
Synthesis and Antimicrobial Activity of Canthin-6-One Alkaloids

 , and
, and
Abstract
1. Introduction
2. Results and Discussion
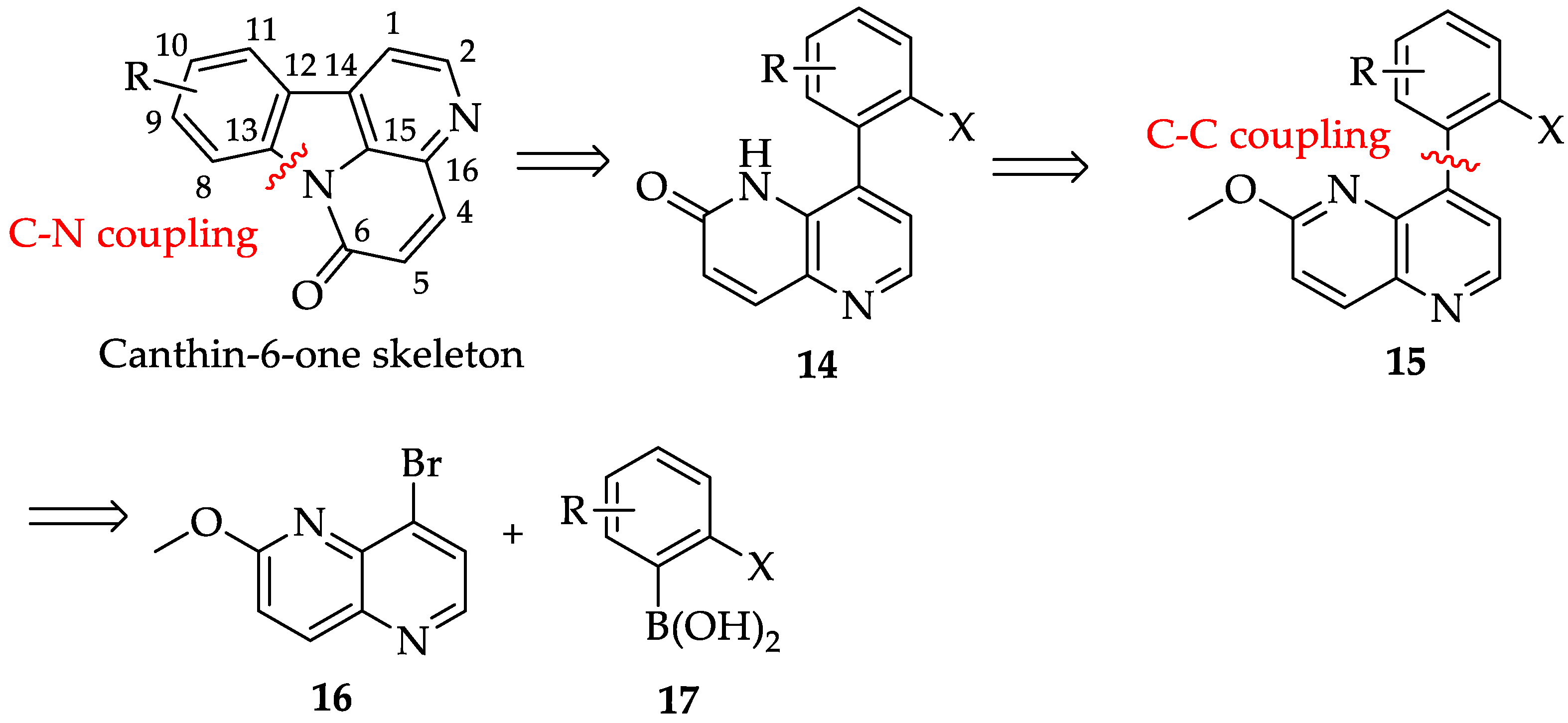
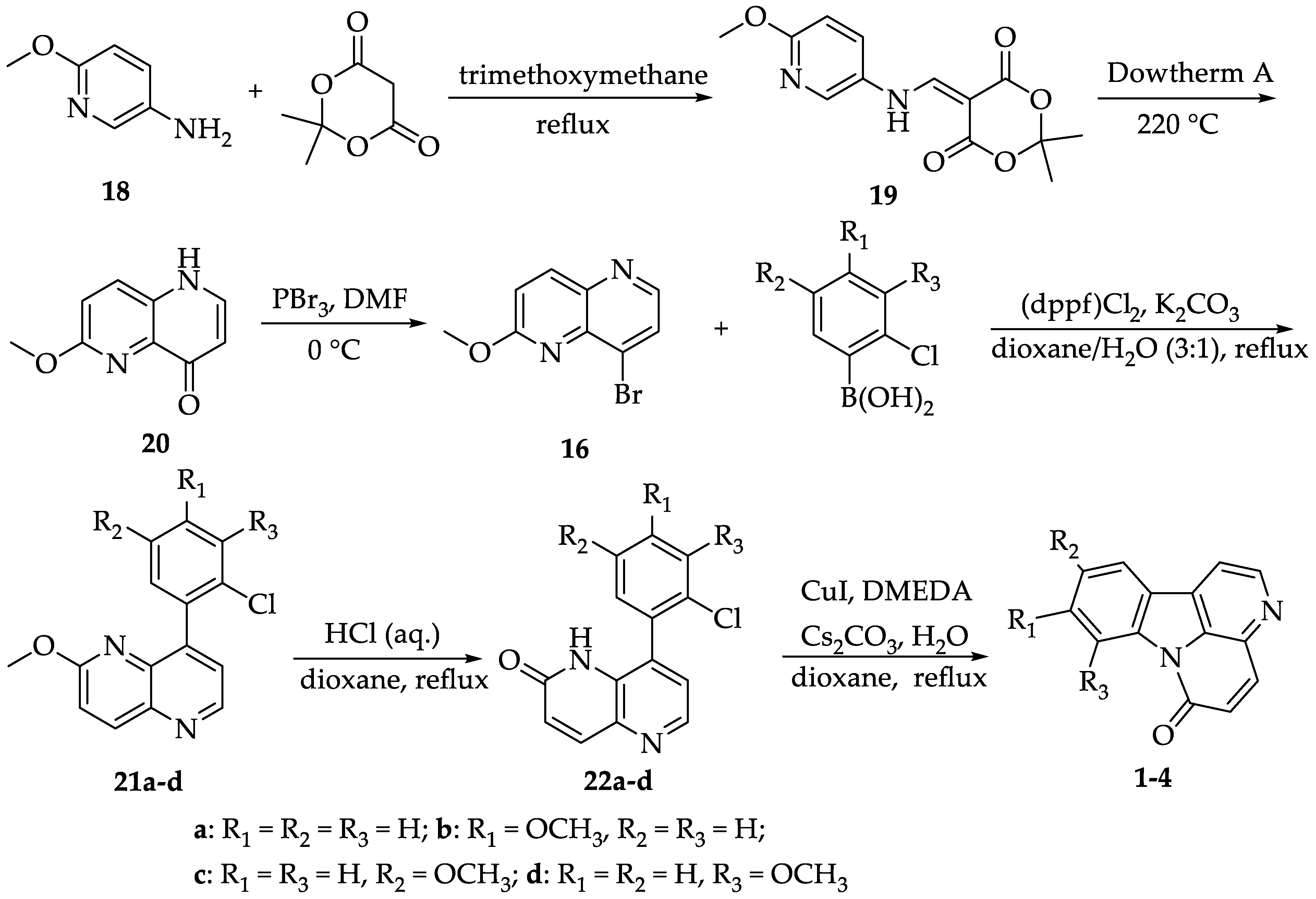
2.1. Synthesis of Canthin-6-Ones 1-4
2.2. Synthesis of Compounds 27-28
2.3. Synthesis of Compounds 5-7, 12-13 and 30-31
2.4. Comparison of NMR Data
2.5. Initial Screening for Antimicrobial Activity
3. Materials and Methods
3.1. General Chemical Procedures
3.2. Synthesis of Target Compounds
3.2.1. Synthesis of Compound 19 [30]
3.2.2. Synthesis of Compound 20 [30]
3.2.3. Synthesis of Compound 16 [30]
3.2.4. Synthesis of Compounds 21
3.2.5. Synthesis of Compounds 22
3.2.6. Synthesis of Compounds 1-4
3.2.7. Synthesis of Compound 25 [31]
3.2.8. Synthesis of Compounds 27 and 28 [32]
3.2.9. Synthesis of Compounds 5-7
3.2.10. Synthesis of Compounds 30
3.2.11. Synthesis of Compounds 12, 13 and 31(c-e)
3.3. Microbroth Dilution Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Dai, W.Q.; He, J.F.; Ye, F.; Xu, J.; Liu, W.Y.; Feng, F.; Qu, W. Design, Synthesis and in Vitro Biological Evaluation of Novel 4-Methoxy-5-Hydroxycanthin-6-One Derivatives as Potential Anti-Tumor Agents. Nat. Prod. Res. 2020, 34, 2289–2294. [Google Scholar] [PubMed]
- Qiu, S.; Sun, H.; Zhang, A.H.; Xu, H.Y.; Yan, G.L.; Han, Y.; Wang, X.J. Natural Alkaloids: Basic Aspects, Biological Roles, and Future Perspectives. Chin. J. Nat. Med. 2014, 12, 401–406. [Google Scholar] [CrossRef]
- Ng, Y.P.; Or, T.C.T.; Ip, N.Y. Plant Alkaloids as Drug Leads for Alzheimer’s Disease. Neurochem. Int. 2015, 89, 260–270. [Google Scholar]
- Thawabteh, A.; Juma, S.; Bader, M.; Karaman, D.; Scrano, L.; Bufo, S.A.; Karaman, R. The biological activity of natural alkaloids against herbivores, cancerous cells and pathogens. Toxins 2019, 11, 656. [Google Scholar] [CrossRef]
- Haynes, H.F.; Nelson, E.R.; Price, J.R. Alkaloids of the Australian Rutaceae—Pentaceras-Australis Hook F. I. Isolation of the Alkaloids and Identification of Canthin-6-One. Aust. J. Sci. Res. Ser. A-Phys. Sci. 1952, 5, 387–400. [Google Scholar] [CrossRef]
- Farouil, L.; Sylvestre, M.; Fournet, A.; Cebrián-Torrejón, G. Review on Canthin-6-One Alkaloids: Distribution, Chemical Aspects and Biological Activities. Eur. J. Med. Chem. Rep. 2022, 5, 21. [Google Scholar] [CrossRef]
- Brahmbhatt, K.G.; Ahmed, N.; Sabde, S.; Mitra, D.; Singh, I.P.; Bhutani, K.K. Synthesis and Evaluation of Β-Carboline Derivatives as Inhibitors of Human Immunodeficiency Virus. Bioorg. Med. Chem. Lett. 2010, 20, 4416–4419. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liu, Y.B.; Li, Y.; Ma, S.G.; Li, L.; Qu, J.; Zhang, D.; Chen, X.G.; Jiang, J.D.; Yu, S.S. Sesquiterpenes and Alkaloids from the Roots of Alangium Chinense. J. Nat. Prod. 2013, 76, 1058–1063. [Google Scholar] [CrossRef]
- Xu, Z.H.; Chang, F.R.; Wang, H.K.; Kashiwada, Y.; McPhail, A.T.; Bastow, K.F.; Tachibana, Y.; Cosentino, M.; Lee, K.H. Anti-Hiv Agents 45 and Antitumor Agents 205: Two New Sesquiterpenes, Leitneridanins A and B, and the Cytotoxic and Anti-Hiv Principles from Leitneria floridana. J. Nat. Prod. 2000, 63, 1712–1715. [Google Scholar] [CrossRef]
- Bharitkar, Y.P.; Hazra, A.; Poduri, N.S.A.; Ash, A.; Maulik, P.R.; Mondal, N.B. Isolation, Structural Elucidation and Cytotoxicity Evaluation of a New Pentahydroxy-Pimarane Diterpenoid Along with Other Chemical Constituents from Aerva Lanata. Nat. Prod. Res. 2015, 29, 253–261. [Google Scholar] [CrossRef]
- Dejos, C.; Voisin, P.; Bernard, M.; Régnacq, M.; Bergès, T. Canthin-6-One Displays Antiproliferative Activity and Causes Accumulation of Cancer Cells in the G2/M Phase. J. Nat. Prod. 2014, 77, 2481–2487. [Google Scholar]
- Rivero-Cruz, J.F.; Chai, H.B.; Kardono, L.B.S.; Setyowati, F.M.; Afriatini, J.J.; Riswan, S.; Farnsworth, N.R.; Cordell, G.A.; Pezzuto, J.M.; Swanson, S.M.; et al. Cytotoxic Constituents of the Twigs and Leaves of Aglaia Rubiginosa. J. Nat. Prod. 2004, 67, 343–347. [Google Scholar]
- Hussain, H.; Al-Harrasi, A.; Al-Rawahi, A.; Green, I.R.; Gibbons, S. Fruitful Decade for Antileishmanial Compounds from 2002 to Late 2011. Chem. Rev. 2014, 114, 10369–10428. [Google Scholar] [PubMed]
- Ferreira, M.E.; Nakayama, H.; de Arias, A.R.; Schinini, A.; de Bilbao, N.V.; Serna, E.; Lagoutte, D.; Soriano-Agatón, F.; Poupon, E.; Hocquemiller, R.; et al. Effects of Canthin-6-One Alkaloids from Zanthoxylum Chiloperon on Trypanosoma Cruzi-Infected Mice. J. Ethnopharmacol. 2007, 109, 258–263. [Google Scholar] [CrossRef]
- Ferreira, M.E.; de Arias, A.R.; de Ortiz, S.T.; Inchausti, A.; Nakayama, H.; Thouvenel, C.; Hocquemiller, R.; Fournet, A. Leishmanicidal Activity of Two Canthin-6-One Alkaloids, Two Major Constituents of Zanthoxylum Chiloperon Var. Angustifolium. J. Ethnopharmacol. 2002, 80, 199–202. [Google Scholar] [PubMed]
- Jiao, W.H.; Gao, H.; Zhao, F.; Lin, H.W.; Pan, Y.M.; Zhou, G.X.; Yao, X.S. Anti-Inflammatory Alkaloids from the Stems of Picrasma Quassioide Bennet. Chem. Pharm. Bull. 2011, 59, 359–364. [Google Scholar]
- Siveen, K.S.; Kuttan, G. Modulation of Humoral Immune Responses and Inhibition of Proinflammatory Cytokines and Nitric Oxide Production by 10-Methoxycanthin-6-One. Immunopharmacol. Immunotoxicol. 2012, 34, 116–125. [Google Scholar] [CrossRef]
- Dai, J.K.; Dan, W.J.; Li, N.; Du, H.T.; Zhang, J.W.; Wang, J.R. Synthesis, in Vitro Antibacterial Activities of a Series of 3-N-Substituted Canthin-6-Ones. Bioorg. Med. Chem. Lett. 2016, 26, 580–583. [Google Scholar] [CrossRef]
- Fournet, A.R.F.M.; Lagoutte, D.; Poupon, E.; Soriano-Agaton, F. Use of Canthin-6-One and Its Analogs in the Treatment of Mycobacteria-Linked Pathologier. U.S. Patent 0059977 A1, 10 March 2011. [Google Scholar]
- O’Donnell, G.; Gibbons, S. Antibacterial Activity of Two Canthin-6-One Alkaloids from Allium Neapolitanum. Phytother. Res. 2007, 21, 653–657. [Google Scholar]
- Soriano-Agatón, F.; Lagoutte, D.; Poupon, E.; Roblot, F.; Fournet, A.; Gantier, J.C.; Hocquemiller, R. Extraction, Hemisynthesis, and Synthesis of Canthin-6-One Analogues.: Evaluation of Their Antifungal Activities. J. Nat. Prod. 2005, 68, 1581–1587. [Google Scholar]
- Lagoutte, D.; Nicolas, V.; Poupon, E.; Fournet, A.; Hocquemiller, R.; Libong, D.; Chaminade, P.; Loiseau, P.M. Antifungal Canthin-6-One Series Accumulate in Lipid Droplets and Affect Fatty Acid Metabolism in Saccharomyces Cerevisiae. Biomed. Pharmacother. 2008, 62, 99–103. [Google Scholar] [PubMed]
- Wang, J.R.; Dai, J.K.; Zhao, F.; Dan, W.J.; Yin, D.Y.; Gao, Y.; Qin, W.J.; Zhang, J.W. Preparation of Canthin-6-One Quaternary Ammonium Salts as Antibacterial Agents. C.N. Patent 104530047A, 16 December 2014. [Google Scholar]
- Zhao, F.; Dai, J.K.; Liu, D.; Wang, S.J.; Wang, J.R. Synthesis and Evaluation of Ester Derivatives of 10-Hydroxycanthin-6-One as Potential Antimicrobial Agents. Molecules 2016, 21, 390. [Google Scholar] [CrossRef]
- Li, N.; Liu, D.; Dai, J.K.; Wang, J.Y.; Wang, J.R. Synthesis and in Vitro Antibacterial Activity of Quaternized 10-Methoxycanthin-6-One Derivatives. Molecules 2019, 24, 1553. [Google Scholar] [CrossRef] [PubMed]
- Dai, J.K.; Dan, W.J.; Li, N.A.; Wang, R.X.; Zhang, Y.; Li, N.N.; Wang, R.Z.; Wang, J.R. Synthesis and Antibacterial Activity of C2 or C5 Modified and D Ring Rejiggered Canthin-6-One Analogues. Food Chem. 2018, 253, 211–220. [Google Scholar] [CrossRef] [PubMed]
- Dai, J.K.; Dan, W.J.; Zhang, Y.Y.; He, M.F.; Wang, J.R. Design and Synthesis of C10 Modified and Ring-Truncated Canthin-6-One Analogues as Effective Membrane-Active Antibacterial Agents. Bioorg. Med. Chem. Lett. 2018, 28, 3123–3128. [Google Scholar]
- Lindsley, C.W.; Wisnoski, D.D.; Wang, Y.; Leister, W.H.; Zhao, Z. A ‘one pot’microwave-mediated synthesis of the basic canthine skeleton: Expedient access to unnatural β-carboline alkaloids. Tetrahedron lett. 2003, 44, 4495–4498. [Google Scholar]
- Gollner, A.; Koutentis, P.A. Two-Step Total Syntheses of Canthin-6-One Alkaloids: New One-Pot Sequential Pd-Catalyzed Suzuki-Miyaura Coupling and Cu-Catalyzed Amidation Reaction. Org. Lett. 2010, 12, 1352–1355. [Google Scholar]
- Kandepedu, N.; Cabrera, D.G.; Eedubilli, S.; Taylor, D.; Brunschwig, C.; Gibhard, L.; Njoroge, M.; Lawrence, N.; Paquet, T.; Eyermann, C.J.; et al. Identification, Characterization, and Optimization of 2,8-Disubstituted-1,5-Naphthyridines as Novel Plasmodium Falciparum Phosphatidylinositol-4-Kinase Inhibitors with in Vivo Efficacy in a Humanized Mouse Model of Malaria. J. Med. Chem. 2018, 61, 5692–5703. [Google Scholar] [CrossRef]
- Michihata, N.; Kaneko, Y.; Kasai, Y.; Tanigawa, K.; Hirokane, T.; Higasa, S.; Yamada, H. High-Yield Total Synthesis of (-)-Strictinin through Intramolecular Coupling of Gallates. J. Org. Chem. 2013, 78, 4319–4328. [Google Scholar]
- Hartman, A.M.; Jumde, V.R.; Elgaher, W.A.; Te Poele, E.M.; Dijkhuizen, L.; Hirsch, A.K. Potential Dental Biofilm Inhibitors: Dynamic Combinatorial Chemistry Affords Sugar-Based Molecules that Target Bacterial Glucosyltransferase. ChemMedChem 2021, 16, 113–123. [Google Scholar]
- Kanchanapoom, T.; Kasai, R.; Chumsri, P.; Hiraga, Y.; Yamasaki, K. Canthin-6-One and Β-Carboline Alkaloids from Eurycoma Harmandiana. Phytochemistry 2001, 56, 383–386. [Google Scholar] [PubMed]
- Song, X.; Gaascht, F.; Schmidt-Dannert, C.; Salomon, C.E. Discovery of Antifungal and Biofilm Preventative Compounds from Mycelial Cultures of a Unique North American Hericium sp. Fungus. Molecules 2020, 25, 963. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| NO. | Compound Names |
|---|---|
| 12 | canthin-6-one-9-O-β-D-glucopyranoside |
| 13 | canthin-6-one-10-O-β-D-glucopyranoside |
| 30a | canthin-6-one-9-O-tetraacetyl-β-D-glucopyranoside |
| 30b | canthin-6-one-10-O-tetraacetyl-β-D-glucopyranoside |
| 30c | canthin-6-one-9-O-tetraacetyl-α-D-galactopyranoside |
| 30d | canthin-6-one-10-O-tetraacetyl-α-D-galactopyranoside |
| 30e | canthin-6-one-10-O-heptaacetyl-β-D-maltoside |
| 31c | canthin-6-one-9-O-α-D-galactopyranoside |
| 31d | canthin-6-one-10-O-α-D-galactopyranoside |
| 31e | canthin-6-one-10-O-β-D-maltoside |
| No. | Natural | Synthetic | Δδ |
|---|---|---|---|
| 1 | 114.3 | 114.4 | 0.1 |
| 2 | 145.9 | 146.1 | 0.2 |
| 4 | 139.9 | 140.1 | 0.2 |
| 5 | 128.2 | 128.4 | 0.2 |
| 6 | 158.9 | 159.0 | 0.1 |
| 8 | 104.5 | 104.6 | 0.1 |
| 9 | 159.7 | 159.8 | 0.1 |
| 10 | 116.5 | 116.7 | 0.2 |
| 11 | 124.4 | 124.6 | 0.2 |
| 12 | 118.2 | 118.4 | 0.2 |
| 13 | 139.9 | 140.1 | 0.2 |
| 14 | 129.3 | 129.5 | 0.2 |
| 15 | 131.8 | 132.0 | 0.2 |
| 16 | 135.3 | 135.4 | 0.1 |
| G-1 | 101.0 | 101.1 | 0.1 |
| G-2 | 73.2 | 73.3 | 0.1 |
| G-3 | 77.2 | 77.2 | 0 |
| G-4 | 69.5 | 69.5 | 0 |
| G-5 | 76.5 | 76.5 | 0 |
| G-6 | 60.5 | 60.5 | 0 |
| Compounds | Average Inhibition Rate (%) | |||
|---|---|---|---|---|
| No. | C. albicans | C. neoformans | S. aureus | E. coli |
| 1 | 95.7 | 96.9 | 99.7 | 15.8 |
| 2 | 58.4 | 76.3 | 99.8 | 14.0 |
| 3 | 39.7 | 57.9 | 88.1 | 4.1 |
| 4 | 55.9 | 96.6 | 46.3 | 1.2 |
| 5 | −13.5 | −37.8 | 10.8 | 5.2 |
| 6 | 7.3 | 35.0 | 14.8 | −0.1 |
| 7 | 81.3 | 83.2 | 98.3 | 13.9 |
| 12 | −3.2 | 22.2 | −9.8 | 5.6 |
| 13 | 1.2 | 4.7 | −5.1 | 1.8 |
| 31c | −2.6 | 13.4 | 0.4 | 4.2 |
| 31d | 0.2 | 0.2 | −3.3 | 4.2 |
| 31e | 1.3 | 0.5 | 0.8 | 1.2 |
| Ketoconazole | 99.9 | 98.2 | ||
| Vanconomycin | 94.6 | |||
| Chloromycin | 99.2 | |||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qi, X.; Jaiswal, Y.; Xie, X.; Fan, Y.; Wu, R.; Su, S.; Guan, Y.; Williams, L.; Song, X. Synthesis and Antimicrobial Activity of Canthin-6-One Alkaloids. Molecules 2025, 30, 1546. https://doi.org/10.3390/molecules30071546
Qi X, Jaiswal Y, Xie X, Fan Y, Wu R, Su S, Guan Y, Williams L, Song X. Synthesis and Antimicrobial Activity of Canthin-6-One Alkaloids. Molecules. 2025; 30(7):1546. https://doi.org/10.3390/molecules30071546
Chicago/Turabian StyleQi, Xubing, Yogini Jaiswal, Xinrong Xie, Yu Fan, Rongping Wu, Shaoyang Su, Yifu Guan, Leonard Williams, and Xun Song. 2025. "Synthesis and Antimicrobial Activity of Canthin-6-One Alkaloids" Molecules 30, no. 7: 1546. https://doi.org/10.3390/molecules30071546
APA StyleQi, X., Jaiswal, Y., Xie, X., Fan, Y., Wu, R., Su, S., Guan, Y., Williams, L., & Song, X. (2025). Synthesis and Antimicrobial Activity of Canthin-6-One Alkaloids. Molecules, 30(7), 1546. https://doi.org/10.3390/molecules30071546