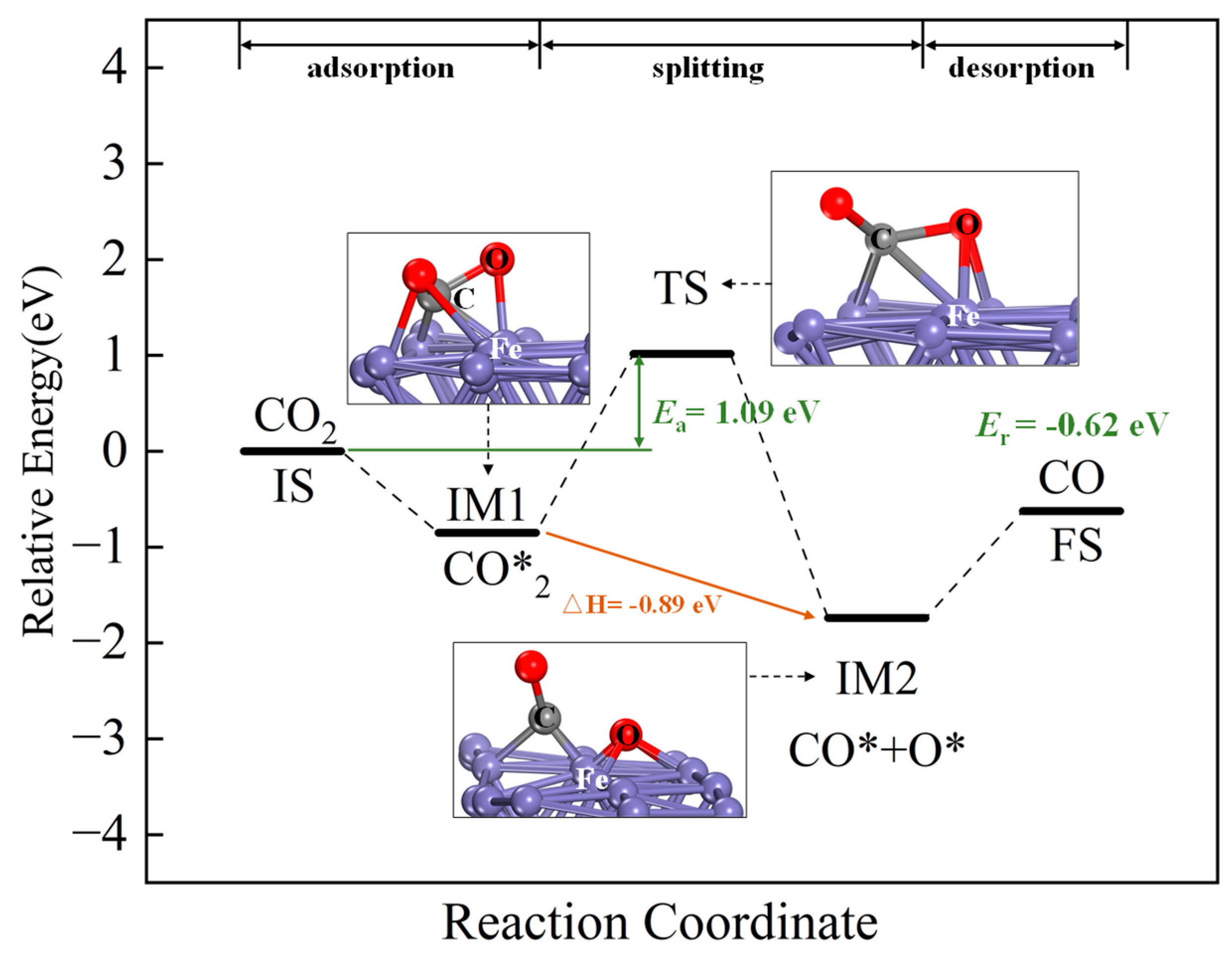
2.3. CO2 Adsorption and Splitting
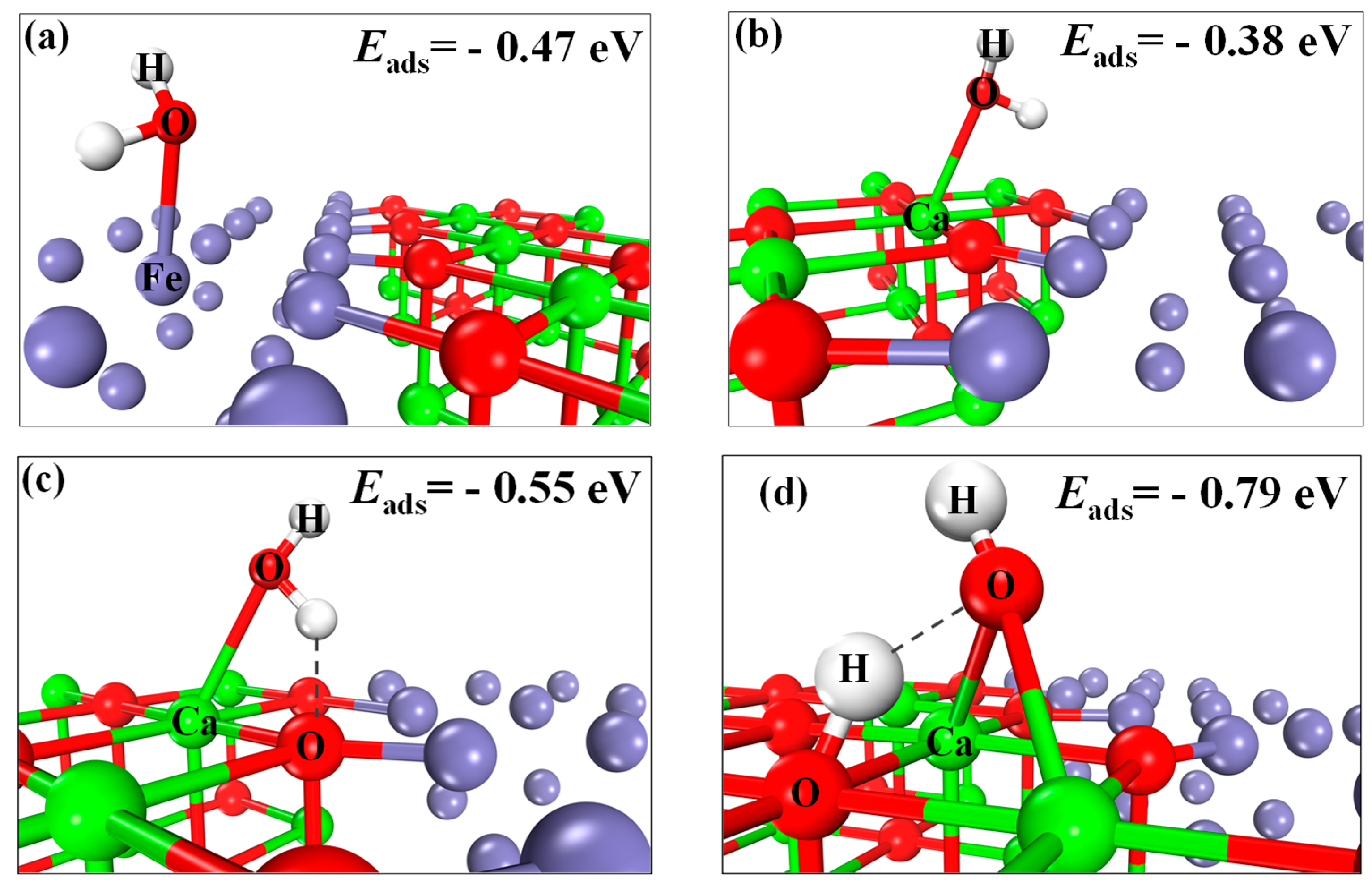
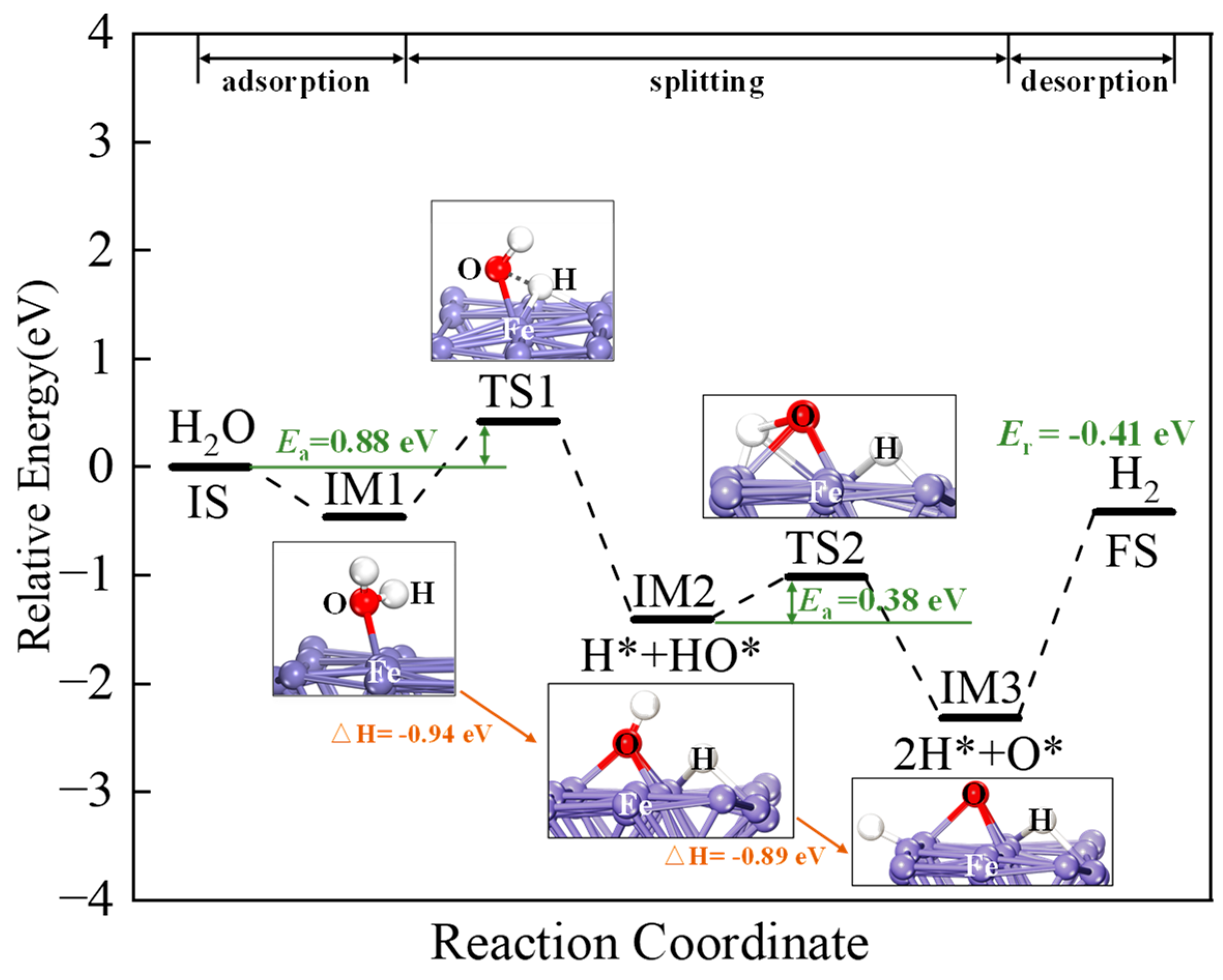
2.4. H2O Adsorption and Splitting
3. Materials and Methods
3.1. Material Preparation
3.2. Material Characterization
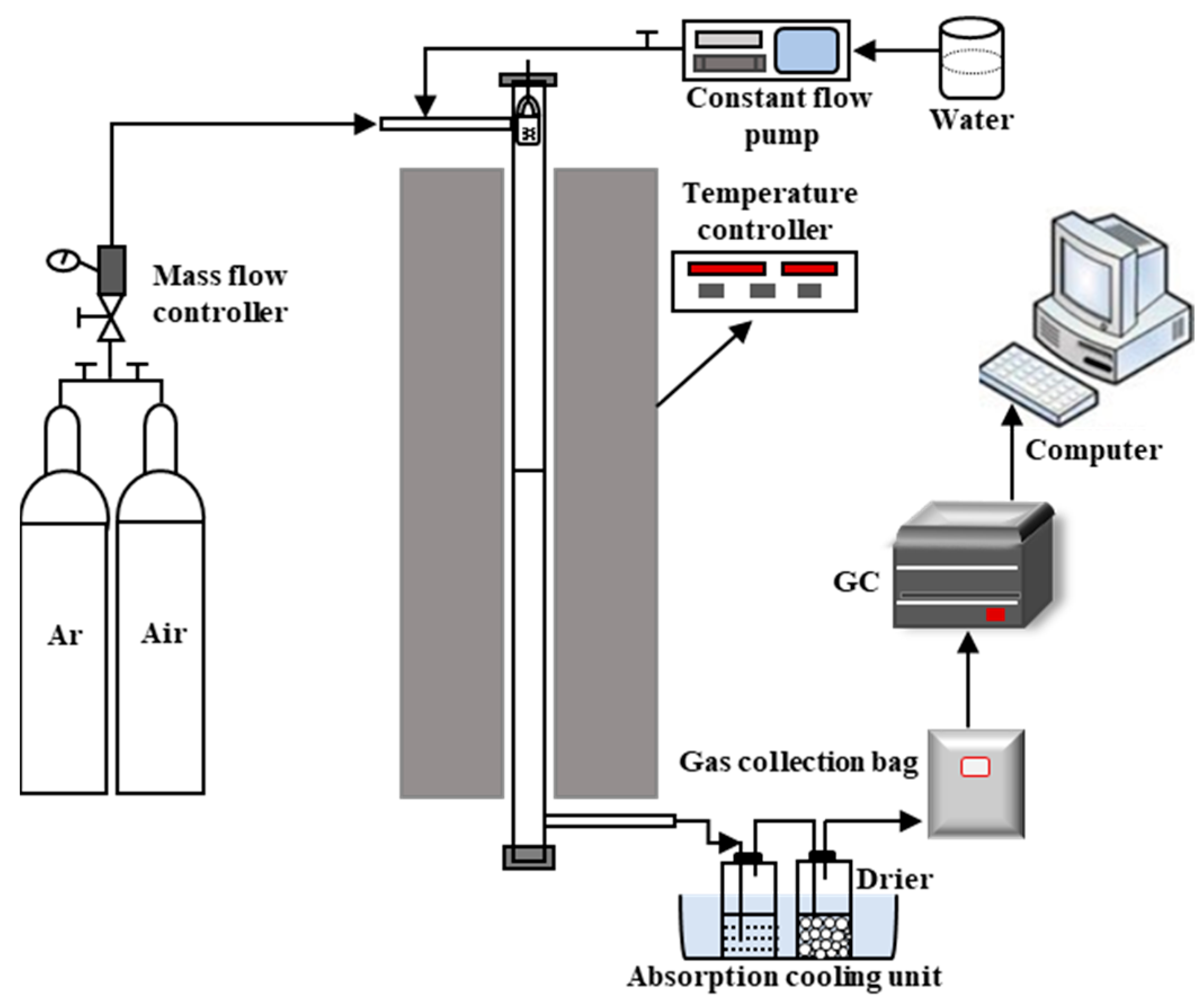
3.3. Chemical Looping Gasification Experiment
3.4. Gasification Performance Evaluation Indicators
3.5. Computational Details
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Adeniyi, O.M.; Azimov, U.; Burluka, A. Algae biofuel: Current status and future applications. Renew. Sustain. Energy Rev. 2018, 90, 316–335. [Google Scholar] [CrossRef]
- Meng, S.; Li, W.; Li, Z.; Song, H. Recent progress of the transition metal-based catalysts in the catalytic biomass gasification: A mini-review. Fuel 2023, 353, 129169. [Google Scholar] [CrossRef]
- Sathya, A.B.; Thirunavukkarasu, A.; Nithya, R.; Nandan, A.; Sakthishobana, K.; Kola, A.K.; Sivashankar, R.; Tuan, H.A.; Deepanraj, B. Microalgal biofuel production: Potential challenges and prospective research. Fuel 2023, 332, 126199. [Google Scholar] [CrossRef]
- Martins, F.; Felgueiras, C.; Smitkova, M.; Caetano, N. Analysis of Fossil Fuel Energy Consumption and Environmental Impacts in European Countries. Energies 2019, 12, 964. [Google Scholar] [CrossRef]
- Ubando, A.T.; Rivera, D.R.T.; Chen, W.-H.; Culaba, A.B. A comprehensive review of life cycle assessment (LCA) of microalgal and lignocellulosic bioenergy products from thermochemical processes. Bioresour. Technol. 2019, 291, 121837. [Google Scholar] [CrossRef]
- Gholkar, P.; Shastri, Y.; Tanksale, A. Renewable hydrogen and methane production from microalgae: A techno-economic and life cycle assessment study. J. Clean. Prod. 2021, 279, 123726. [Google Scholar] [CrossRef]
- Lin, Y.; Wang, H.; Wang, Y.; Huo, R.; Huang, Z.; Liu, M.; Wei, G.; Zhao, Z.; Li, H.; Fang, Y. Review of Biomass Chemical Looping Gasification in China. Energy Fuels 2020, 34, 7847–7862. [Google Scholar] [CrossRef]
- Dieringer, P.; Marx, F.; Alobaid, F.; Stroehle, J.; Epple, B. Process Control Strategies in Chemical Looping Gasification-A Novel Process for the Production of Biofuels Allowing for Net Negative CO2 Emissions. Appl. Sci. 2020, 10, 4271. [Google Scholar] [CrossRef]
- Zhu, X.; Imtiaz, Q.; Donat, F.; Mueller, C.R.; Li, F. Chemical looping beyond combustion—A perspective. Energy Environ. Sci. 2020, 13, 772–804. [Google Scholar] [CrossRef]
- Goel, A.; Moghaddam, E.M.; Liu, W.; He, C.; Konttinen, J. Biomass chemical looping gasification for high-quality syngas: A critical review and technological outlooks. Energy Convers. Manag. 2022, 268, 116020. [Google Scholar] [CrossRef]
- Jiang, H.; Huo, R.; Zhang, Z.; Lin, Y.; Zhao, Z.; Huang, Z.; Fang, Y.; Li, H. Removal of pollution from the chemical looping process: A mini review. Fuel Process. Technol. 2021, 221, 106937. [Google Scholar] [CrossRef]
- Zeng, L.; Cheng, Z.; Fan, J.A.; Fan, L.-S.; Gong, J. Metal oxide redox chemistry for chemical looping processes. Nat. Rev. Chem. 2018, 2, 349–364. [Google Scholar] [CrossRef]
- Yu, L.; Zhou, W.; Luo, Z.; Wang, H.; Liu, W.; Yin, K. Developing Oxygen Carriers for Chemical Looping Biomass Processing: Challenges and Opportunities. Adv. Sustain. Syst. 2020, 4, 2000099. [Google Scholar] [CrossRef]
- Zeng, J.; Hu, J.; Qiu, Y.; Zhang, S.; Zeng, D.; Xiao, R. Multi-function of oxygen carrier for in-situ tar removal in chemical looping gasification: Naphthalene as a model compound. Appl. Energy 2019, 253, 113502. [Google Scholar] [CrossRef]
- De Vos, Y.; Jacobs, M.; Van der Voort, P.; Van Driessche, I.; Snijkers, F.; Verberckmoes, A. Development of Stable Oxygen Carrier Materials for Chemical Looping Processes-A Review. Catalysts 2020, 10, 926. [Google Scholar] [CrossRef]
- Huang, Z.; Zheng, A.; Deng, Z.; Wei, G.; Zhao, K.; Chen, D.; He, F.; Zhao, Z.; Li, H.; Li, F. Insitu removal of toluene as a biomass tar model compound using NiFe2O4 for application in chemical looping gasification oxygen carrier. Energy 2020, 190, 116360. [Google Scholar] [CrossRef]
- O'Malley, K.; Donat, F.; Whitty, K.J.; Sohn, H.Y. Scalable Preparation of Bimetallic Cu/Ni-Based Oxygen Carriers for Chemical Looping. Energy Fuels 2020, 34, 11227–11236. [Google Scholar] [CrossRef]
- Miller, D.D.; Smith, M.; Shekhawat, D. Interaction of manganese with aluminosilicate support during high temperature (1100 °C) chemical looping combustion of the Fe-Mn-based oxygen carrier. Fuel 2020, 263, 116738. [Google Scholar] [CrossRef]
- Cai, Y.; Wang, C.; Zhang, Z.; Zhong, M.; Wu, Q.; Xiao, B.; Xu, T.; Wang, X. Performance optimization of Ca2Fe2O5 oxygen carrier by doping different metals for coproduction syngas and hydrogen with chemical looping gasification and water splitting. J. Energy Inst. 2023, 111, 101391. [Google Scholar] [CrossRef]
- Sun, H.; Wang, Z.; Fang, Y.; Liu, Z.; Dong, L.; Zhou, X.; Yu, Z.; Li, X.; Bai, J.; Huang, J. A novel system of biomass for the generation of inherently separated syngas by combining chemical looping CO2-gasification and steam reforming process. Energy Convers. Manag. 2022, 251, 114876. [Google Scholar] [CrossRef]
- Wei, G.-q.; Feng, J.; Hou, Y.-L.; Li, F.-Z.; Li, W.-Y.; Huang, Z.; Zheng, A.-q.; Li, H.-b. Ca-enhanced hematite oxygen carriers for chemical looping reforming of biomass pyrolyzed gas coupled with CO2 splitting. Fuel 2021, 285, 119125. [Google Scholar] [CrossRef]
- Hu, Q.; Mao, Q.; Ren, X.; Yang, H.; Chen, H. Inert chemical looping conversion of biochar with iron ore as oxygen carrier: Products conversion kinetics and structural evolution. Bioresour. Technol. 2019, 275, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Wei, G.; Wang, H.; Zhao, W.; Huang, Z.; Yi, Q.; He, F.; Zhao, K.; Zheng, A.; Meng, J.; Deng, Z.; et al. Synthesis gas production from chemical looping gasification of lignite by using hematite as oxygen carrier. Energy Convers. Manag. 2019, 185, 774–782. [Google Scholar] [CrossRef]
- Wang, X.; Gong, Y.; Wang, X.; Jin, B. Experimental and kinetics investigations of separated-gasification chemical looping combustion of char with an iron ore as the oxygen carrier. Fuel Process. Technol. 2020, 210, 106554. [Google Scholar] [CrossRef]
- Xu, F.; Xing, X.; Gao, S.; Zhang, W.; Zhu, L.; Wang, Y.; Chen, J.; Chen, H.; Zhu, Y. Direct chemical looping gasification of pine sawdust using Fe2O3-rich sludge ash as an oxygen carrier: Thermal conversion characteristics, product distributions, and gasification performances. Fuel 2021, 304, 121499. [Google Scholar] [CrossRef]
- Huang, X.; Wu, J.; Wang, M.; Ma, X.; Jiang, E.; Hu, Z. Syngas production by chemical looping gasification of rice husk using Fe-based oxygen carrier. J. Energy Inst. 2020, 93, 1261–1270. [Google Scholar] [CrossRef]
- Feng, Y.; Wang, N.; Guo, X.; Zhang, S. Dopant screening of modified Fe2O3 oxygen carriers in chemical looping hydrogen production. Fuel 2020, 262, 116489. [Google Scholar] [CrossRef]
- Zeng, D.; Qiu, Y.; Zhang, S.; Ma, L.; Li, M.; Cui, D.; Zeng, J.; Xiao, R. Synergistic effects of binary oxygen carriers during chemical looping hydrogen production. Int. J. Hydrogen Energy 2019, 44, 21290–21302. [Google Scholar] [CrossRef]
- Wu, J.; Hu, Z.; Miao, Z.; Wu, W.; Jiang, E. Effect of alkaline earth metal Ca in rice husk during chemical looping gasification process. Fuel 2021, 299, 120902. [Google Scholar] [CrossRef]
- Yan, X.; Li, Y.; Zhang, C.; Wang, Y.; Zhao, J.; Wang, Z. Understanding the enhancement of CaO on water gas shift reaction for H2 production by density functional theory. Fuel 2021, 303, 121257. [Google Scholar] [CrossRef]
- Si, T.; Huang, K.; Lin, Y.; Gu, M. ReaxFF Study on the Effect of CaO on Cellulose Pyrolysis. Energy Fuels 2019, 33, 11067–11077. [Google Scholar] [CrossRef]
- Chen, X.; Li, S.; Liu, Z.; Chen, Y.; Yang, H.; Wang, X.; Che, Q.; Chen, W.; Chen, H. Pyrolysis characteristics of lignocellulosic biomass components in the presence of CaO. Bioresour. Technol. 2019, 287, 121493. [Google Scholar] [CrossRef]
- Chan, M.S.C.; Liu, W.; Ismail, M.; Yang, Y.; Scott, S.A.; Dennis, J.S. Improving hydrogen yields, and hydrogen: Steam ratio in the chemical looping production of hydrogen using Ca2Fe2O5. Chem. Eng. J. 2016, 296, 406–411. [Google Scholar] [CrossRef]
- Wang, Y.; Niu, P.; Zhao, H. Chemical looping gasification of coal using calcium ferrites as oxygen carrier. Fuel Process. Technol. 2019, 192, 75–86. [Google Scholar] [CrossRef]
- Sun, Z.; Wu, X.; Russell, C.K.; Dyar, M.D.; Sklute, E.C.; Toan, S.; Fan, M.; Duan, L.; Xiang, W. Synergistic enhancement of chemical looping-based CO2 splitting with biomass cascade utilization using cyclic stabilized Ca2Fe2O5 aerogel. J. Mater. Chem. A 2019, 7, 1216–1226. [Google Scholar] [CrossRef]
- Feng, Y.; Wang, N.; Guo, X.; Zhang, S. Reaction mechanism of Ca2Fe2O5 oxygen carrier with CO in chemical looping hydrogen production. Appl. Surf. Sci. 2020, 534, 147583. [Google Scholar] [CrossRef]
- Feng, Y.; Wang, N.; Guo, X. Reaction mechanism of methane conversion over Ca2Fe2O5 oxygen carrier in chemical looping hydrogen production. Fuel 2021, 290, 120094. [Google Scholar] [CrossRef]
- Sun, Z.; Liu, H.; Bai, H.; Yu, S.; Russell, C.K.; Zeng, L.; Sun, Z. The crucial role of deoxygenation in syngas refinement and carbon dioxide utilization during chemical looping-based biomass gasification. Chem. Eng. J. 2022, 428, 132068. [Google Scholar] [CrossRef]
- Shah, V.; Cheng, Z.; Baser, D.S.; Fan, J.A.; Fan, L.-S. Highly Selective Production of Syngas from Chemical Looping Reforming of Methane with CO2 Utilization on MgO-supported Calcium Ferrite Redox Materials. Appl. Energy 2021, 282, 116111. [Google Scholar] [CrossRef]
- Shah, V.; Cheng, Z.; Mohapatra, P.; Fan, L.-S. Enhanced methane conversion using Ni-doped calcium ferrite oxygen carriers in chemical looping partial oxidation systems with CO2 utilization. React. Chem. Eng. 2021, 6, 1928–1939. [Google Scholar] [CrossRef]
- Rassouli, L.; Dupuis, M. Electronic Structure of Excitons in Hematite Fe2O3. J. Phys. Chem. C 2024, 128, 743–758. [Google Scholar] [CrossRef]
- Liu, S.; Tian, X.; Wang, T.; Wen, X.; Li, Y.-W.; Wang, J.; Jiao, H. Coverage dependent water dissociative adsorption on Fe(110) from DFT computation. Phys. Chem. Chem. Phys. 2015, 17, 8811–8821. [Google Scholar] [CrossRef] [PubMed]
- Jung, S.C.; Kang, M.H. Adsorption of a water molecule on Fe(100): Density-functional calculations. Phys. Rev. B 2010, 81, 115460. [Google Scholar] [CrossRef]
- Govind, N.; Petersen, M.; Fitzgerald, G.; King-Smith, D.; Andzelm, J. A generalized synchronous transit method for transition state location. Comput. Mater. Sci. 2003, 28, 250–258. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Biomass | Oxygen Carrier | Steam (mL/min) | Temperature (°C) | Time (min) | |
|---|---|---|---|---|---|
| A1 | Chlorella vulgaris | SiO2 | / | 800 | 30 |
| A2 | Chlorella vulgaris | Ca2Fe2O5 | / | 800 | 30 |
| A3 | Chlorella vulgaris | Ca2Fe2O5 | 0.04 | 800 | 30 |
| Configuration | C | O1 | O2 | Fe1O | Fe2C | |
|---|---|---|---|---|---|---|
| a | Before adsorption | 1.626 | −0.812 | −0.814 | −0.057 | 0.172 |
| After adsorption | 0.918 | −0.907 | −0.963 | −0.109 | 0.503 | |
| b | Before adsorption | 1.236 | −1.087 | −0.814 | −0.057 | 0.172 |
| After adsorption | 0.421 | −0.974 | −0.868 | −0.605 | 0.433 |
| Configuration | H1 | H2 | O | Fe1 | Fe2 | |
|---|---|---|---|---|---|---|
| a | Before adsorption | 0.581 | 0.551 | −1.132 | 0.019 | −0.011 |
| After adsorption | 0.329 | 0.231 | −1.172 | 0.198 | 0.391 | |
| b | Before adsorption | 0.513 | 0.551 | −1.132 | 0.018 | −0.011 |
| After adsorption | 0.431 | −0.467 | −0.871 | 0.927 | 0.198 |
| Proximate Analysis (wt.%, ad) | Ultimate Analysis (wt.%, ad) | Low Heating Value (MJ/kg) | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Moisture | Volatiles | FC | Ash | C | H | N | S | O a | |
| 4.40 | 76.63 | 14.55 | 6.42 | 48.66 | 6.96 | 9.26 | 0.65 | 34.47 | 19.23 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.; Li, J.; Zhu, S.; Hitch, M. Electronic–Oxygen Synergy at Ca-Fe Dual-Metal Interfaces for Selective Syngas Regulation in Biomass Chemical Looping Gasification. Molecules 2025, 30, 1471. https://doi.org/10.3390/molecules30071471
Wang Y, Li J, Zhu S, Hitch M. Electronic–Oxygen Synergy at Ca-Fe Dual-Metal Interfaces for Selective Syngas Regulation in Biomass Chemical Looping Gasification. Molecules. 2025; 30(7):1471. https://doi.org/10.3390/molecules30071471
Chicago/Turabian StyleWang, Yijie, Jiajie Li, Sitao Zhu, and Michael Hitch. 2025. "Electronic–Oxygen Synergy at Ca-Fe Dual-Metal Interfaces for Selective Syngas Regulation in Biomass Chemical Looping Gasification" Molecules 30, no. 7: 1471. https://doi.org/10.3390/molecules30071471
APA StyleWang, Y., Li, J., Zhu, S., & Hitch, M. (2025). Electronic–Oxygen Synergy at Ca-Fe Dual-Metal Interfaces for Selective Syngas Regulation in Biomass Chemical Looping Gasification. Molecules, 30(7), 1471. https://doi.org/10.3390/molecules30071471