

Decarboxylation-Driven Double Annulations: Innovative Multi-Component Reaction Pathways


Abstract
1. Introduction
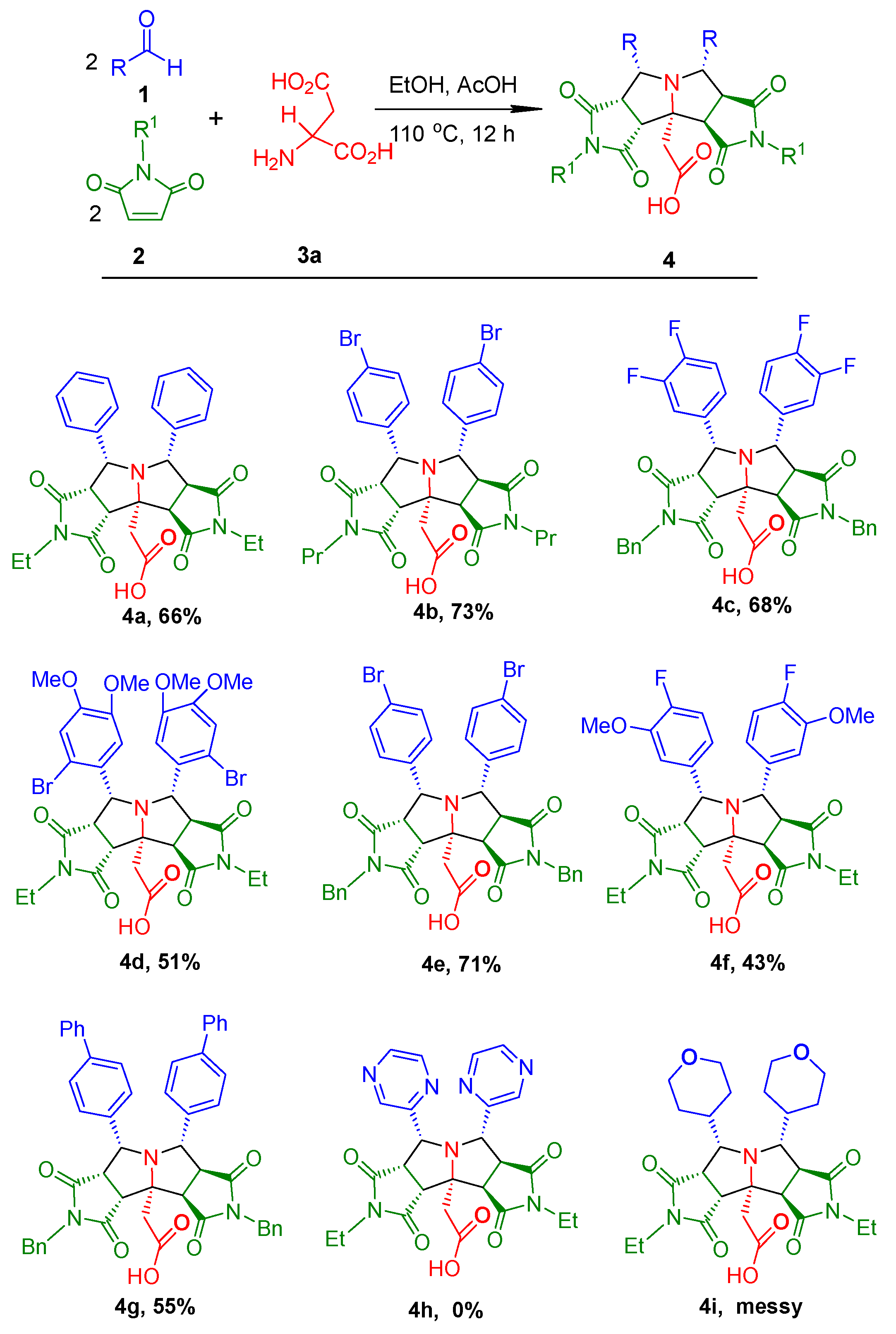
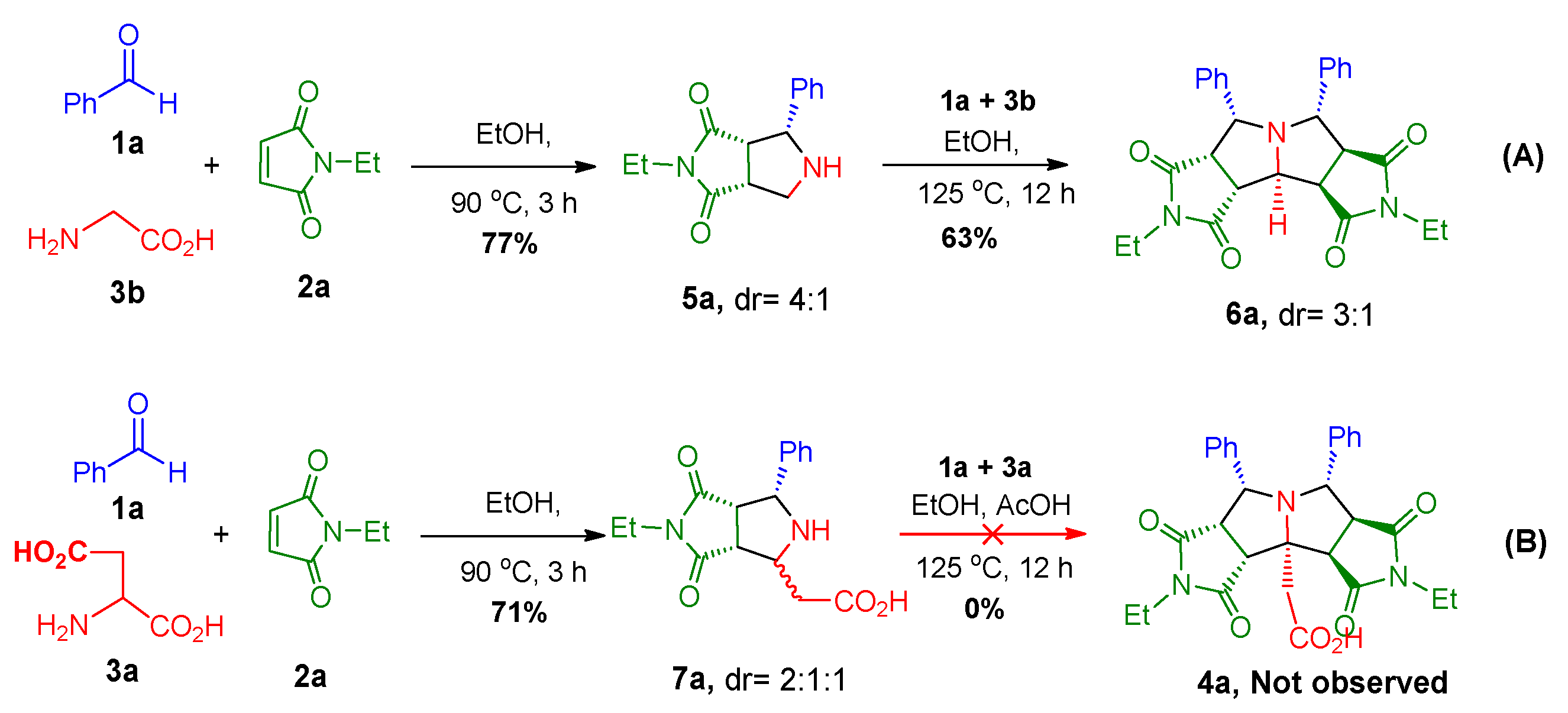
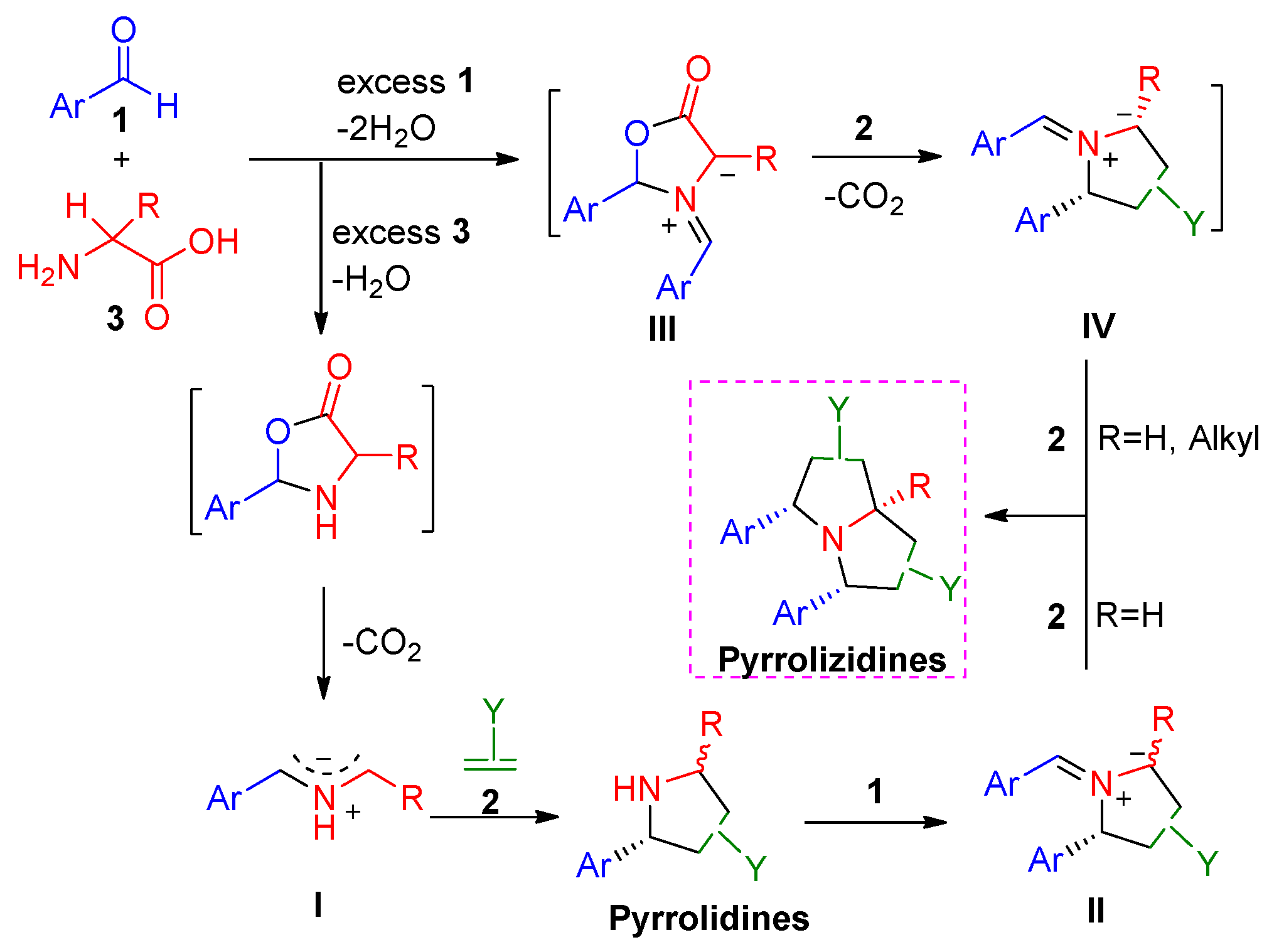
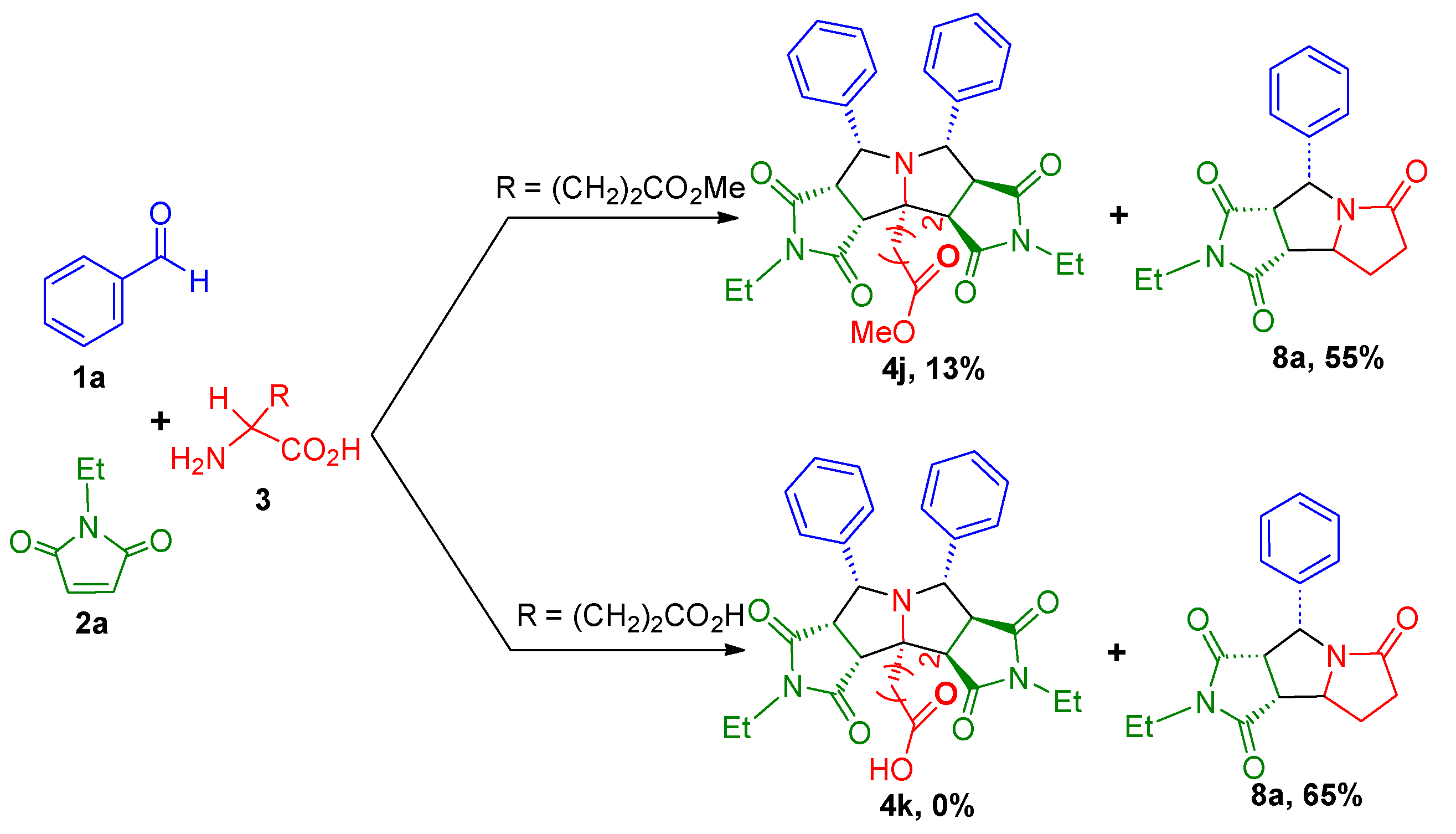
2. Results and Discussion
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Dömling, A.; Wang, W.; Wang, K. Chemistry and Biology of Multicomponent Reactions. Chem. Rev. 2012, 112, 3083–3135. [Google Scholar] [CrossRef] [PubMed]
- Ganem, B. Strategies for Innovation in Multicomponent Reaction Design. Acc. Chem. Res. 2009, 42, 463–472. [Google Scholar] [CrossRef]
- Yadav, M.S.; Rajkhowa, S.; Singh, S.K.; Jaiswal, M.K.; Tiwari, V.K. Multicomponent Click Reaction: An Indispensable Tool for Easy Access of Functionalized 1,2,3-Triazoles. ChemistrySelect 2024, 9, e202400776. [Google Scholar] [CrossRef]
- Ruijter, E.; Orru, R.V.A. Discovery of MCRs. In Multicomponent Reactions in Organic Synthesis, 1st ed.; Zhu, J.P., Wang, Q., Wang, M.X., Eds.; Wiley: Amsterdam, The Netherlands, 2014; pp. 13–38. [Google Scholar] [CrossRef]
- Preeti; Singh, K.N. Multicomponent Reactions: A Sustainable Tool to 1,2- and 1,3-Azoles. Org. Biomol. Chem. 2018, 16, 9084–9116. [Google Scholar] [CrossRef]
- Jiang, B.; Rajale, T.; Wever, W.; Tu, S.J.; Li, G. Multicomponent Reactions for the Synthesis of Heterocycles. Chem. Asian J. 2010, 5, 2318–2335. [Google Scholar] [CrossRef]
- Sikandar, S.; Zahoor, A.F.; Ghaffar, A.; Anjum, M.N.; Noreen, R.; Irfan, A.; Munir, B.; Kotwica-Mojzych, K.; Mojzych, M. Unveiling the Chemistry and Synthetic Potential of Catalytic Cycloaddition Reaction of Allenes: A Review. Molecules 2023, 28, 704. [Google Scholar] [CrossRef] [PubMed]
- Benjamin, H.; Rotstein, B.H.; Zaretsky, S.; Rai, V.; Yudin, A.K. Small Heterocycles in Multicomponent Reactions. Chem. Rev. 2014, 114, 8323–8359. [Google Scholar] [CrossRef]
- John, S.E.; Gulati, S.; Shankaraiah, N. Recent Advances in Multi-Component Reactions and Their Mechanistic Insights: A Triennium Review. Org. Chem. Front. 2021, 8, 4237–4287. [Google Scholar] [CrossRef]
- Moyano, A.; Rios, R. Asymmetric Organocatalytic Cyclization and Cycloaddition Reactions. Chem. Rev. 2011, 111, 4703–4832. [Google Scholar] [CrossRef]
- Appukkuttan, P.; Mehta, V.P.; Van der Eycken, E.V. Microwave-assisted Cycloaddition Reactions. Chem. Soc. Rev. 2010, 39, 1467–1477. [Google Scholar] [CrossRef]
- Nájera, C.; Sansano, J.M.; Yus, M. 1,3-Dipolar Cycloadditions of Azomethine Imines. Org. Biomol. Chem. 2015, 13, 8596–8636. [Google Scholar] [CrossRef] [PubMed]
- Perreault, S.; Rovis, T. Multi-Component Cycloaddition Approaches in the Catalytic Asymmetric Synthesis of Alkaloid Targets. Chem. Soc. Rev. 2009, 38, 3149–3159. [Google Scholar] [CrossRef]
- Zhang, X.; Qiu, W.; Evans, J.; Kaur, M.; Jasinski, J.P.; Zhang, W. Double 1,3-Dipolar Cycloadditions of Two Nonstabilized Azomethine Ylides for Polycyclic Pyrrolidines. Org. Lett. 2019, 21, 2176–2179. [Google Scholar] [CrossRef]
- Zhang, X.; Qiu, W.; Murray, S.A.; Zhan, D.; Evans, J.; Jasinski, J.P.; Wang, X.; Zhang, W. Pseudo-Five-Component Reaction for Diastereoselective Synthesis of Butterfly Shaped Bispiro[Oxindole-Pyrrolidine]s. J. Org. Chem. 2021, 86, 17395–17403. [Google Scholar] [CrossRef] [PubMed]
- Zhan, D.; Yao, B.; Huan, C.; Lu, J.; Ji, Y.; Zhang, X. A Step-Economical and Diastereoselective Synthesis of Dispirooxindole-Pyrrolizines bearing Seven Stereocenters. Tetrahedron Lett. 2023, 126, 154638. [Google Scholar] [CrossRef]
- Cui, P.; Xu, L.; Shi, Z.; Gan, L. Synthesis of Decahydropyrrolo [2,1,5-cd]indolizine through Consecutive [2+3] Cycloadditions and 6-Exo-Trig Cyclization. J. Org. Chem. 2011, 76, 4210–4212. [Google Scholar] [CrossRef]
- Lu, Q.; Song, G.; Jasinski, J.P.; Keeley, A.C.; Zhang, W. One-pot Double [3+2] Cycloaddition for Diastereoselective Synthesis of Tetracyclic Pyrrolidine Compounds. Green Chem. 2012, 14, 3010–3012. [Google Scholar] [CrossRef]
- Veronica Selva, V.; Larrañaga, O.; Castello, L.M.; Najera, C.; Sansano, J.M.; Cozar, A. Diastereoselective [3+2] vs. [4+2] Cycloadditions of Nitroprolinates with α,β-Unsaturated Aldehydes and Electrophilic Alkenes: An Example of Total Periselectivity. J. Org. Chem. 2017, 82, 6298–6312. [Google Scholar] [CrossRef]
- Zhang, X.; Qiu, W.; Ma, X.; Evans, J.; Kaur, M.; Jasinski, J.P.; Zhang, W. One-Pot Double [3+2] Cycloadditions for Diastereoselective Synthesis of Pyrrolidine-Based Polycyclic Systems. J. Org. Chem. 2018, 83, 13536–13542. [Google Scholar] [CrossRef]
- Cantero, T.M.; Silva Junior, P.I.; Negri, G.; Nascimento, R.M.; Mendonca, R.Z. Antimicrobial Activity of Flavonoids Glycosides and Pyrrolizidine Alkaloids from Propolis of Scaptotrigona aff. Postica. Toxin Rev. 2023, 42, 300–315. [Google Scholar] [CrossRef]
- Schramm, S.; Kohler, N.; Rozhon, W. Pyrrolizidine Alkaloids: Biosynthesis, Biological Activities and Occurrence in Crop Plants. Molecules 2019, 24, 498. [Google Scholar] [CrossRef]
- Fayed, M.A.A. Heliotropium; a Genus Rich in Pyrrolizidine Alkaloids: A Systematic Review Following its Phytochemistry and Pharmacology. Phytomed. Plus 2021, 1, 100036. [Google Scholar] [CrossRef]
- Chen, T.; Mei, N.; Fu, P.P. Genotoxicity of pyrrolizidine alkaloids. J. Appl. Toxicol. 2010, 30, 183–196. [Google Scholar] [CrossRef]
- Jayawickreme, K.; Swistak, D.; Ozimek, E.; Reszczynska, E.; Rysiak, A.; Maluch-Kocka, A.; Hanaka, A. Pyrrolizidine Alkaloids—Pros and Cons for Pharmaceutical and Medical Applications. Int. J. Mol. Sci. 2023, 24, 16972. [Google Scholar] [CrossRef] [PubMed]
- Robertson, J.; Stevens, K. Pyrrolizidine Alkaloids: Occurrence, Biology, and Chemical Synthesis. Nat. Prod. Rep. 2017, 34, 62–89. [Google Scholar] [CrossRef]
- Moreira, R.; Pereira, D.M.; Valentão, P.; Andrade, P.B. Pyrrolizidine Alkaloids: Chemistry, Pharmacology, Toxicology and Food Safety. Int. J. Mol. Sci. 2018, 19, 1668. [Google Scholar] [CrossRef]
- Fan, Y.; Wang, M.; Zhang, Q.; Ouyang, S.; Mao, W.; Xu, C.; Wang, M.; Long, C. Traditional Uses, Phytochemistry, Pharmacology, Toxicity and Clinical Application of Traditional Chinese Medicine Cynoglossum Amabile: A Review. Front. Pharmacol. 2024, 15, 1325283. [Google Scholar] [CrossRef]
- Ma, X.; Qiu, W.; Liu, L.; Zhang, X.; Awad, J.; Evans, J.; Zhang, W. Synthesis of Tetrahydropyrrolothiazoles through One-Pot and Four-Component N,S-Acetalation and Decarboxylative [3+2] Cycloaddition. Green Synth. Catal. 2021, 2, 74–77. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, W. PASE Synthesis of Pyrrolidine-Containing Heterocycles Through [3+2] Cycloaddition-Initiated Reactions. Curr. Opin. Green Sustain. Chem. 2018, 11, 65–69. [Google Scholar] [CrossRef]
- Yi, W.B.; Zhang, W. Pot, Atom, and Step Economy (PASE) Synthesis; Springer: Cham, Switzerland, 2019. [Google Scholar]
- Zhang, X.; Ma, X.; Zhang, W. Decarboxylative 1,3-Dipolar Cycloaddition of Amino Acids for the Synthesis of Heterocyclic Compounds. Beilstein J. Org. Chem. 2023, 19, 1677–1693. [Google Scholar] [CrossRef]
- Zhou, T.; Zhang, X.; Zhan, D.; Zhang, W. Glycine-Based [3+2] Cycloaddition for the Synthesis of Pyrrolidine-Containing Polycyclic Compounds. Molecules 2024, 29, 5726. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Ma, L.; Paul, A.; Seidel, D. Direct α-C–H Bond Functionalization of Unprotected Cyclic Amines. Nat. Chem. 2018, 10, 165–169. [Google Scholar] [CrossRef] [PubMed]
- Seidel, D. The Azomethine Ylide Route to Amine C–H Functionalization: Redox-Versions of Classic Reactions and a Pathway to New Transformations. Acc. Chem. Res. 2015, 48, 317–328. [Google Scholar] [CrossRef]
- Pearson, W.H.; Walters, M.A.; Oswell, K.D. Intramolecular 2-Azaallyl Anion Cycloadditions. Application to the Synthesis of Fused Bicyclic Pyrrolidines. J. Am. Chem. Soc. 1986, 108, 2769–2771. [Google Scholar] [CrossRef]
- Vedejs, E.; West, F.G. Ylides by the Desilylation of α.-Silyl Onium Salts. Chem. Rev. 1986, 86, 941–955. [Google Scholar] [CrossRef]
- Clark, R.B.; Pearson, W.H. Nonstabilized N-Unsubstituted Azomethine Ylides: A Synthesis of Indolizidine 239CD. Org. Lett. 1999, 1, 349–351. [Google Scholar] [CrossRef] [PubMed]
- Takano, Y.; Herranz, M.Á.; Martín, N.; Radhakrishnan, S.G.; Guldi, D.M.; Tsuchiya, T.; Nagase, S.; Akasaka, T. Donor−Acceptor Conjugates of Lanthanum Endohedral Metallofullerene and π-Extended Tetrathiafulvalene. J. Am. Chem. Soc. 2010, 132, 8048–8055. [Google Scholar] [CrossRef]
- Hölzel, H.; Haines, P.; Kaur, R.; Lungerich, D.; Jux, N.; Guldi, D.M. Probing Charge Management across the π-Systems of Nanographenes in Regioisomeric Electron Donor–Acceptor Architectures. J. Am. Chem. Soc. 2022, 144, 8977–8986. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
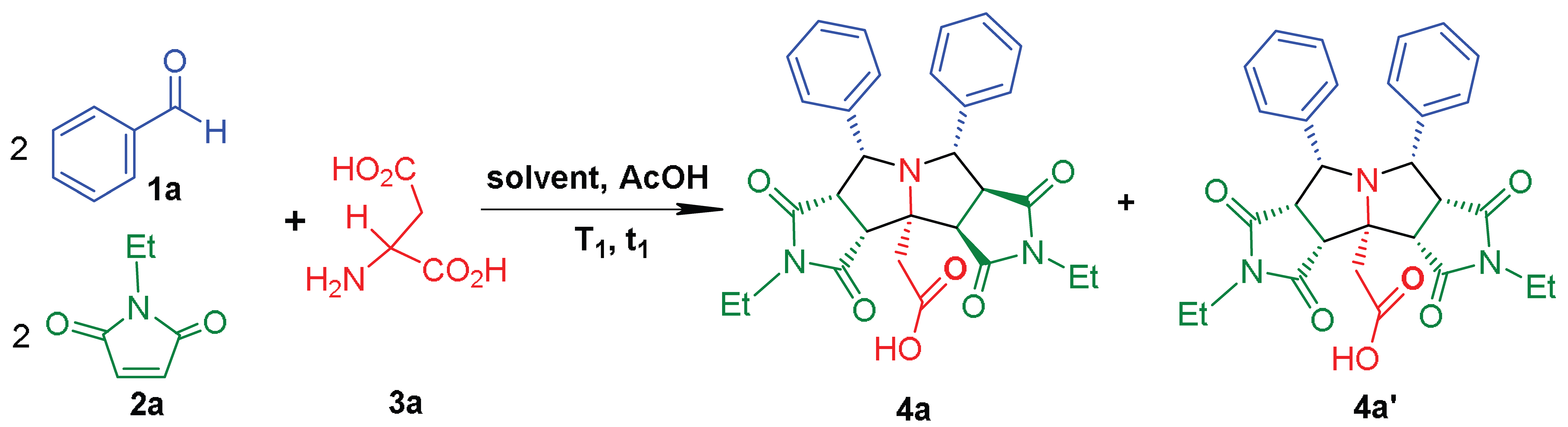
| Entry | Solvent | T1 (°C) | t1 (h) | 4a (%) b | Dr c |
|---|---|---|---|---|---|
| 1 | EtOH | 90 | 6 | 53 | 4:1 |
| 2 | EtOH | 90 | 12 | 60 | 4:1 |
| 3 | EtOH | 110 | 12 | 71 (66) | 4:1 |
| 4 | EtOH | 125 | 12 | 63 | 4:1 |
| 5 d | EtOH | 110 | 12 | 55 | 3:1 |
| 6 | MeCN | 110 | 12 | 46 | 4:1 |
| 7 | MeOH | 85 | 24 | 39 | 3:1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhan, D.; Yang, G.; Zhou, T.; Nallapati, S.; Zhang, X. Decarboxylation-Driven Double Annulations: Innovative Multi-Component Reaction Pathways. Molecules 2025, 30, 1594. https://doi.org/10.3390/molecules30071594
Zhan D, Yang G, Zhou T, Nallapati S, Zhang X. Decarboxylation-Driven Double Annulations: Innovative Multi-Component Reaction Pathways. Molecules. 2025; 30(7):1594. https://doi.org/10.3390/molecules30071594
Chicago/Turabian StyleZhan, Desheng, Gang Yang, Tieli Zhou, Sashirekha Nallapati, and Xiaofeng Zhang. 2025. "Decarboxylation-Driven Double Annulations: Innovative Multi-Component Reaction Pathways" Molecules 30, no. 7: 1594. https://doi.org/10.3390/molecules30071594
APA StyleZhan, D., Yang, G., Zhou, T., Nallapati, S., & Zhang, X. (2025). Decarboxylation-Driven Double Annulations: Innovative Multi-Component Reaction Pathways. Molecules, 30(7), 1594. https://doi.org/10.3390/molecules30071594